






 , Khizra Jabeen 1, Tariq Aziz 2,*, Muhammad Saad Mughal 1, Hammad Arif 1, Metab Alharbi 3, Thamer H Albakeiri 3, Abdullah F. Alasmari 3
, Khizra Jabeen 1, Tariq Aziz 2,*, Muhammad Saad Mughal 1, Hammad Arif 1, Metab Alharbi 3, Thamer H Albakeiri 3, Abdullah F. Alasmari 31 Department of Biotechnology, Faculty of Science and Technology, University of Central Punjab, 54590 Lahore, Pakistan
2 Department of Agriculture University of Ioannina, 47100 Arta, Greece
3 Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia
Abstract
Background: Pneumocystis jirovecii is the most emerging
life-threating health problem that causes acute and fatal pneumonia infection. It
is rare and more contagious for patients with leukemia and immune-deficiency
disorders. Until now there is no treatment available for this infection
therefore, it is needed to develop any treatment against this pathogen.
Methods: In this work, we used comparative proteomics, robust
immune-informatics, and reverse vaccinology to create an mRNA vaccine against
Pneumocystis jirovecii by targeting outer and transmembrane proteins.
Using a comparative subtractive proteomic analysis of two Pneumocystis
jirovecii proteomes, a distinct non-redundant Pneumocystis jirovecii
(strain SE8) proteome was chosen. Seven Pneumocystis jirovecii
transmembrane proteins were chosen from this proteome based on hydrophilicity,
essentiality, virulence, antigenicity, pathway interaction, protein-protein
network analysis, and allergenicity. Objective: The reverse vaccinology
approach was used to predict the immunogenic and antigenic epitopes of major
histocompatibility complex (MHC) I, II and B-cells from the selected proteins on
the basis of their antigenicity, toxicity and allergenicity. These immunogenic
epitopes were linked together to construct the mRNA-based vaccine. To enhance the
immunogenicity, suitable adjuvant, linkers (GPGPG, KK, and CYY), and PRDRE
sequences were used. Results: Through predictive modeling and
confirmation via the Ramachandran plot, we assessed secondary and 3D structures.
The adjuvant RpfE was incorporated to enhance the vaccine construct’s
immunogenicity (GRAVY index: –0.271, instability index: 39.53, antigenicity:
1.0428). The physiochemical profiling of vaccine construct was predicted it an
antigenic, efficient, and potential vaccine. Notably, strong interactions were
observed between the vaccine construct and TLR-3/TLR-4 (–1301.7 kcal/mol
Keywords
- proteome
- immune-deficiency
- transmembrane
- pathway
- reverse vaccinology
- mRNA
- profiling
- in-vitro
Pneumocystis jirovecii a unicellular protozoon that predominantly a yeast like fungus belongs to Ascomycota phylum and Pneumocystidaceae family. According to phylogenetic studies Pneumocystis jirovecii previously analyzed as Pneumocystis carinii. The first genome sequence of Pneumocystis jirovecii without virulence factor was assembled in 2012. Pneumocystis jirovecii caused acute and fatal pneumonia infection. Mostly in premature infants that have weak immune system due to malnutrition. Pneumocystis jirovecii is rare but a life threatening for the patients of leukemia and immunodeficiency disorders. From the literature it is confirmed that the main target of causes Pneumocystis pneumonia (PCP) is immune compromised host. This pathogen is specialized to colonize in human lungs in alveoli. Pneumocystis priory it was first recognized in 1940 cause pneumonia in immunocompromised individuals but this disease awakend during 1980 worldwide epidemic of HIV [1].
This an airborne transmission disease propagate person to person. The researcher
finds out that after the experiment in which they detect the air sample 5 million
from the infected person. The risk of spreading increase as the colonized
pneumocystis travel through nosocomial space [2]. The research finds out that the
main reservoir of Pneumocystis jirovecii is infected human beings. Some
researchers noted that by pneumocystis also attack in real region during
Transplant. Mostly it found in the pulmonary alveoli in human lungs [3]. PCP
divided into different stages trophozoite means the fungus situate in clusters
[4, 5]. The pre cystic form called as sporozoite means which pneumocystis present
in spores form with a cyst. The CD4
Anti-pneumocystis agents like Trimethoprim/sulfamethoxazole (TMP-SMX), it is
considered as the first line agent that used intravenously in hospital in
specialized room [9, 10]. Alternative treatments include dapsone, pentamidine,
atovaquone, clindamycin, primaquine, and echinocandins [11]. For HIV-positive
patients, adjunctive corticosteroids have proven beneficial in reducing mortality
[12]. Molecular techniques identify mutations in the dihydropteroate synthase
gene, indicating potential sulfa drug resistance. But the pills are also taken by
patient who is mildly affected by fungus [13]. These all medicine are taken
during the diagnosis. The role of CD4
Currently, the therapeutic strategy targets the host organism’s membrane since it contains several antigens and there is little chance of the pathogen reverting to its virulent state, as well as few unwanted effects as compared to live attenuated or dead whole cell vaccination [17]. Subtractive proteomics and reverse vaccinology are powerful strategies for identifying antigenic and immunogenic proteins that might be employed as chimeric vaccine candidates. The subtractive technique eliminates pathogen transmembrane proteins that play a role in virulence or include an essentiality component that is necessary for Pneumocystis jirovecii survival but not present in the human host. This work is based on a key purpose to develop a novel mRNA-based vaccination against N. fowleri. Although, to the best of our knowledge, an mRNA-based vaccine against Pneumocystis jirovecii has not been reported yet, these candidates have amazing potential against pathogenic infections. mRNA vaccines offer rapid development potential against evolving pathogens. Additionally, they don’t integrate into the genome, posing no risk of altering genetic information. Antigenic and immunogenic epitopes of all vaccine targets will be examined using various approaches once they have been shortlisted. The structural configuration of the vaccine is predicted, followed by its docking with the host’s immune molecules, specifically TLR-3 and TLR-4. To ensure the stability of the vaccine complex, molecular dynamic modeling techniques are employed.
We downloaded proteomes of pneumocystis pneumonia of 2 different strains Pneumocystis jirovecii (Human pneumocystis pneumonia agent) (SE8) and Pneumocystis jirovecii (strain RU7) (Human pneumocystis pneumonia agent) (RU7) from the UNIPROT server (www.uniprot.org). For both of these strains, redundancy and non-redundancy analyses were carried out [18]. The Reference genome of Pneumocystis jirovecii (strain SE8) consists of 3419 proteins which are then analyzed to shortlist antigen, allergen, and immunogenic proteins.
In addition to the role of proteins in active transport, outer membrane proteins (OMPs) also play an important role in the pathogenicity of fungus, the permeability barrier of the outer membrane (OM), efflux pumps, and the diffusion of nutrients as well as outer membrane proteins (OMPs) have a role in the preservation of the structural integrity of the membrane. Due to their antigenic and immunogenic properties, fungal OMPs are very versatile and have been selected for use in the production of mRNA vaccines. To get a complete subtractive analysis, the first thing we did was predict the cellular localization of 3419 proteins by using PSORT-II. This allowed us to have a more in-depth look at the data. This software is able to classify the localization of proteins as being extracellular, outer membrane, cytoplasmic, periplasmic, or inner membrane.
The candidates for the vaccination that are homologous to proteins found in other organisms are being investigated as a potential target. It is possible that the selection of these protein targets will reduce the likelihood of cross-reactivity in the host cell. We carried out a BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) study of 982 proteins in the National Center for Biotechnology Information (NCBI) database against the proteome of the host Homo sapiens in order to remove homologous sequences between the human host and the pathogen [19]. If the E-value (expectation value) was 104 or less, the hits were deemed to be homologous proteins, and they were discarded from the study.
We were able to validate the proteins localization within the cell by utilizing two different servers at the same time (PSORT-II). Through the utilization of Signal P 4.037, we were able to generate hypotheses on the secretary function of 918 non-human comparable proteins [20]. Those forecasts turned out to be spot on. The presence of N-terminal signal sequences allowed for the prediction of proteins that were going to be exported from the inner membrane to the periplasm in accordance with the sec pathway, which is also known as the secretory pathway. This prediction was made possible because the sec pathway is also known as the secretory pathway. Because the sec route is also known as the secretory pathway, this prediction was made possible by the fact that it has both names. Following the narrowing down of the available alternatives, the proteins that generated a strong signal were selected for further analysis.
The non-classical pathway is responsible for transporting proteins from the periplasm to the extracellular region, whereas the classical system moves proteins from the cytosol to the periplasm. Proteins that are released through non-classical pathways do not have signal peptides, although they can still be identified in the extracellular region of fungus. With the assistance of the programme Secretome P 2.0 (https://services.healthtech.dtu.dk/services/SecretomeP-2.0/), 50 proteins were omitted from the analysis due to the non-classical secreted extracellular proteins they contained [21]. It was determined that the proteins with a SecP score of 0.5 or higher were non-classically secreted. Using the Ensembl database (https://www.ensembl.org/index.html), BLAST was performed on the finalized 10 transmembrane proteins chosen, non-redundant proteomes.
The experimentally identified essential proteins are included in the database of
essential genes (DEG), which was used to establish which pathogens require which
essential proteins [22]. In DEG, a BLASTp search was carried out using a cut-off
of 1*10
Virulence factors that are connected with transmembrane proteins play a
significant part in the interaction between the host and the pathogen and affect
the host’s defence system. The VFDB database (http://www.mgc.ac.cn/VFs/search_VFs.htm) is utilised in order to identify
OMPs that are linked with virulence [24]. The 7 Pneumocystis jirovecii
proteins that were identified were BLASTp against the VFDB protein core dataset
(R1) with an E-value of 1 × 10
The Kyoto encyclopaedia of genes and genome’s (KEGG) pathway database provides an explanation of the function of proteins within several metabolic pathways [25]. In order to determine which metabolic pathways are specific to the human host and which are shared with pneumocystis pneumonia, we have manually compared all of the enumerated metabolic pathways. The 7 proteins were separated into two groups, one for their role in unique, pathogen-specific pathways, and the other for their role in common, shared pathways between the pathogen and the host. We have also determined the fundamental role of these 7 proteins using either analysis relying on pathways or analysis independent of pathways.
Using a comparative subtractive proteomics technique, we were able to exclude 7 of P. jirovecii most pathogenic proteins from its 3419 total protein sequences, as previously studied. These seven OMPs were taken into consideration as possible targets for the construction of an mRNA vaccine. To begin, an antigenicity analysis was performed with the use of the VaxiJen server [26]. Antigenic proteins were determined to be those that had a score value greater than 0.5, and these proteins were taken into consideration for further investigation.
The diverse protein expressions in the fungal cell will be affected by the mRNA vaccination that is directed against the proteins of the outer- and trans membrane. These vaccinations may have an effect on the intra-species interaction partners, which would result in a reduction in the pathogenesis of a Pneumocystis jirovecii infection. The entire outer membrane protein-protein intra-species interaction analysis was carried out with the help of the protein interaction database STRING (https://string-db.org/) in order to locate the proteins network for identified OMPs [27].
When pathogenic proteins enter the proteasomal complex of the host cell, they naturally form epitopes. T-cells respond to MHC class I epitopes to activate the immune response against the pathogen. In order to find MHC -Peptide complexes that can generate an immune response in the host cell, we used major histocompatibility (MHC) class I (MHC I) immunogenicity prediction tool, the IEDB website (http://tools.immuneepitope.org/immunogenicity/). The epitopes that were finally selected for further investigation were examined using this server with the default settings. Prediction tools’ other parameters were left at their default values. For further investigation, we focused on the epitopes that received a high score for their immunogenicity. Every single one of the outer membrane and transmembrane proteins was evaluated using IEDB prediction algorithms separately. The best-predicted protein epitopes were chosen for further analysis.
Epitopes that attach to MHC class II molecules can be identified using the MHC Class II prediction tool http://tools.iedb.org/mhcii/[28]. The Helper T-Lymphocytes epitopes were predicted under the MHC-II IEDB server NetMHCpan BA 4.1 module by selecting full set human allele with the default values. Stabilization matrix alignment and the average relative binding matrix were the two consensus methods used to determine which epitopes of MHC II would be the most effective.
Antigenicity and toxicity were investigated in MHC I and II epitopes. Both
analyses made use of the VaxiJen 2.0 and ToxinPred tools (https://webs.iiitd.edu.in/raghava/toxinpred/) [29]. The
physicochemical features of VaxiJen servers (
The recognition of epitopes by B cells is necessary to start the process of humoral immunity. ABCPred server, were used to make predictions on B-cell epitopes [30]. The shortlisted proteins were delivered with threshold of 0.51. The epitopes that were chosen were further take into consideration in the vaccine construction process.
In order to build an mRNA vaccine that is targeted to OMPs and transmembrane proteins, we connected all of the selected epitopes (HTL, CTL, and B epitopes) using amino acid linkers GGGS, GPGPG, HEYGAEALERAG, and CYY Adjuvant ‘EAAK’ was added at the N-terminus of the vaccine construct which improved their immunogenicity. In order for the mRNA vaccine to be effective, it needs to have a Kozak sequence. In addition, two structures known as tPA (P00750) and MITD (Q8WV92) were inserted into the construct’s 5’ region and the mRNA vaccine’s 3’ locus, respectively. All of the proteins that make up adjuvants act as agonists to various TLR complexes, which polarises CTL responses and has a powerful immunomodulating effect [31].
The vaccine construct was evaluated on the basis of their antigenicity,
allergenicity, and solubility prediction methods in order to check that vaccine
design would be most appropriate for use. The vaccine was run through the AlgPred
server so that we could determine how allergic the vaccine was. Both ANTIGENpro
and the VaxiJen 2.0 server were used to make the antigenicity prediction for
vaccine constructions. Using the SOLpro service, we were able to forecast the
solubility of vaccine construction probability (
The Expasy ProtParam server was utilised in order to define the physiochemical qualities of the vaccine constructions [32]. This site is utilised by a large number of people in order to calculate the number of amino acids, PI values, molecular weight, aliphatic index, instability index, and hydropathicity GRAVY values. The protein’s instability index serves as a predictor of the protein’s stability. The thermostability of vaccines can be understood through their aliphatic index values. The hydrophilic or hydrophobic properties of a protein can be deduced from its GRAVY [33].
For the prediction of the secondary structure of the vaccine, PSIPRED server is used [34]. This server predicts the alpha, beta helix and the coil structure of the protein with the accuracy of 81.6%.
For Molecular Docking, the vaccine three-dimensional structure using Robetta. The Procheck, ERRAT, and Ramachandran Plot, at the saves website of University of California Los Angeles (UCLA, https://saves.mbi.ucla.edu/) were utilized to verify the proteins’ estimated structural analyses. The alleles were downloaded from Protein Data Bank PDB RCSB. Cluspro was used for the docking of vaccine with TLR-3 and TLR-4 complex [35]. For the Molecular Dynamics Simulation, I-Mod was used [36].
The proteomes of several strains of Pneumocystis jirovecii were retrieved through the UniPort service, which has a list of two strains. Manual redundancy and non-redundancy assessments were performed on the proteomes of both Pneumocystis jirovecii strains. In this work, the proteome of Pneumocystis jirovecii (strain SE8) is used as a reference proteome.
The UniPort server was used to download the protein sequences from the whole proteome of Pneumocystis jirovecii (strain SE8). There are a total of 3419 proteins in this strain of the Pneumocystis jirovecii. The UniPort Id for this Pneumocystis jirovecii strain SE8 is UP000010422. Table 1 shows the proteins that are extracted from the whole proteome of this strain.
| Protein name | Protein ID | Antigenicity | Cellular component | Function | Pathway |
| Uncharacterized protein | L0PI24* | 0.9982 | Small ribosomal subunit | Ribosome binding | - |
| Uncharacterized protein | L0PAX6** | 0.6030 | Nuclear | - | - |
| ER membrane protein complex subunit 2 | L0PCV9* | 0.7660 | EMC complex | Enables the energy-independent insertion into ER | - |
| tRNA (guanine-N (7)-)-methyltransferase | L0PCI0* | 0.5484 | Nucleus | Phosphatidylinositol binding, tRNA (guanine-n7-)-methyltransferase activity, trna binding | tRNA modification; n7-methylguanine-trna biosynthesis |
| Phosphatidylserine decarboxylase proenzyme 1 | L0PAD9* | 0.7257 | Integral component of mitochondrial inner membrane | Phosphatidylserine decarboxylase activity, phosphatidylethanolamine biosynthetic process, protein auto processing | Lipid metabolism |
| Cytochrome c oxidase subunit 1 | L0PH87* | 0.5143 | Integral component of membrane, mitochondrial inner membrane, respiratory chain complex IV | Cytochrome-c oxidase activity, heme binding, metal ion binding, oxidative phosphorylation | Energy metabolism; oxidative phosphorylation |
| Deoxyhypusine hydroxylase | L0PGD6* | 0.5054 | Cytoplasm, nucleus | Deoxyhypusine monooxygenase activity, metal ion binding, peptidyl-lysine modification to peptidyl-hypusine | Protein modification; eif5a hypusination |
Allergenicity: non-allergen. Virulence Factor: Yes. DEG Analysis. *Yes. **No.
From the reference proteome, the potential proteins for vaccine design can be selected by filtering the 3419 proteins on the base of their subcellular location. By using PSORT-II, we filtered 884 proteins out of 3419 proteins of the Pneumocystis jirovecii (strain SE8). These proteins are present in the sub-cellular location of the fungal cell. To avoid vaccination cross-reactivity with human host cells, the effective vaccine should be assembled against non-human homologous proteins. These non-human homologous fungal proteins were used to construct a vaccine design that only interacts with Pneumocystis jirovecii proteins.
The selection of potential proteins depends on their antigenicity and allergenicity of the proteins from Pneumocystis jirovecii fungus were considered as displayed in Table 1. As vaccine targets, essential proteins will have a significant impact on the fungus, hence we examined targeting proteins using the DEG server as displayed in Table 1. One protein, L0PAX6, an uncharacterized protein, is thought to be crucial for the survival of the Pneumocystis jirovecii strain.
STRING predictions of these selected proteins of Pneumocystis jiroveciishow various intra-species interactions between the proteins were revealed. We may be able to grasp the protein networking of proteins with the aid of all interaction studies. An mRNA vaccine against these proteins may affect all interacting network proteins, increasing the vaccine’s efficacy, all protein networking is presented in Fig. 1, except the second uncharacterized protein with UniPort Id L0PAX6.
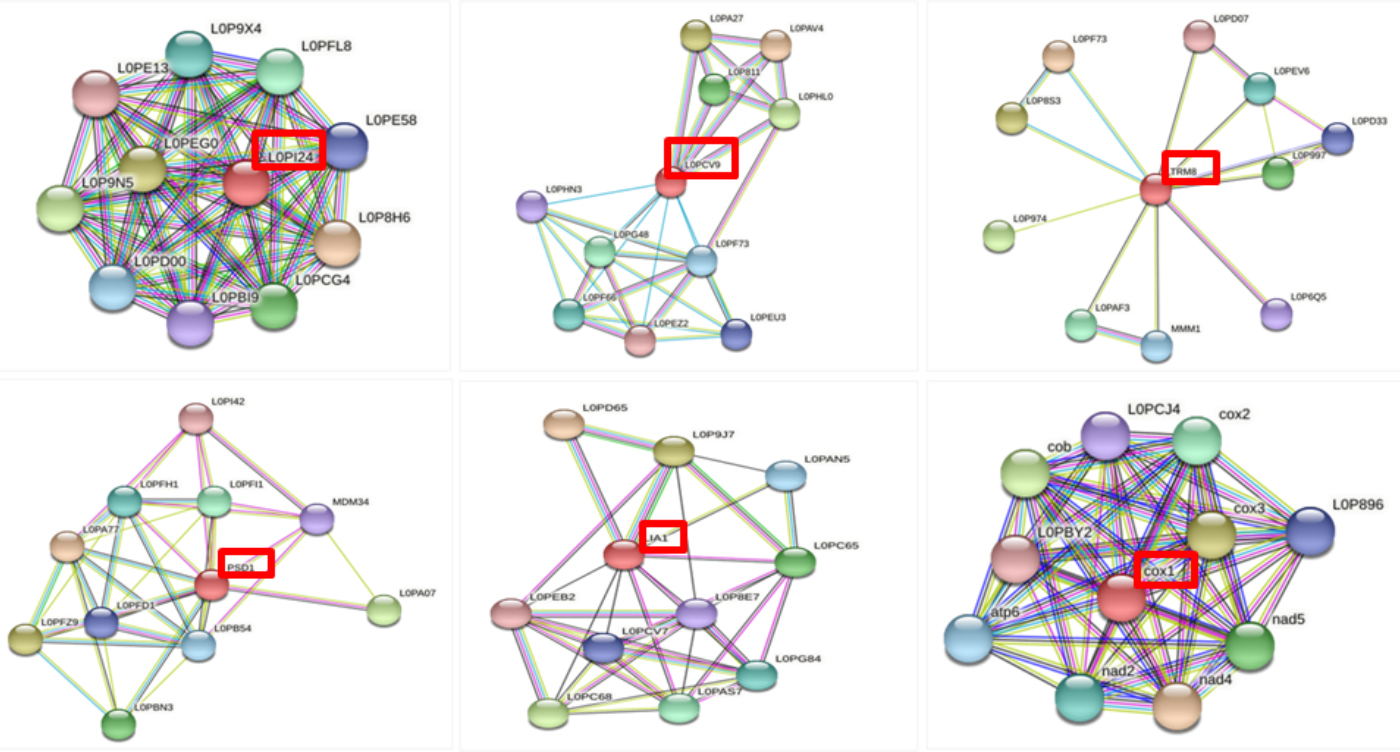 Fig. 1.
Fig. 1.STRING interaction of targeted proteins.
An mRNA vaccine against the virulence factor-associated proteins will boost the protein’s potency and effectiveness. The DFVF (Database of Virulence Factors in Fungal Pathogens) analysis in Table 1 revealed that all selected proteins are associated with Pneumocystis jirovecii virulence. As a result, these proteins are regarded as a crucial target for inhibiting Pneumocystis jirovecii pathogenesis.
To shortlist the identified proteins involved in various pathways, a manual comparison was performed. These findings uncover seven distinct pathogen pathways prevalent in both pathogen and host Pneumocystis jirovecii. As demonstrated in Table 1, four of the seven identified proteins were found to be metabolic pathway dependent.
The IEDB server was used for the binding prediction of MHC-I of 7 selected proteins. For analysis, epitopes with a greater affinity (IC 50 nM) and a good percentile rank (0.2) were chosen along with ANN predicted method. To increase accuracy, we eliminated epitopes with Inhibitory Concentration 50 (IC50) values smaller than 500. 80 MHC class I epitopes from various sources were tested for class I immunogenicity, allergenicity, and toxicity. A high immunogenicity score indicated the ability to excite T cells as well as promote cell-mediated immunity. In the immunogenicity prediction, the antigenicity score of selected 14 epitopes ranges from 3.0104 to 0.5226 as shown in Table 2.
| Protein ID | Epitopes | Antigenicity | Alleles |
| L0PI24 | ALVFSPNRYW | 2.5163 | HLA-B*57:01, HLA-B*58:01, HLA-A*32:01, HLA-B*53:01, HLA-A*30:02, HLA-B*15:01, HLA-B*44:02, HLA-B*44:03, HLA-A*26:01, HLA-A*30:01, HLA-A*23:01, HLA-A*31:01, HLA-A*68:01, HLA-A*24:02 |
| LVFSPNRYW | 3.0104 | HLA-B*57:01, HLA-B*58:01, HLA-A*32:01, HLA-B*53:01, HLA-A*26:01, HLA-B*15:01, HLA-A*23:01, HLA-A*30:02, HLA-B*35:01, HLA-A*30:01, HLA-B*51:01 | |
| L0PAX6 | RQYTPALRRF | 1.6189 | HLA-B*15:01, HLA-A*24:02, HLA-A*23:01, HLA-A*32:01, HLA-A*30:02, HLA-B*57:01, HLA-A*03:01, HLA-B*58:01, HLA-A*31:01, HLA-A*30:01, HLA-A*02:06, HLA-B*44:03, HLA-B*44:02, HLA-B*40:01, HLA-A*26:01, HLA-A*11:01, HLA-A*01:01 |
| DTSLMGSWMR | 1.581 | HLA-A*68:01, HLA-A*33:01, HLA-A*31:01, HLA-A*26:01, HLA-A*11:01, HLA-A*01:01, HLA-A*68:02, HLA-A*03:01 | |
| L0PCV9 | LYNNFLQGF | 0.9157 | HLA-A*24:02, HLA-A*23:01, HLA-A*30:02, HLA-B*57:01, HLA-A*32:01, HLA-B*15:01, HLA-B*58:01, HLA-A*26:01, HLA-A*33:01, HLA-B*08:01, HLA-A*31:01 |
| AELADIYFSF | 0.8934 | HLA-B*44:03, HLA-B*44:02, HLA-B*40:01, HLA-A*32:01, HLA-B*15:01, HLA-A*23:01, HLA-A*24:02, HLA-A*26:01, HLA-B*57:01, HLA-A*30:02, HLA-A*02:06, HLA-A*02:01 | |
| L0PCI0 | LYIIEIQKF | 1.0115 | HLA-A*24:02, HLA-A*23:01, HLA-B*57:01, HLA-B*58:01, HLA-B*53:01 |
| HPAVTFFSF | 0.7660 | HLA-B*35:01, HLA-B*53:01, HLA-B*07:02, HLA-B*51:01, HLA-B*08:01, HLA-A*26:01, HLA-A*23:01, HLA-A*24:02, HLA-B*57:01, HLA-B*58:01, HLA-B*15:01 | |
| L0PAD9 | RMISGRLMW | 1.2732 | HLA-A*32:01, HLA-B*57:01, HLA-B*58:01, HLA-A*23:01, HLA-B*15:01, HLA-A*24:02, HLA-A*30:02, HLA-A*30:01, HLA-B*53:01, HLA-A*31:01, HLA-A*03:01 |
| WVVERRRYF | 2.7421 | HLA-B*08:01, HLA-B*15:01, HLA-B*57:01, HLA-A*32:01, HLA-B*07:02, HLA-B*35:01, HLA-A*30:02, HLA-A*30:01, HLA-B*58:01, HLA-B*53:01, HLA-A*33:01 | |
| L0PH87 | DEKLGHLHFW | 0.5226 | HLA-B*44:03, HLA-B*44:02, HLA-B*57:01, HLA-B*53:01, HLA-A*26:01, HLA-B*35:01, HLA-B*08:01, HLA-B*15:01, HLA-A*01:01 |
| YLQGDNQLY | 1.0261 | HLA-A*01:01, HLA-B*15:01, HLA-A*30:02, HLA-B*35:01, HLA-A*26:01, HLA-A*32:01, HLA-B*58:01, HLA-A*02:01, HLA-A*02:06, HLA-B*57:01, HLA-A*03:01 | |
| L0PGD6 | SEQCKTETL | 0.745 | HLA-B*40:01, HLA-B*44:02, HLA-B*44:03, HLA-B*08:01, HLA-B*07:02, HLA-A*02:06, HLA-B*51:01, HLA-A*24:02, HLA-B*15:01, HLA-A*23:01, HLA-B*35:01, HLA-A*26:01, HLA-A*68:02 |
| KEFPLGERF | 2.7011 | HLA-B*44:03, HLA-B*44:02, HLA-B*40:01, HLA-B*15:01, HLA-A*23:01, HLA-A*30:02, HLA-A*32:01, HLA-B*57:01, HLA-A*24:02, HLA-B*53:01, HLA-B*58:01, HLA-B*35:01, HLA-A*02:06 |
Allergenicity: non-allergen.
Pneumocystis jirovecii seven selected proteins were evaluated to predict the potential MHC-II epitopes. The IEDB server was used to test MHC-II binding predictions for all seven identified proteins. 14 epitopes were chosen for vaccine design based on their percentile rank and IC50 value (50 nM). As stated in Table 3, only those epitopes that were studied as antigenic, non-toxic, and non-allergenic were chosen.
| Protein ID | Epitopes | Antigenicity | Alleles |
| L0PI24 | SKHLYSLDACDIINA | 1.2014 | HLA-DRB3*01:01, HLA-DRB1*12:01, HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DRB1*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB1*04:05, |
| CISLCWSADGQTLFS | 2.2866 | HLA-DRB3*01:01, HLA-DRB1*03:01, HLA-DRB1*15:01, HLA-DRB1*09:01 | |
| L0PAX6 | SKHLYSLDACDIINA | 1.2014 | HLA-DRB3*01:01, HLA-DRB1*12:01, HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DRB1*01:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB1*04:05 |
| KHLYSLDACDIINAL | 1.0204 | HLA-DRB3*01:01, HLA-DRB1*12:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB1*01:01, HLA-DRB1*04:05 | |
| L0PCV9 | YSELLLLQPFSHLIH | 0.7997 | HLA-DPA1*03:01/DPB1*04:02, HLA-DRB4*01:01, HLA-DRB1*04:05, HLA-DPA1*01:03/DPB1*04:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DRB1*15:01, HLA-DRB1*12:01, HLA-DRB1*01:01 |
| SELLLLQPFSHLIHA | 1.2347 | HLA-DPA1*03:01/DPB1*04:02, HLA-DPA1*03:01/DPB1*04:02, HLA-DRB1*04:05, HLA-DPA1*01:03/DPB1*04:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DRB1*15:01, HLA-DRB1*01:01, HLA-DRB1*12:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DRB1*07:01, HLA-DRB1*11:01 | |
| L0PCI0 | ELRIFLSQQNSTLPS | 1.6884 | HLA-DRB1*04:01, HLA-DRB4*01:01, HLA-DRB1*04:05, HLA-DRB1*01:01, HLA-DRB1*15:01, HLA-DRB5*01:01, HLA-DRB1*08:02, HLA-DRB3*02:02, HLA-DPA1*02:01/DPB1*01:01, HLA-DRB1*07:01, HLA-DRB1*13:02 |
| NISVIRMNAMKFLPN | 0.7368 | HLA-DRB1*13:02, HLA-DRB1*12:01, HLA-DRB3*02:02, HLA-DRB4*01:01, HLA-DRB1*15:01, HLA-DRB1*09:01, HLA-DRB5*01:01, HLA-DPA1*02:01/DPB1*05:01, HLA-DRB1*11:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DRB1*07:01 | |
| L0PAD9 | ERRRYFSGELFSVSP | 2.1696 | HLA-DPA1*01:03/DPB1*04:01, HLA-DPA1*02:01/DPB1*14:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DPA1*02:01/DPB1*05:01 |
| EFEFLVSKGSRILMG | 1.5542 | HLA-DRB1*07:01, HLA-DRB3*02:02, HLA-DRB1*09:01, HLA-DRB1*01:01, HLA-DPA1*02:01/DPB1*14:01, HLA-DRB5*01:01, HLA-DQA1*01:02/DQB1*06:02, HLA-DRB1*13:02 | |
| L0PH87 | PRWIPDYPDAFAQWN | 1.3535 | HLA-DRB3*01:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DRB1*03:01, HLA-DQA1*01:01/DQB1*05:01 |
| AHFHYVLSMGAVFAL | 0.7846 | HLA-DRB1*01:01, HLA-DRB1*09:01, HLA-DRB3*02:02, HLA-DPA1*02:01/DPB1*14:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*11:01 | |
| L0PGD6 | PRWIPDYPDAFAQWN | 1.3535 | HLA-DRB3*01:01, HLA-DQA1*05:01/DQB1*02:01, HLA-DRB1*03:01 |
| AHFHYVLSMGAVFAL | 0.7846 | HLA-DRB1*01:01, HLA-DRB1*09:01, HLA-DRB3*02:02, HLA-DPA1*02:01/DPB1*14:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*11:01, HLA-DRB1*15:01, HLA-DRB3*01:01 |
Allergenicity: non-allergen.
Using the ABCpred server, we identified the top two epitopes from each targeted protein sequence. Furthermore, we solely considered antigenic, non-toxic, and non-allergenic epitopes for vaccine development. Table 4 indicates that we have chosen 14 B-cell epitopes to include in the vaccine design, beginning with the targeted proteins.
| Protein ID | Epitopes | Position | Score | Antigenicity score |
| L0PI24 | NAVTVSPDGSLCASGGKDGT | 37 | 0.85 | 1.0174 |
| GKDGTVMLWDLNESKHLYSL | 52 | 83 | 2.1442 | |
| L0PAX6 | MGSGGLCADRAVGGGAACGL | 80 | 0.83 | 1.3838 |
| SVSGAISPLCVEPLIQLRDT | 3 | 0.81 | 0.8821 | |
| L0PCV9 | DLLHIATKEYLRSIELYNNF | 135 | 0.82 | 0.8525 |
| MNESNIPISKRRIALLRSLG | 43 | 0.79 | 0.7262 | |
| L0PCI0 | EMSTDNLRLPIHDSQLISEA | 281 | 0.94 | 1.5147 |
| KEDVSTVFETYFSENLLNIS | 661 | 0.93 | 1.1422 | |
| L0PAD9 | IFEAPQEFEFLVSKGSRILM | 439 | 0.89 | 0.5693 |
| YFAKRISNLFVLNERVVLLG | 343 | 0.89 | 0.7478 | |
| L0PH87 | YVLSMGAVFALLAAWYFWSP | 379 | 0.92 | 0.7310 |
| SLDVALHDTYYVVAHFHYVL | 362 | 0.87 | 0.952 | |
| L0PGD6 | DKNRVIRESCLLGNNRHVSL | 282 | 0.86 | 1.0017 |
| QICSPLSVPSLSKILANPSE | 233 | 0.83 | 0.8769 |
The proposed construct of mRNA vaccine illustrated in Fig. 2, displayed the
vaccine from N-terminal to C-terminal is: 5
 Fig. 2.
Fig. 2.The layout of mRNA vaccine construct.
The VaxiJen server was used to predict the antigenicity of Vaccine design. The
antigenicity of Vaccine constructs 1.0428, and non-allergen and non-toxic. The
vaccine is expected to be antigenic, non-allergenic, non-toxic, and soluble.
Using SOLpro, a vaccine design demonstrated excellent solubility (
| Physiochemical Profiling | Measurement | Indication |
| Number of Amino Acid | 798 | Appropriate |
| Number of Atoms | 12,498 | - |
| Molecular Weight | 89,979.85 | Appropriate |
| Formula | C4122H6178N1046O1109S43 | - |
| Theoretical PI | 8.88 | Basic |
| Total number of negatively charged residues (Asp + Glu) | 61 | - |
| Total number of positively charged residues (Arg + Lys) | 82 | - |
| Instability Index (II) | 39.53 | Stable |
| Aliphatic index | 80.68 | Thermostable |
| Grand average of hydropathicity (GRAVY) | –0.271 | Hydrophilic |
| Antigenicity (by VaxiJen) | 1.0428 | Antigenic |
| Allergenicity | Non-Allergenic | Non-Allergen |
| Toxicity | Non-Toxic | Non-Toxic |
| Solubility | 0.965145 | Soluble |
PSIPRED was used to predict the secondary structures of the final vaccine constructions. Fig. 3 depicts the secondary structure of the vaccine design. The vaccine construct has an alpha-helix, a beta-sheet, and a beta-turn structure. The Rosetta server predicted the vaccination model’s tertiary structure, which was verified by the Ramachandran plot.
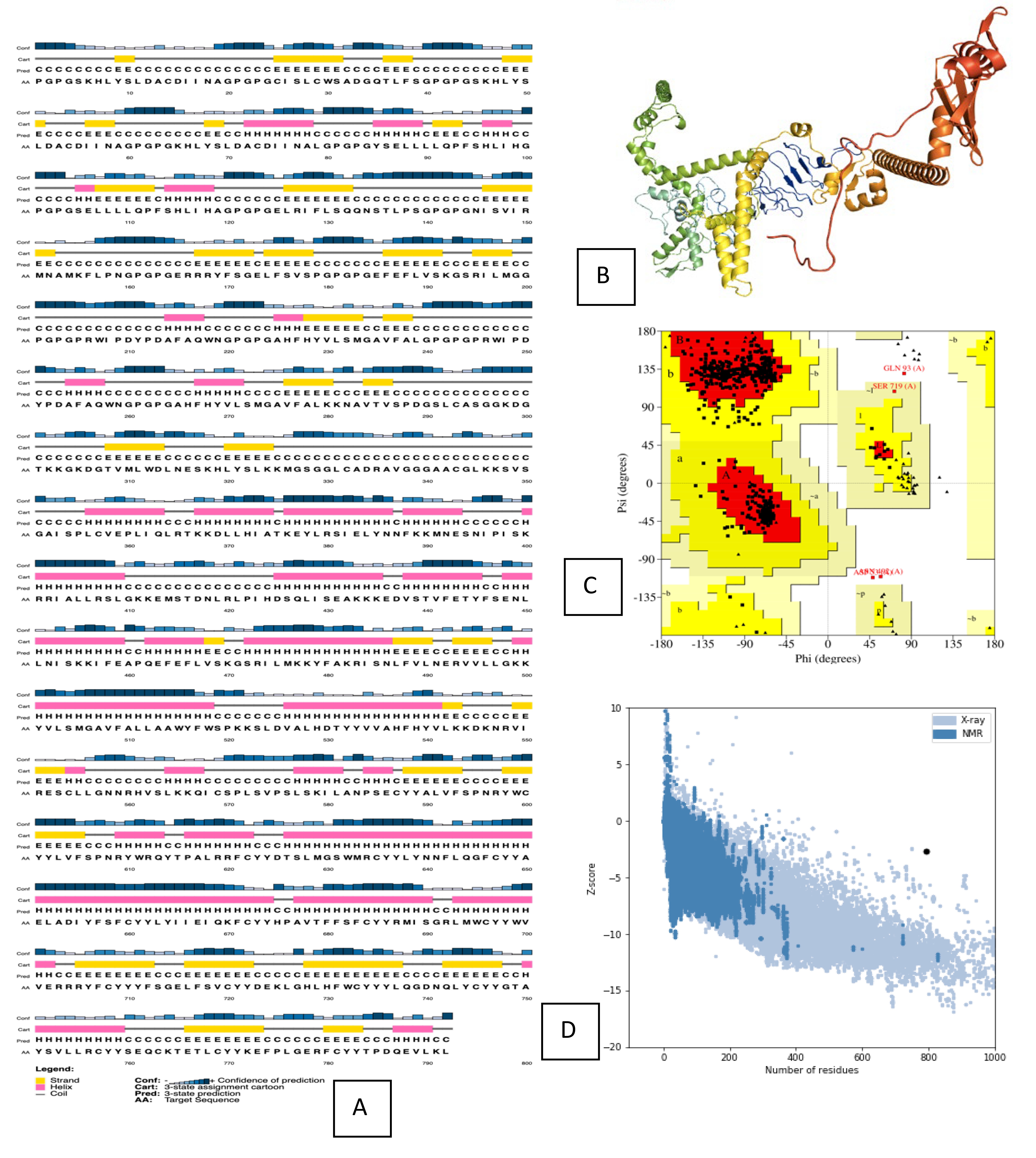 Fig. 3.
Fig. 3.Structure prediction and validation of vaccine construct. (A) PSIPRED server results of secondary structure of vaccine. (B) The Robetta server used to predict the Tertiary structure of vaccine. (C) The PROCHECK server used to analyze the Ramachandran Plot. (D) Z-score analysed by the Pro-SA webserver.
A vaccine can interact with various HLA-alleles found in human populations. The final vaccine design was docked with 5 distinct HLA-allele proteins, as indicated in Table 6. We investigated the vaccine design created against Pneumocystis jirovecii and established its applicability.
| Type of Lymphocytes | MHC Alleles | Epitope | PDB ID |
| CLT | HLA-B*57:01 | ALVFSPNRYW | 5VUF |
| HLA-A*01:01 | RQYTPALRRF | 4NQX | |
| HLA-B*44:03 | AELADIYFSF | 3KPN | |
| HLT | HLA-DRB1*01:01 | SKHLYSLDACDIINA | 2FSE |
| HLA-DRB1*15:01 | CISLCWSADGQTLFS | 1BX2 |
The projected protein model folding produces a conformational B-cell epitope. The discontinuous B-cell Epitopes were analyzed using the ElliPro server. The prediction score for all 392 residues ranged from 0.557 to 0.824. Fig. 4 shows 2D and 3D representations of conformational B-cell epitopes.
 Fig. 4.
Fig. 4.The ElliPro server of IEBD database for the prediction of seven conformational B-cell epitopes. 3D models of B-cells epitopes where yellow spheres present the conformational B-cell epitopes. (A) 10 residues with a score of 0.675. (B) 42 residues with a score of 0.802. (C) 35 residues with a score of 0.819. (D) 20 residues with a score of 0.568. (E) 13 residues with a score of 0.634. (F) 51 residues with a score of 0.615. (G) 11 residues with a score of 0.579.
We used ClusPro tool, for the determination of molecular docking and analyzing
the interaction among the vaccine construct and receptors of TLR-3 and TLR-4. For
the TLR-3-vaccine complex, the binding affinity was –1301.7 kcal/mol
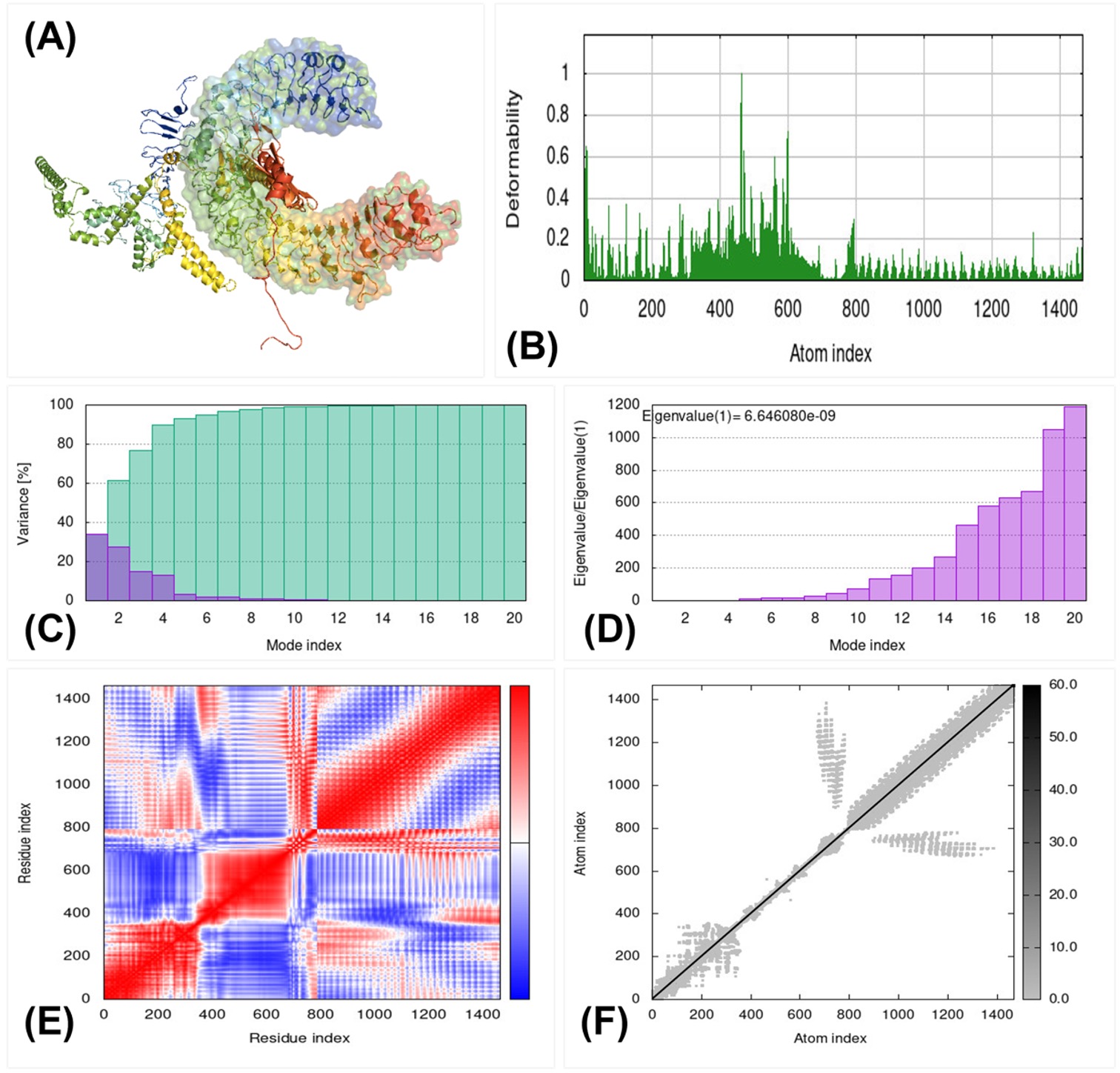 Fig. 5.
Fig. 5.Molecular dynamics simulation, Normal Mode Analysis, and receptor-ligand interactions. (A) Vaccine-TLR4 docked complex using the Cluspro server. (B) Deformability graph. (C) Eigenvalue of vaccine-TLR4 complex. (D) B-factor graph. (E) Covariance matrix. (F) Elastic network model using the iMODS server.
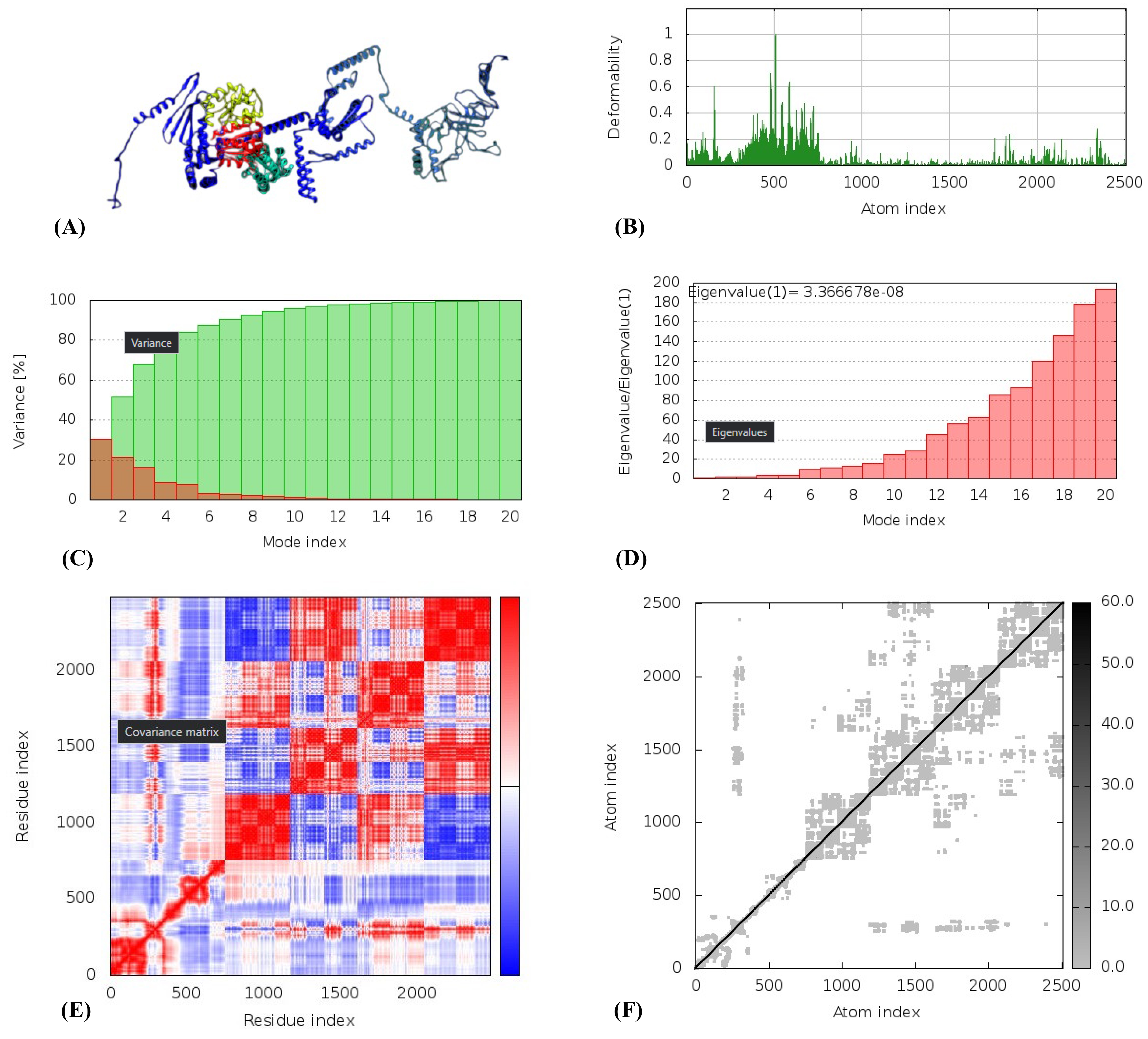 Fig. 6.
Fig. 6.Molecular dynamics simulation of TLR-4 vaccine complex, Normal Mode Analysis, and receptor-ligand interactions. (A) Vaccine-TLR4 docked complex using the Cluspro server. (B) Deformability graph. (C) Eigenvalue of vaccine-TLR4 complex. (D) B-factor graph. (E) Covariance matrix. (F) Elastic network model using the iMODS server.
The molecular dynamic simulation of docked vaccine-TLR-3 and TLR-4 was performed using the iMODS server. The deformability graph, as shown in Fig. 5B and Fig. 6B, examines the deformable loci. The graphic depicts the B-factor graph, which was used to evaluate protein flexibility using the Normal Mode Analysis (NMA) technique. Fig. 5C and Fig. 6C depicts the complex eigenvalues, which signify a low deformation index, increased stiffness, and increased stability. The Fig. 5D and Fig. 6D depicts the link between the amino acid duplets that disperse in the dynamical area. The white color in Fig. 5E and Fig. 6E represents anti-correlated amino acids, the red color represents correlated amino acids, the blue color indicates atom pairs joined by springs, and the grey color in Fig. 5F and Fig. 6F represents the stiffer area.
Pneumocystis jirovecii Pneumonia is the most malignant disease for the infants and elder patients that are immune-compromised due to malnutrition. The mortality rate of this deadly infection is 25% on total average but 74% in patient that are treated in intense care ward. Previously, individuals with AIDS or other immune deficiencies have been highly susceptible to this lethal fungal infection. Currently, treatment options are limited antibiotic, Trimethoprim/sulfamethoxazole, with the risk of antibiotic resistance due to overuse or misuse. This situation underscores the urgent need for a vaccine to prevent PCP. Despite this, the situation is critical, particularly for infants and elderly patients with weakened immune systems due to malnutrition. Unlike drugs, a preventive vaccine could potentially offer long-term immunity by inducing the production of antibodies that can last for several months. In this context, the development of a preventive vaccine against PCP is crucial, as it would offer more effective and lasting protection compared to existing drug treatments by pre-emptively stimulating the immune system [37].
According to pervious research immune-informatics tools and dry lab activities proved that there is big way to design a multiple epitope vaccine under the shed of mRNA-based vaccine designing principal. Prior researches noted that the experiment of mRNA-based vaccine development against many diseases like zika virus, rabies and HIV etc. This approach is really a good inattentive. But alter on the there is one issue associated with mRNA-based vaccine [38]. The manuscript has been updated to reflect that the antibodies induced by RNA vaccines, including the one in our study, may exhibit varying degrees of specificity. While primarily targeted against the specific antigen presented by the vaccine, these antibodies can also demonstrate cross-reactivity with other antigens. The exact specificity and potential cross-reactivity are subject to further experimental validation, as indicated in our studyf [39].
The vaccine design focuses on controlling outbreaks and reducing disease severity. T-cell epitopes play a vital role in this regard, enhancing the immune system’s ability to respond effectively to the pathogen. We joined predicted B-cell and T-cell epitopes using linkers like EAAAK, GPGPG, and AAY, based on previous studies. Physiochemical properties, antigenicity, allergenicity, and toxicity were analyzed [40]. The RpfE adjuvant, a TLR3 and TLR4 agonist, was used to boost immunogenicity, enhancing antigen processing by APCs. RpfE’s protein nature allows for structural modifications for optimal immunogenicity and reduced toxicity, ensuring efficient adjuvant-antigen co-delivery and robust activation of immune responses [41].
The reasons behind the development of vaccine are to establish reliable memory. This memory will activate B lymphocytes and T cell quickly, when the antigen tries to enter in the host body [42]. Epitope are the crucial region that, have to select from the certain antigen. Efficacy of every vaccine depending on it. Here the importance of hyper T lymphocytes is increase because they generate the IL-4 and IL-10 anti-inflammatory Cytokines, chemokines shown by HT-lymphocytes on antigen presenting cells eradication of parasite, after the eradication all immune cell except memory cells undergoes death [43].
In this study, we compared the non-redundant SE8 strain to the Pneumocystis jirovecii reference proteome. Using the Pneumocystis jirovecii SE8 strain as a reference proteome, 3419 proteins were examined using several subtractive methods. We narrowed down the proteome in the subtractive analysis based on cellular localization and transmembrane analysis. Following the shortlisting, we discovered seven antigenic proteins. These seven proteins were characterized as essential using the DEG server, as virulence factors using VFDB, and as pathway-dependent and independent using the KEGG database. These seven proteins were chosen for reverse vaccinology-based mRNA-based vaccine design. Various servers were used to identify antigenic, non-allergenic, nontoxic MHC I, MHC II, and B cell epitopes. To boost immune responses, all of the identified epitopes were combined with various adjuvants and linkers. These vaccine designs were then tested for antigenicity, toxicity, and allergenicity. The vaccine, which is non-toxic and non-allergenic, might be effective immunotherapy against the pathogenic Pneumocystis jirovecii. To boost immune response, we used several adjuvants [44]. We also included adjuvant L7/L12 ribosomal protein, Pan-DR sequence, and linker sequences, as well as multi-epitope base mRNA sequences, which may boost the considerable Pneumocystis jirovecii-specific immune responses. Dunne emphasized that the Immunosuppressed patients had higher TLR4 surface expression compared to non-immunosuppressed adults [45]. Consequently, our approach to molecular docking with TLR3 and TLR4 involves computationally predicting and validating these binding interactions as part of the vaccine design process. In summary, we have considered probable critical aspects that may promote immunogenicity and feasibility of the mRNA vaccine design.
The current investigation found that the vaccine design had excellent physicochemical properties and immunological responses to Pneumocystis jirovecii. Using acknowledged immune-informatics techniques, it was discovered that this vaccination would induce an immunological response against Pneumocystis jirovecii in the host. The result provided show that by employing diverse comparative proteomics, robust immunoinformatics, and reverse vaccinology methodologies, progressive prioritizing of the proteome is an efficient means of rapidly overcoming the adverse effect of Pneumocystis jirovecii. The hypothetical proteins and uncharacterized proteins might lead to the identification of promising candidates of the vaccine and drive there in the annotation. As a result, we merged this method into an mRNA-based vaccine shown as a potential antigenic vaccination. Immunogenic, physicochemical, and structural features imply that our vaccine candidate might produce encouraging results in in vitro and in vivo studies in the near future.
All the data generated in this research work has been included in this manuscript.
Conceptualization, MN, and TA; methodology, KJ, MSM, and HA; software, KJ, MSM, and HA; validation, MN, and MA; formal analysis, KJ, MSM, and HA; investigation, KJ, MSM, THA, and HA; resources, MN, and MA; data curation, MN, and MA; writing—original draft preparation, KJ, MSM, and THA; writing—review and editing, MN, and MA; visualization, MA and AFA; supervision, MN; project administration, MA and AFA. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors greatly acknowlegde and express their gratitude to Researchers Supporting Project number (RSPD2024R568), King Saud University, Riyadh, Saudi Arabia.
The research is supported by Researchers Supporting Project number (RSPD2024R568), King Saud University, Riyadh, Saudi Arabia.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.