








1 Laboratory of Psychiatry and Experimental Alzheimer’s Research, Medical University of Innsbruck, 6020 Innsbruck, Austria
Abstract
Background: Alzheimer’s disease is characterized by extracellular beta-amyloid plaques, intraneuronal tau neurofibrillary tangles and excessive neurodegeneration. The mechanisms of neuron degeneration and the potential of these neurons to form new nerve fibers for compensation remain elusive. The present study aimed to evaluate the impact of beta-amyloid and tau on new formations of nerve fibers from mouse organotypic brain slices connected to collagen-based microcontact prints. Methods: Organotypic brain slices of postnatal day 8–10 wild-type mice were connected to established collagen-based microcontact prints loaded with polyornithine to enhance nerve fiber outgrowth. Human beta-amyloid(42) or P301S mutated aggregated tau was co-loaded to the prints. Nerve fibers were immunohistochemically stained with neurofilament antibodies. The physiological activity of outgrown neurites was tested with neurotracer MiniRuby, voltage-sensitive dye FluoVolt, and calcium-sensitive dye Rhod-4. Results: Immunohistochemical staining revealed newly formed nerve fibers extending along the prints derived from the brain slices. While collagen-only microcontact prints stimulated nerve fiber growth, those loaded with polyornithine significantly enhanced nerve fiber outgrowth. Beta-amyloid(42) significantly increased the neurofilament-positive nerve fibers, while tau had only a weak effect. MiniRuby crystals, retrogradely transported along these newly formed nerve fibers, reached the hippocampus, while FluoVolt and Rhod-4 monitored electrical activity in newly formed nerve fibers. Conclusions: Our data provide evidence that intact nerve fibers can form along collagen-based microcontact prints from mouse brain slices. The Alzheimer’s peptide beta-amyloid(42) stimulates this growth, hinting at a neuroprotective function when physiologically active. This “brain-on-chip” model may offer a platform for screening bioactive factors or testing drug effects on nerve fiber growth.
Keywords
- microcontact printing
- organotypic brain slices
- neurite guidance
- collagen
- polyornithine
- beta-amyloid
- tau
- Alzheimer's disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder
representing one of the most prevalent forms of dementia [1]. A critical aspect
of AD pathology is the vulnerability of neurons to dysfunction and degeneration,
ultimately contributing to the characteristic cognitive decline observed in
affected individuals. The exact etiology of AD remains complex and
multifactorial, involving genetic, environmental and age-related factors [2]. At
a microscopic level, AD is identified by the combined presence of two abnormally
aggregated proteins: extracellular beta-amyloid (A
For studying neurodegenerative processes, in vivo mouse models [14] or
in vitro cell cultures are of relevance. Induced pluripotent stem cells
have greatly advanced in vitro models of neurological diseases over the
past decade, particularly in relation to human cells [15]. However, ex
vivo organotypic brain slice cultures offer a valuable approach that combines
the manipulability of in vitro models with the physiologic integrity of
in vivo models. Unlike other in vitro cultures, organotypic
brain slice cultures provide access to all cell types within a three-dimensional,
cytoarchitecturally intact 150 µm thick brain slice [16]. In terms
of the 3R’s of animal research, organotypic brain slice cultures substantially
reduce the need of animal experiments because multiple slices can be obtained
from one brain, depending on slice thickness and brain region of interest.
Additionally, it enables to investigate multiple variables within a single
system, potentially reducing variability in experimental setups. Numerous
research groups [17], including our own [16], commonly employ the membrane
interface technique in neuroscience research. This technique involves culturing
brain tissue regions of interest on a semipermeable membrane interface between a
humidified atmosphere (maintained at 37 °C and 5% CO
Organizing neurons in a spatial manner in vitro hold even broader significance across various applications, encompassing fundamental research, toxicology testing, pharmaceutical screening, and the development of neuronal implant interfaces. Microcontact printing is an attractive patterning technique to engineer neurons and neurites along defined patterns in µm size [18]. This technique is referred to as soft lithography method since a soft elastomeric and hence more biocompatible stamp with a pattern is used to mirror the corresponding pattern onto a substrate [19]. Various biological substances can be precisely patterned and used for the delivery of diverse proteins. Collagen hydrogel crosslinked with 4arm-poly(ethylene glycole) (PEG) is a well-established system [20]. Despite collagen being found only in basement membrane of the vasculature in the brain, it is widely utilized as a biomaterial to promote the growth of neurites [20, 21, 22]. The extracellular matrix protein collagen exerts both biochemical and physical guidance to shape the trajectory of neural circuits and formation of synaptic connections with target cells. Polyamines, such as polyornithine (pORN), augment neuronal adhesion electrostatically, but also provide an ideal condition for neurite outgrowth and formation of neuronal networks in culture [23, 24, 25]. Due to this fact, pORN is commonly used to coat coverslips or cell culture dishes before seeding neurons [26].
In the present study, our aim was to induce neurite outgrowth along
microcontact-printed lanes and subsequently utilize this model to test whether
loaded human A
Microcontact printing transfers a pattern consisting of biomolecules onto another surface. This technique, which is well-established in our laboratory [18], allows biomolecule deposition in µm-sized patterns. Polydimethylsiloxane (PDMS) stamps were produced using a micropatterned template from a silicon wafer referred to as a “master plate”. The surface relief of the elastomer stamp is formed by casting and curing liquid PDMS against the master plate. The raised and lowered regions of the master plate are replicated into the cured stamp. Elastomer is chosen as material for its ability to ensure conformal contact to non-planar surfaces and its hydrophobic nature, which is ideal for transferring biomolecules. In our experimental setup, the PDMS stamp adsorbs a collagen hydrogel solution loaded with peptides or proteins of our interest and transfers it onto a semipermeable extra membrane upon contact.
Numerous stamps were generated from one master plate. The creation of the master plate follows a previously described protocol [18]. The PDMS prepolymer (Sylgard 184 Silicone Elastomer Kit, Dow, Seneffe, Belgium, 01673921) consists of two components. The elastomer curing agent was mixed with the elastomer base solution in a ratio of 1:10. The blended PDMS was then poured onto the master plate positioned in the center of a petri dish. Air bubbles were removed using a desiccator connected to a vacuum pump. After curing overnight at 60 °C, the solid PDMS was peeled off the master plate, and was cut to size with a scalpel, resulting in stamps.
In this study, we loaded pORN (poly-DL-ornithine, Sigma-Aldrich, St. Louis, MO,
USA, P-0671), human recombinant A
Following collagen hydrogel preparation, 15 µL of the solution was applied onto each micropatterned PDMS stamp. A coverslip (R. Langenbrinck, Emmendingen, Germany, 01-2126/1) was placed on top to distribute the ink solution on the stamp and incubated for 15 min at 37 °C to allow crosslinking of the collagen hydrogel solution. After removing the coverslip, striking off the excess solution, the stamp was inverted, and the collagen hydrogel solution was transferred onto the semipermeable extra membrane (Isopore, Merck Millipore, Darmstadt, Germany, HTTP02500) by applying pressure with 18 g coins on top overnight at 4 °C. The position of the µCP was marked with small dots using a permanent marker to aid in arranging the brain slices [18], and the membranes were sterilized under UV light for 20 min.
Organotypic brain slice cultures serve as a physiologically relevant three-dimensional ex vivo model of the brain, a technique well-established in our laboratory [16]. Organotypic brain slices were taken from postnatal day 8–10 C57BL/6 WT mice randomized between the groups, irrespective of sex. All experimental procedures were approved by the Austrian Ministry of Science and Research, and complied with Austrian guidelines on animal welfare and experimentation, following the ethical principles of the three Rs (replace, reduce, refine).
Brains were dissected under sterile conditions. After carefully removing the
cerebellum, the brains were affixed onto the sample holder platform of a
water-cooled vibratome (Leica, Nussloch, Germany, VT1000S) with adhesive (Loctite
401, Henkel, Düsseldorf, Germany 231435) on their newly formed caudal
surface. Coronal slices, 150 µm thick at the hippocampus level, were cut in
a sterile preparation medium cooled down to approx. 5 °C (pH 7.2–7.3,
autoclaved, 1
Fluorescently labeled dextrans are frequently employed in live cell imaging
studies to track both anterograde and retrograde neuronal pathways, a technique
firmly established in our laboratory [28]. MiniRuby (Invitrogen, Thermo Fisher
Scientific, Waltham, MA, USA, D3312) is a dextran conjugate combined with the
red-fluorescent dye tetramethylrhodamine. The MiniRuby crystal was positioned on
the microcontact-printed surface, two to three mm away from the two-week-old
brain slice, using the tip of a fine needle. The slices were then observed for a
time period ranging from 1 to 7 days under a microscope (Leica, Nussloch,
Germany, DMIRB) within a chamber equipped with temperature (PeCon, Erbach,
Germany, tempcontrol 37-2 digital) and CO
Voltage-sensitive and calcium-sensitive dyes are used to monitor the electrical
activity in neurons. Voltage-sensitive dyes change their spectral properties with
membrane potential shifts. Conversely, calcium-sensitive dyes alter spectral
properties upon binding intracellular calcium ions (Ca
Immunostaining was employed to assess the outgrowth of neurites along the
microcontact-printed surfaces using the cytoskeleton markers neurofilament (NF)
and microtubule-associated protein 2 (MAP2), as well as the myelin sheath marker
myelin oligodendrocyte glycoprotein (MOG). Additionally, the interaction with
astrocytes was visualized through glial fibrillary acidic protein (GFAP) and the
neuronal activity via membrane bound Na,K-ATPase (ATP1A1). Furthermore, an
immunostaining for SIRT1 was conducted, which is an intracellular regulatory
protein related to aging and age-associated diseases. Fixed slices on the
membrane were transferred to a six-well plate and washed 3
Confocal microscopy was performed using LSM 980 with Airyscan 2 (Zeiss,
Oberkochen, Germany). The 63
Propidium iodide (PI) is a widely utilized red-fluorescent nuclear stain
employed to identify dead cells, as it does not permeate through living cell
membranes. The inserts containing viable brain slices were gently immersed in
slice culture medium containing 2 µg/mL of PI (Sigma-Aldrich, St. Louis,
MO, USA, P4170) and were then incubated for 30 min at 37 °C and 5 %
CO
Human recombinant peptides of monomeric A
An unbiased image evaluation was conducted to determine the quantity and length
of neurites that had extended. Slices were considered for inclusion if they
exhibited at least one protruding neurite. Only neurites surpassing a length of
100 µm and clearly situated within the microcontact-printed lanes
were included in the quantitative examination. Images were captured using a
widefield microscope (Olympus, Tokyo, Japan, BX61 and Leica, Nussloch, Germany,
DMIRB) at a 20
Hippocampal organotypic half brain slices were arranged on microcontact-printed extra membranes, which were centrally laid into a cell culture insert in a six well plate filled with slice culture medium (Fig. 1A). These half brain slices were connected to the µCPs with the horizontal cutting side and straight lanes arranged at a 90° angle (Fig. 1B). Our area of interest was located along brain slice’s horizontal cutting side (Frame in Fig. 1B). µCPs were identified as 800 µm long and 30 µm in width sharp and clear lines using a red-fluorescent Alexa-546 antibody (Fig. 1C). The brain slice’s position on the µCP was easily recognized using the blue-fluorescent nuclear marker DAPI (Fig. 1D). To evaluate brain slice viability on the µCP, 2-week-old cultures were incubated with PI, revealing minimal nuclear staining (Fig. 1E), in contrast to a hydrogen peroxide-treated control (Fig. 1F,G).
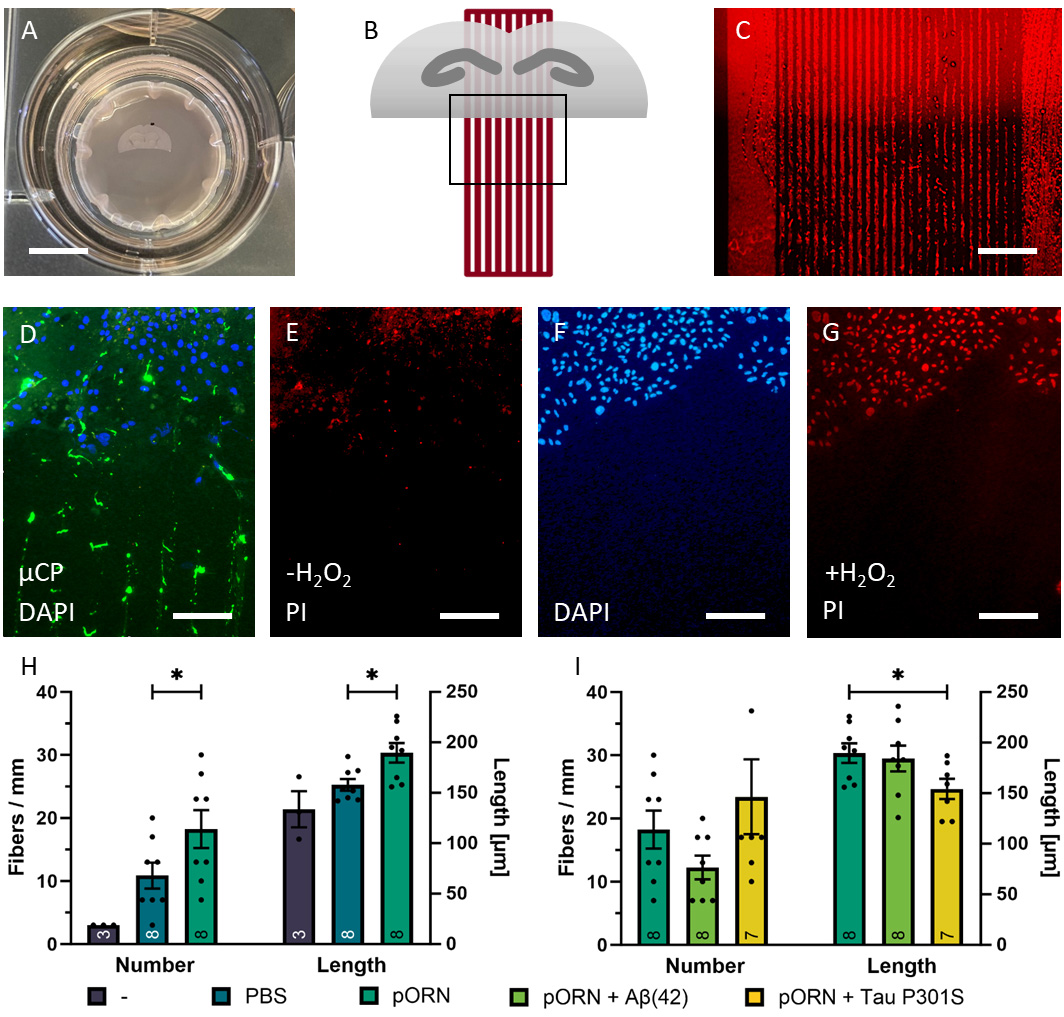 Fig. 1.
Fig. 1.Coupling of collagen-based microcontact prints
(µCPs) with organotypic brain slices. (A) Coronal half slice of 150
µm in thickness at the hippocampus level arranged on the
microcontact-printed extra membrane in a cell culture insert. (B) Top view
schematic representation of the coupling between an organotypic brain slice
(grey) and a µCP (red). The frame shows the area of interest located
along brain slice’s horizontal cutting side. (C) Alexa Fluor 546 (comprised in
the collagen hydrogel solution) visualizes the µCP pattern and brain
slice position can be recognized through brighter background fluorescence (D)
Nuclear DAPI staining shows the location of the brain slice (blue) connected to
the µCP (green, Alexa Fluor 488). (E) Viability of brain cells is
examined by propidium iodide (PI) staining under normal conditions. Since PI is a
membrane impermeable DNA binding dye, intact and viable cells do not show any
nuclear staining. (F) For control purposes, brain slices (blue, DAPI) were
exposed to oxidative stress (+ H
Nearly no spontaneous outgrowth of neurites was visible in slices without µCPs (Fig. 1H). Since we excluded all slices that had no neurites or neurites less than 100 µm in length for quality standard reasons, we had a sample number of only 3 in the “no µCP” group (Fig. 1H). Collagen-only µCPs without any additional load but PBS already promoted nerve fiber growth along the microcontact-printed lanes when stained for NF (Fig. 1H). When pORN was loaded to the collagen-based µCP, the number of NF-positive outgrown neurites was significantly enhanced compared to collagen-only µCPs (Fig. 1H). The length of these outgrown NF-positive nerve fibers was also significantly longer when pORN was applied to the collagen-based µCP (Fig. 1H).
NF was used to stain and quantify outgrown neurites. NF effectively stained nerve fibers, demonstrating their growth along the collagen-based microcontact-printed lanes (Fig. 2A–C). To further confirm the specificity of the neurites, cultures were stained for MAP2, MOG, and GFAP. MAP2-positive nerve fibers were identified growing along the collagen-based microcontact-printed lanes (Fig. 2D). MOG-positive staining was observed along the collagen-based microcontact-printed lanes, however, the MOG staining appeared to be patchier (Fig. 2E). GFAP was utilized to visualize astrocytes, which were exclusively found in the brain slices, with some processes rearranging and extending towards the microcontact-printed lanes (Fig. 2F).
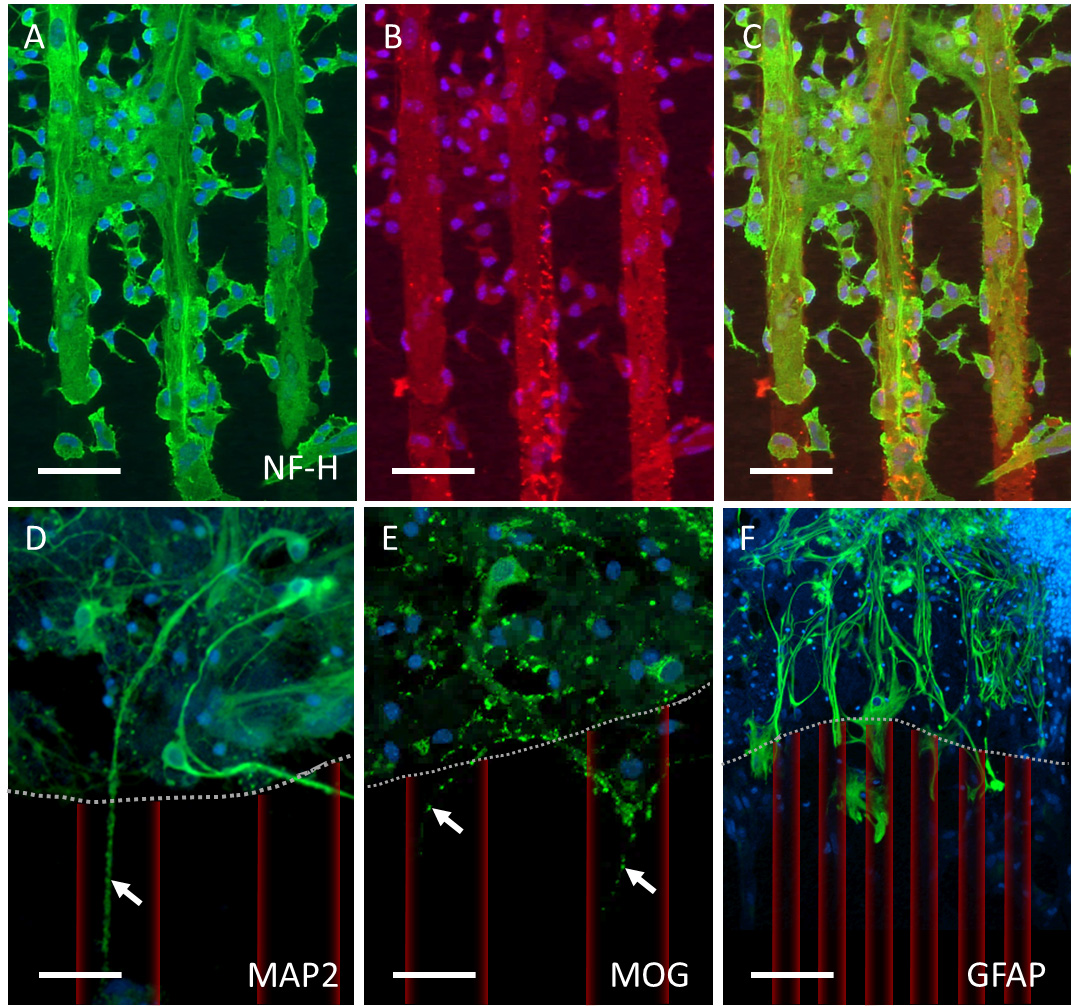 Fig. 2.
Fig. 2.Characterization of neurite outgrowth. (A–C) Neurofilament-heavy (NF-H)-positive neurites (green, Alexa Fluor 488) extended beyond the brain slice along the microcontact print (µCP) pattern (red, Alexa Fluor 546). (D) Microtubule-associated protein 2 (MAP2) and (E) myelin oligodendrocyte glycoprotein (MOG) also stains outgrown neurites (arrows in D and E, dotted line marks the brain slice boarder, artificially overlaid red µCP). (F) Glial fibrillary acidic protein (GFAP)-positive astrocytes primarily remain within the brain slice but organize themselves along microcontact-printed lanes, too. Nuclei are stained with DAPI (blue). Scale bar in A–C = 100 µm, in D–E = 75 µm, in F = 120 µm.
To examine the effect of A
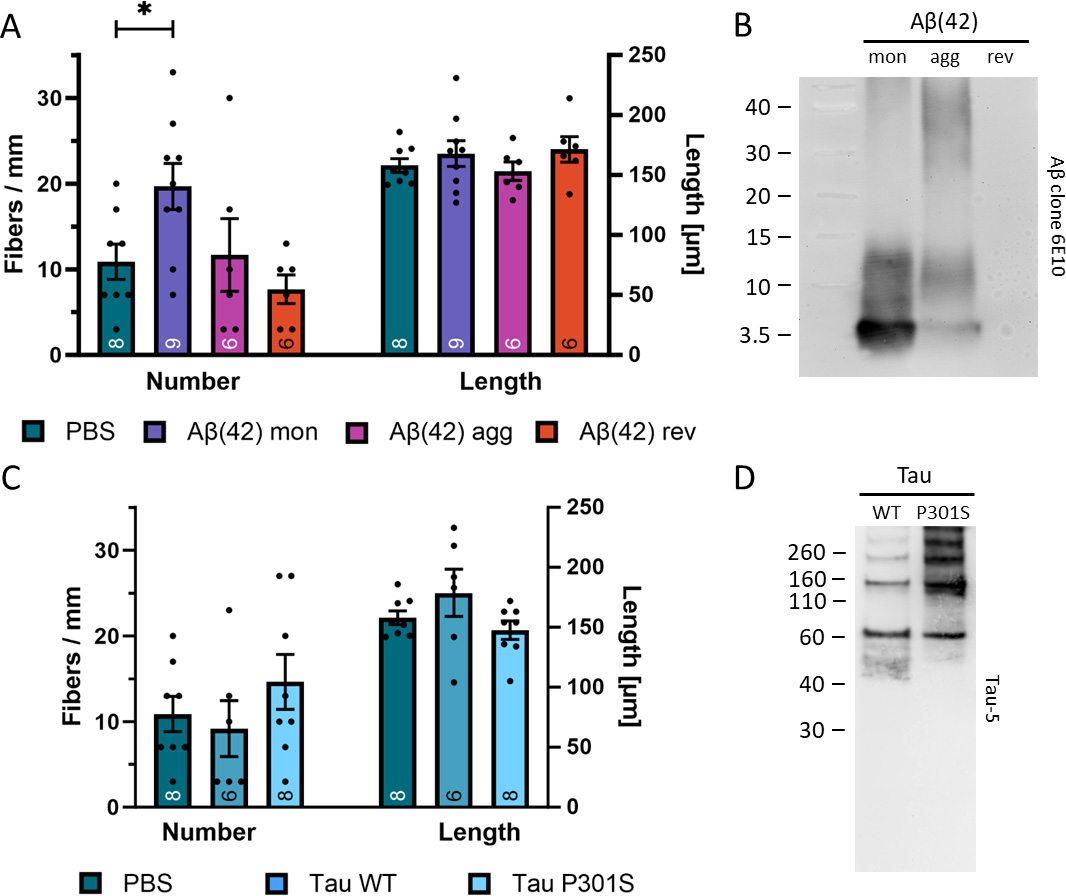 Fig. 3.
Fig. 3.Effects of human beta-amyloid (A
Confocal imaging revealed several long intact nerve fibers running in parallel bundles after two weeks of incubation on µCPs (Fig. 4A,B). Long NF-positive nerve fibers formed exclusively along the microcontact-printed lanes (Fig. 4C).
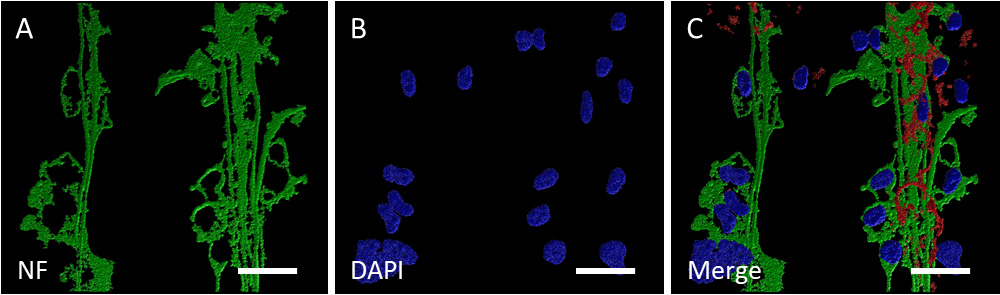 Fig. 4.
Fig. 4.Confocal imaging of neurites outgrown along microcontact-printed lanes. (A) Confocal zoomed-in image of several neurofilament (NF)-positive long side-by-side nerve fibers (green, Alexa Fluor 488) in three-dimensional representation (processed with the Imaris 10.0.1 software). (B) Associated nuclei (blue, DAPI) (C) NF-positive nerve fibers (green, Alexa Fluor 488) form exclusively within the two microcontact-printed lanes (µCP, red, Alexa Fluor 546). Scale bar in A–C = 40 µm.
To investigate the functional activity of the newly formed nerve fibers, MiniRuby crystals were introduced to the µCPs close to the brain slices (Fig. 5A). The MiniRuby appeared as a large red-fluorescent dot and was transported to the brain slice after 1 day of incubation, though not in direct contact with the brain slice (Fig. 5B). A clear red-fluorescent MiniRuby-positive area was observed at the green-fluorescent collagen-based µCPs (Fig. 5C). Some MiniRuby-positive immunoreactive spots were visible directly along the NF-positive nerve fibers on the µCP (Fig. 5D–F). Interestingly, after 7 days of incubation with MiniRuby, MiniRuby-positive cells were identified in the hippocampus (Fig. 5G–I).
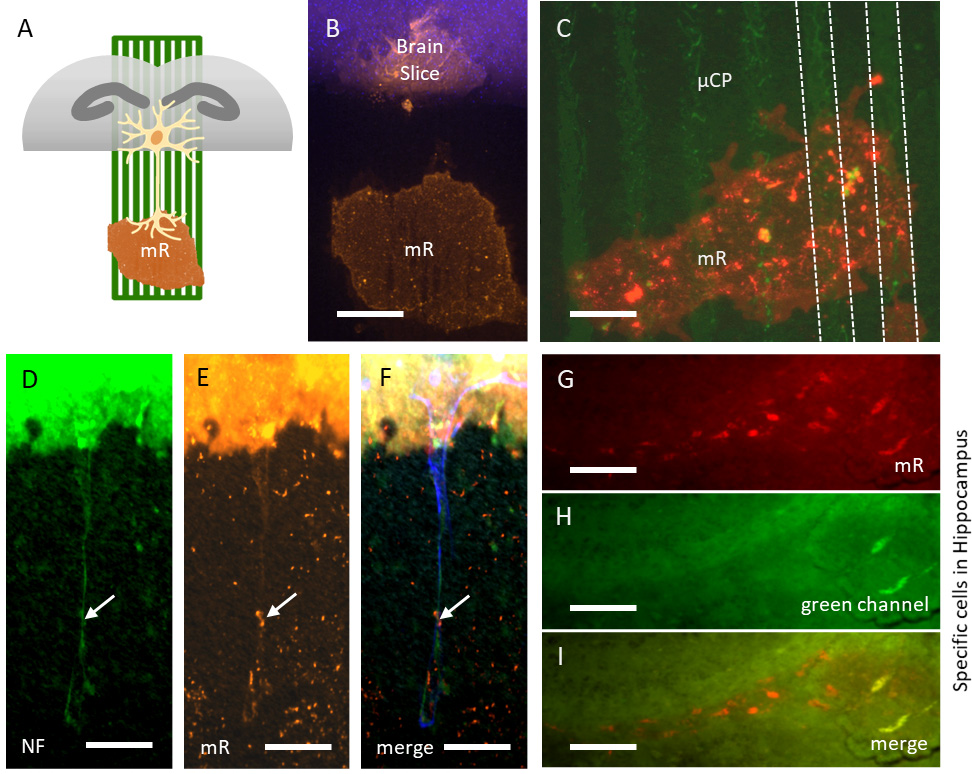 Fig. 5.
Fig. 5.Live cell imaging using the neurotracer MiniRuby. (A) Schematic representation of the MiniRuby (mR) neurotracer placement on a green microcontact print (µCP) connected to a brain half slice (grey). (B) MiniRuby (mR, red-fluorescent) is retrogradely transported to the nuclei-stained brain slice (blue, DAPI), though not in direct contact to the brain slice. (C) MiniRuby (mR, red-fluorescent) is well positioned on the µCP (green, Alexa Fluor 488). (D–F) When fixed sections are stained for neurofilament (NF) revealing outgrown neurites (green, Alexa Fluor 488), MiniRuby (mR, red-fluorescent) is taken up by the neurons (arrows) and transported retrogradely to the brain slice (blue, DAPI). (G–I) Within the brain slice, MiniRuby (mR, red-fluorescent) specifically stained neurons in the hippocampal area, which were not visible in the green channel. Scale bar in B,C = 100 µm, in D–I = 75 µm.
Voltage-sensitive dye FluoVolt alters its spectral properties in response to
membrane potential shifts, while calcium-sensitive dye Rhod-4 changes its
spectral properties upon binding intracellular calcium ions (Ca
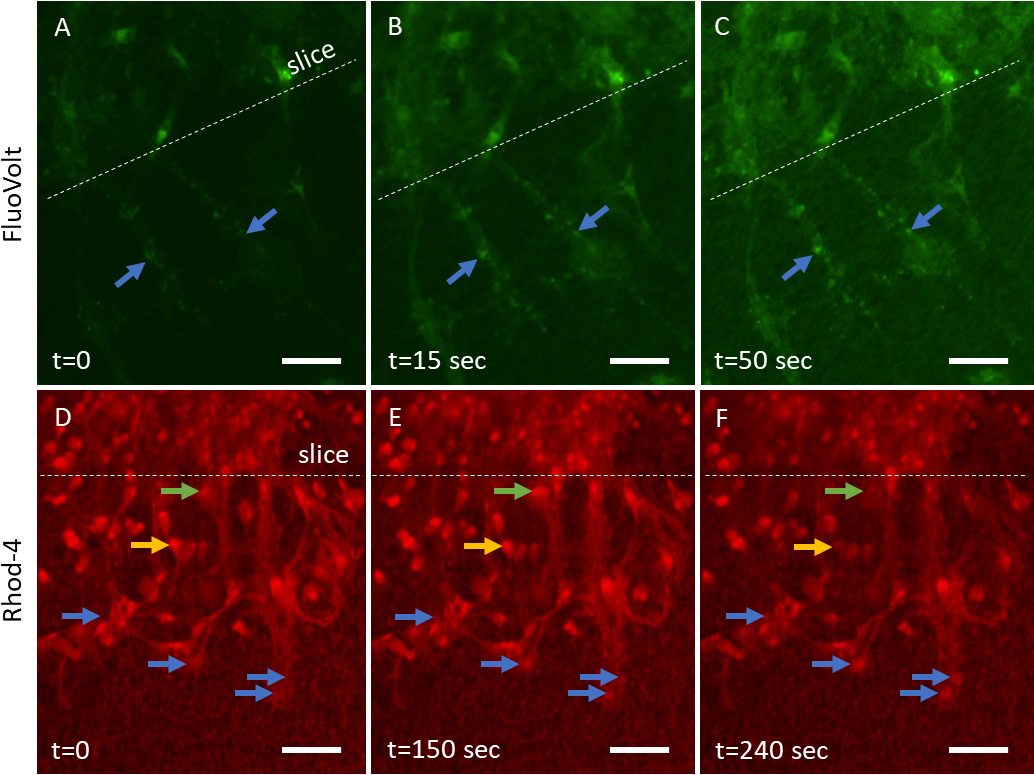 Fig. 6.
Fig. 6.Voltage-sensitive (FluoVolt) and calcium-sensitive (Rhod-4) dyes
to monitor the electrical activity in neurons. Two-week-old organotypic brain
slices, incubated on collagen-based microcontact prints (µCPs)
loaded with monomeric beta-amyloid (A
The presence of the membrane bound Na,K-ATPase serves as an indicator of
neuronal activity. Neurites extending from organotypic brain slices cultured for
2 weeks on collagen-based µCPs loaded with A
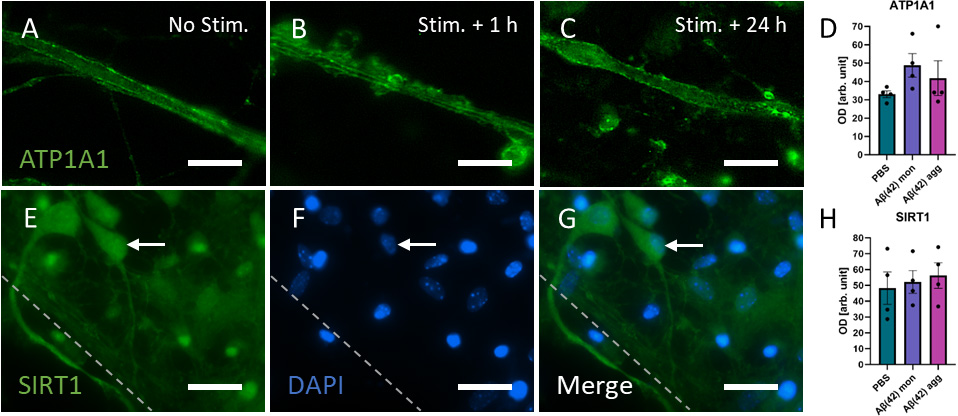 Fig. 7.
Fig. 7.Expression levels of Na,K-ATPase (ATP1A1) and SIRT1 showed no
difference between collagen-based microcontact prints (µCPs) devoid
of load (PBS) and those loaded with monomeric or aggregated beta-amyloid
(A
Finally, an immunostaining for SIRT1 was conducted, which is an intracellular
regulatory protein related to aging and age-associated diseases and may be
involved in A
In the present study, we demonstrate that organotypic brain slices can be
connected to collagen-based µCPs, and newly formed nerve fibers
developed along these defined microcontact-printed lanes. These emerging nerve
fibers were found to be functionally active since they were capable of
transporting the neurotracer MiniRuby, and demonstrated calcium fluctuations as
well as membrane potential changes. The presence of the Alzheimer’s peptide
A
In our research group, organotypic brain slice cultures are well-established and have been extensively utilized, with a record of more than 60 publications, including a comprehensive review by Humpel [16]. Recently, we introduced and modified the microcontact printing technique in our lab to immobilize bioactive factors in a µm-size defined pattern, which we connected to brain slices [18]. We have successfully demonstrated the microcontact printing efficiency on various aspects, such as microglia [29], vascular structures [30], and NFG-induced nerve fibers [31]. This experience instills confidence in the robust viability of our brain slices cultured on µCPs for two weeks. During this culture period, the brain slice underwent flattening and attained transparence, serving as a reliable indicator of high quality brain slices. Additionally, a viability assay using PI reinforces the excellent brain quality and survival of the brain cells. Finally, positive immunostainings for neuronal markers unequivocally illustrates the survival of neurons in our experimental setting.
NFs are neuron-specific 10-nm filaments recognized as members of the intermediate filament family. They exhibit a relatively sparse and convoluted distribution within dendrites and perikarya. Within axons, however, they tend to be numerous, predominantly straight, unbranched, and extending to considerable length [32]. NFs are predominantly composed of three subunits, distinguished by their apparent molecular weight: NF-H (heavy, ~200 kDa), NF-M (medium, ~160 kDa), and NF-L (light, ~68 kDa) protein [32]. In our present study, we employed a commercial NF-H monoclonal antibody [33], which exhibited robust staining of nerve fibers derived from mouse slices. Especially in the confocal microscopy, this antibody distinctly highlighted nerve fibers that extended from brain slices along the collagen-based µCPs. When axons undergo radial expansion, often increasing their size by up to 10-fold, this is accompanied by a proportional increase in the number of NFs [34]. During the onset of axonal growth, NF-H expression is typically delayed compared to NF-L and NF-M [35], suggesting that positive NF-H reactivity may signify a more mature state of NFs. NF-H and NF-M stand out due to their longer tail domains that extend radially from the filament core, forming side arms. Phosphorylation of these side arms further enhances the stability of the axonal NF cytoskeleton which extensively cross-links to actin filaments and microtubules [32].
To reinforce our findings, we employed another neuronal marker, MAP2, confirming the neuronal nature. Additionally, we applied the oligodendrocyte marker MOG revealing a punctuated staining, indicative of potential myelin sheets on the emerging nerve fibers. A GFAP staining displayed reactive astrocytes only within the brain slices but not along the µCPs. Subsequently, confocal microscopy was conducted, revealing numerous long, intact nerve fibers organized in parallel bundles with a tendency toward the borders of microcontact-printed lanes.
Collagen is widely employed in numerous cell culture models due to its bioactive and biodegradable properties. At present, collagen is the foremost extracellular matrix protein employed in three-dimensional cell cultures [36]. Over the course of several years, we have utilized collagen in our experimental approaches, as elucidated in previous reviews [20, 37]. Our proficiency extends to the construction of collagen hydrogels [38, 39] or collagen-based µCPs [18] incorporated in organotypic brain slices with preserved three-dimensional neuronal architecture and microenvironment. Despite collagen being found only in basement membrane of the vasculature in the brain, it is also widely utilized as a biomaterial to promote the growth of neurites [21, 22]. Our current findings clearly demonstrate the capacity of collagen since brain slice-derived nerve fibers selectively extended and aligned along the µCP composed solely of collagen. Neurite outgrowth quantity is known to be influenced by collagen concentration through altering its fibril density and pore size [40]. An optimal concentration of 2 mg/mL collagen was identified [40], aligning with the end concentration achieved in our µCPs, which promotes neuronal differentiation and outgrowth of neurites. Neurons encapsulated in a collagen hydrogel containing decellularized brain extracellular matrix facilitated neurite development and enhanced neurite outgrowth [41]. More experiments are necessary to assess the role of collagen, which was out of focus in this study.
Compelling evidence from existing literature underscores an influence of
polyamines in supporting mediated cell adhesion and nerve fiber growth due to
their intrinsic positive charges [42]. In particular, the capacity of
microcontact-printed polylysine in guiding axons has been extensively
investigated [23, 24, 25, 43, 44, 45]. In the present study, we aimed to examine the impact
of another polyamine, pORN, on neurite outgrowth. Notably, pORN and polylysine
share an almost identical structure, differing only by a single atom in the
length of their primary amino group carbon chain, making them functionally
equivalent regarding neuron adhesion, viability, and pattern compliance [24]. The
non-proteinogenic amino acid ornithine plays a crucial role in synthesizing
various essential cellular products, making L-ornithine a widely utilized
supplement in cell culture media [46] and pORN a widely utilized coating reagent
for neuronal cell lines [26]. Our present study revealed that pORN loaded to the
collagen-based µCPs significantly enhanced nerve fiber growth
compared to collagen-only µCPs, in terms of number of fibers as well
as fiber length. However, pORN had no additive impact on either the effects of
A
A
Our present findings reveal a twofold stronger stimulatory effect of human
A
Sirtuins represent a highly conserved class of nicotinamide adenine dinucleotide
(NAD
A recent study demonstrated that SIRT1 promotes the degradation of oligomeric
A
The microtubule-associated protein tau exemplifies a highly dynamic protein due to posttranslational modifications at more than 50 sites, which comes at a heightened propensity for self-aggregation [65, 66]. The appearance of intracellular tau NFTs mainly composed of such aggregated hyperphosphorylated tau protein is the second defining feature of AD. Tau is encoded by the MAPT gene, comprising 16 exons that generate six alternative splicing isoforms ranging from 351 to 441 amino acids [67]. Tau protein is distinctly divided into the N-terminal part, proline-rich region (PRR), microtubule-binding domain (MTBD), and C-terminus [67]. Each contains either 3 or 4 MTBD repeats (3R or 4R) which have been shown to be essential for the ability of tau to bind to microtubules [67]. However, experiments performed by several other groups revealed that tau serves functions beyond merely stabilizing the microtubules, notably regulation of neuronal network activity [67]. In the present study, we microcontact-printed a human tau variant comprising 441 amino acids, characterized by the formation of active aggregates resulting from a mutation at position 301 (P301S). We previously conducted experiments to assess the spreading of P301S aggTau in brain slices [68]. Although tau primarily functions as a cytosolic microtubule-associated protein, it is also found in extracellular environments under physiological conditions [69]. It appears that tau can be transferred from cell-to-cell through various pathways, including vesicular mechanisms (exosomes, ectosomes, endosomes, cell membrane fusion), vesicle-free mechanisms (direct translocation across the plasma membrane known as unconventional protein secretion I) or tunneling nanotubes [69]. Human tau was employed due to potential differences in tau expression between mice and humans [70]. Although the tau sequences in mice and humans exhibit an 89% amino acid similarity, the human tau variant features an additional 11 amino acids at the N-terminal end [70].
Microcontact printing of aggTau within a collagen matrix did not lead to an increase in outgrown nerve fibers derived from brain slices. For control purposes, collagen-based µCPs were loaded with WT tau, resulting in neurite outgrowth similar to that of baseline collagen-only µCPs. In Western blot analysis, we detected WT tau at the anticipated 60 kDa position, while the P301S mutated aggTau appeared as several bands exceeding the 60 kDa mark in molecular weight, confirming their aggregated state. Upon concurrent co-loading of aggTau and pORN, we observed a synergistic effect in the number of outgrown neurites, although this effect did not reach statistical significance. However, the combined load of aggTau and pORN led to a significant decrease in the length of outgrown neurites. Since aggTau may be bioinactive, it could interfere with normal cellular processes of tau necessary for neurite elongation. Several studies indicate that tau is essential in modulating synaptic plasticity and signaling [71, 72, 73, 74]. Tau mediates changes in the cytoskeleton by binding actin to its PRR, concurrently binding to microtubules through MTBD [75]. Acting as a cross-linker between microtubules and actin filaments, tau contributes to the dynamic reorganization of the cytoskeleton network, potentially playing a crucial role in neurite formation. In fact, several studies have reported the involvement of tau in neurite outgrowth, however, the specific details regarding the implication of tau in axonal guidance remain unclear [9, 10, 11].
In our present study, we show the growth of brain slice-derived nerve fibers
along microcontact-printed lanes, with the most pronounced effects observed when
A
The voltage-sensitive dye FluoVolt indicates changes in its spectral properties
upon membrane potential shifts. At resting potential (–65 mV) of neurons, there
is a higher concentration of K
The calcium dye Rhod-4 enters the cells through passive loading, and once
inside, intracellular esterases cleave the dye to its cell-impermeant, active
form, which exhibits fluorescence upon binding with calcium ions. The
concentration of calcium ions (Ca
To demonstrate that depolarization is linked to the activity of axonal plasma
membrane-associated Na,K-ATPase [77, 78], brain slices were immunohistochemically
stained for Na,K-ATPase 1 hour or 24 hours after depolarization. Neurites indeed
showed increased Na,K-ATPase expression following depolarization compared to
non-depolarized brain slices. This increased expression returned to baseline
levels 24 hours post-stimulation. However, there was no significant difference in
Na,K-ATPase immunoreactivity among neurites along collagen-based
µCPs without load (PBS), monomeric, or aggregated A
This study provides a proof-of-concept demonstrating the utility of
collagen-based microcontact prints as an extracellular matrix for nerve fiber
growth. However, certain limitations must be acknowledged. (i) Firstly, our
previous studies have revealed inconsistent microcontact printing, variable rates
of collagen degradation in culture and variability in organotypic brain slice
culture. This potentially contributed to increased experimental variance,
necessitating the implementation of exclusion criteria (i.e., fibers shorter than
100 µm were excluded) for quantitative analysis. (ii) Secondly, our
current model lacks the ability to print a concentration gradient of different
biomolecules, which could potentially improve the model. (iii) Thirdly, we
exclusively tested the effects of human A
In this study, we describe an ex vivo model that enables the study of selective neurite growth affected by exogenous stimuli. This model extends our previous research, which focused on microglial migration [29] and endothelial cell migration up to vessel formation [30]. Utilizing microcontact printing in combination with organotypic mouse brain slices, we envision the development of a “triple model”, where the effects of exogenous factors can be concurrently investigated across three distinct functional assays: neurons, microglia, and vessels. This integrated approach could be further refined into a “brain-on-a-chip” functional assay, serving as a valuable platform for screening various bioactive factors, novel drugs, or medications. Such a model holds promise in contributing to the principles of the 3Rs in animal research by minimizing the number of animals used and alleviating associated pain.
In the present study, we demonstrate that connecting organotypic brain slices to
collagen-based µCPs enables the growth of newly formed nerve fibers
aligned along these microcontact-printed lanes. These brain slice-derived
emerging neurites exhibited functional activity by retrogradely transporting the
neurotracer MiniRuby and electric activity when exposed to voltage-sensitive dye
(FluoVolt) and calcium-sensitive dye (Rhod-4). Our findings suggest a substantial
enhancement in neurite growth induced by the Alzheimer’s peptide A
AD, Alzheimer’s disease; APP, amyloid precursor protein; aggTau, aggregated tau;
A
The data that support the findings of this study are available upon request from the corresponding author.
CH designed the research study. KS performed the research. KS analyzed the data. KS wrote the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were approved by the Austrian Ministry of Science and Research (2021-0.150.227, approval date 26 August 2021) and conformed to the Austrian guidelines on animal welfare and experimentation. Our study using animals (mice) follows ethical guidelines for sacrificing animals, and our animal work complies with international and national regulations. All work was performed according to the 3Rs (reduce–refine–replace) rules of animal experiments. All our slice experiments are defined as “organ removal” and are not “animal experiments”.
The master mold is a kind gift from Jenny Emnéus and Janko Kajtez, Department of Biotechnology and Biomedicine, DTU Bioengineering, Technical University of Denmark. We sincerely thank Anna Draxl and Mohadeseh Ragerdikashani for excellent technical assistance.
This research was funded by the Austrian Science Funds (FWF), grant number P32558-B.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.