
 , Cynthia M. Simbulan-Rosenthal 2, Dean S. Rosenthal 2, Jeffrey W. Shupp 1,2,3,4,5
, Cynthia M. Simbulan-Rosenthal 2, Dean S. Rosenthal 2, Jeffrey W. Shupp 1,2,3,4,51 Firefighters' Burn and Surgical Research Laboratory, MedStar Health Research Institute, Washington, D.C. 20010, USA
2 Department of Biochemistry and Molecular & Cellular Biology, Georgetown University School of Medicine, Washington, D.C. 20007, USA
3 Department of Surgery, Georgetown University School of Medicine, Washington, D.C. 20007, USA
4 The Burn Center, Department of Surgery, MedStar Washington Hospital Center, Washington, D.C. 20010, USA
5 Department of Plastic and Reconstructive Surgery, Georgetown University School of Medicine, Washington, D.C. 20007, USA
Abstract
Background: Existing animal models for testing therapeutics in the skin
are limited. Mouse and rat models lack similarity to human skin in structure and
wound healing mechanism. Pigs are regarded as the best model with regards to
similarity to human skin; however, these studies are expensive, time-consuming,
and only small numbers of biologic replicates can be obtained. In addition,
local-regional effects of treating wounds that are closely adjacent to
one-another with different treatments make assessment of treatment effectiveness
difficult in pig models. Therefore, here, a novel nude mouse model of xenografted
porcine hypertrophic scar (HTS) cells was developed. This model system was
developed to test if supplying hypo-pigmented cells with exogenous alpha melanocyte stimulating hormone (
Keywords
- hypertrophic scar
- animal modek
- burns
- xenografts
- dyschromia
- melanocytes
There is no perfect animal model for the study of hypertrophic scar (HTS) [1, 2]. Studying patient samples is most likely the best recapitulation of the disease process; however, this strategy has the drawback that it is a snapshot in time. These samples do not provide information as to the natural history of HTS formation, as they must be taken after a scar is already formed. Additionally, HTS samples from patients are most likely at the severe end of the spectrum because samples large enough to graft can most often only be collected from severe HTS that requires excision. Murine species are generally not accepted as models of HTS because of the “loose-skinned” nature of these animals [3]. They also have a panniculus carnosus which allows them to heal by contraction instead of by granulation tissue deposition and re-epithelialization. As such, murine species that are not transgenic do not form robust fibroproliferative HTS.
Non-animal models for wound healing and scar formation have also been suggested [4]. These include in vitro models utilizing co-cultures of HTS-derived cells and organotypic culturing of biopsies of HTS. Ex vivo models utilizing excised human skin have also been proposed. While these models are useful for certain research questions, the drawback of not having the full in vivo system is clear.
Nude mouse models of xenografted normal human skin have been used to create human-like HTS [5, 6, 7]. Although the skin was normal, and not HTS when it was xenografted, the skin forms scar that has many of the morphologic and histologic criteria of HTS. Additional models have been developed where the normal skin is “scratched” to create a wound in the skin that then forms additional HTS [8]. While these models are useful, and they have shown retention of the HTS and pigmentation phenotype even 1-year post-xenografting, the etiology of HTS is not the same as the etiology of full thickness wounding or burn injury. Lastly, this model relies on the availability of large amounts of normal human skin, a resource that is not universally available.
In prior work, it was shown that hypo-pigmented melanocytes derived from human
hypopigmented hypertrophic scars could be stimulated by synthetic alpha
melanocyte stimulating hormone (NDP
There is no previously optimized dose for NDP
Nude mice are ideal for xenografting because they lack an immune system, and hence, do not reject the cells when grafted [28]. A xenograft model that has been used extensively in our lab in prior experiments was modified for use in the current experiment. This model has been used previously to create human skin xenografts with primary human fibroblasts and immortalized primary human keratinocytes derived from neonatal foreskin. In this prior work, the effect of sulfur mustard on the death receptor pathway and apoptotic processes was studied in human xenografts [29]. Another project studied the dysregulation of inhibitor of DNA binding 2 (Id2) in relation to the regulation of genetically modified keratinocytes [30]. The model has also been used to create xenografts from freshly isolated, cultured, or genetically modified human cell lines in work from multiple other groups [31]. These groups have studied a number of epidermal tumor cell lines, as well as freshly isolated cells from neonates. Lastly, this model has been used to study melanoma cells [32].
The work reported here is novel because the cells of interest were derived from
adult pigs, not neonates. Previously reported upon use of the model relied
exclusively on neonatal-derived, genetically immortalized, or cancer cell lines.
The modified model was first developed with normally pigmented cells derived from
un-injured skin. Then, the hypothesis that synthetic
All animal work described was reviewed and approved by the MedStar Health Research Institute’s Institutional Animal Care and Use Committee (IACUC). Juvenile castrated male red Duroc swine (30–55 kg) were received and handled according to facility standard operating procedures under an animal care and use program accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and the Public Health Service.
On the day of surgery, animals (n = 3) were anesthetized with a combination of ketamine (10 mg/kg) and xylazine (2 mg/kg) that were delivered intramuscularly. Animals were intubated, maintained on isoflurane, placed on a warming blanket, and ventilated while heart rate, blood pressure, peripheral oxygen saturation, end-tidal carbon dioxide saturation, and core body temperature were continuously monitored. The body hair was clipped, and the skin was prepped with chlorhexidine gluconate scrub.
To create full-thickness wounds, one 10.16 cm
The nature of the excisional wounding procedure creates wounds that are down to the subcutaneous fat and removes all viable dermal elements, including hair follicles and shafts, and apocrine tissues. Mepilex® Ag dressings (Molnlycke, Gothenburg, Sweden) were applied after wound creation and at each dressing change. Animals were fitted with custom-made neoprene vests [36], which completely covered the applied dressings while still allowing mobility of the animal. Buprenorphine (0.005 mg/kg) and fentanyl (25 mcg/hour) were administered for pain control at the end of each surgical procedure. Each week, through 133 days, animals underwent wound cleansing, imaging, biopsy procurement, dressing changes, and vest replacement. The animals were examined at least twice daily to monitor animal health and to identify any signs of pain, wound infection, or distress, of which there were no serious events.
On day 134 post-injury, porcine HTS was excised, split into distinct regions of
hyper- and hypo-pigmented scar, cut into thin 0.5 cm strips, and processed with
1
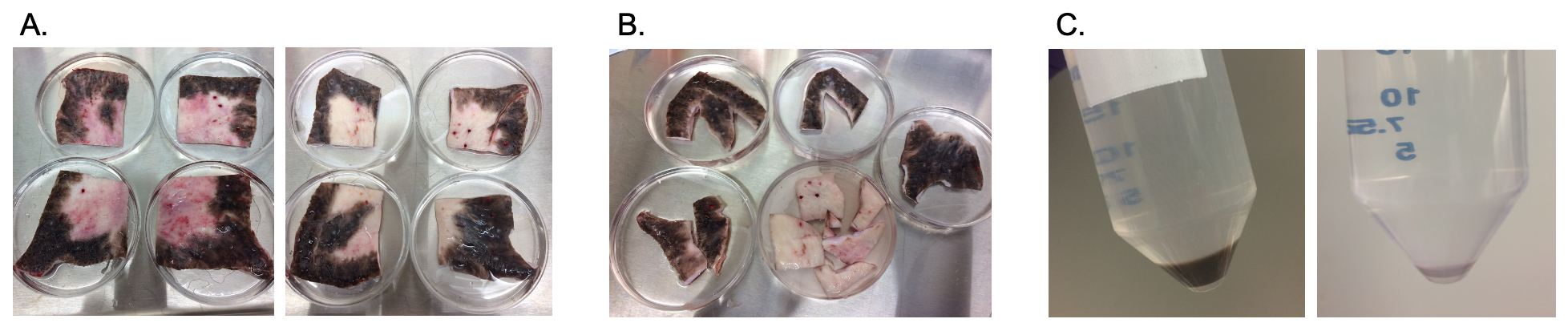
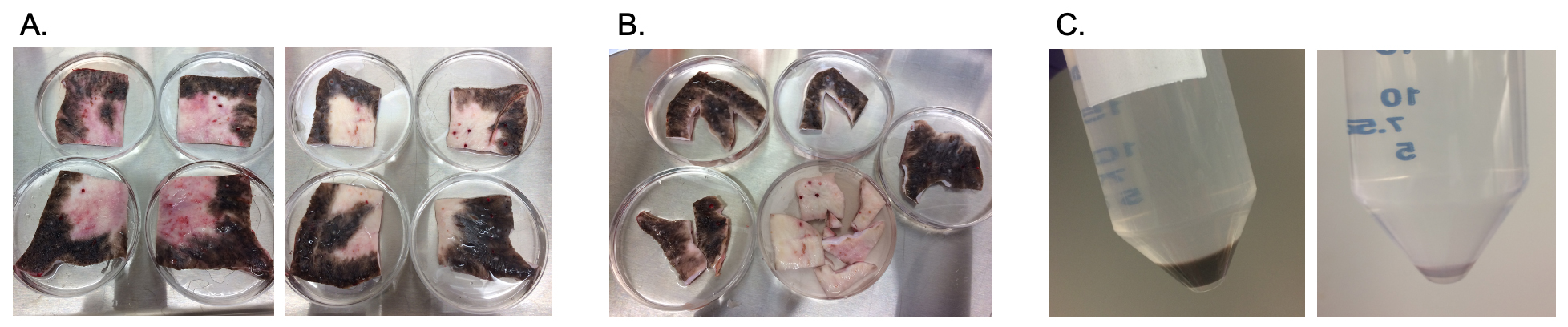 Fig. 1.
Fig. 1.Dyspigmented epidermal cells are isolated from porcine HTS as shown. Scars are excised on the terminal experimental day (A). Scars are cut into pigmentation phenotypes and sorted (B). Scars are further cut and treated with dispase to isolate epidermal cells (C) which were cryopreserved until use in the xenograft model. HTS, hypertrophic scar.
In hyper-, hypo-, and normally-pigmented skin and scar, after the epidermal
sheets were peeled, the dermal pieces were further processed with 1 mg/mL
collagenase (MP Biomedicals, Solon, OH, USA) at 37 °C for 4 hours to obtain
fibroblasts. The resulting cell suspension was raised to a volume of 40 mL with
PBS and was shaken vigorously to release single cells. The final suspension was
then passed through a 45-µm filter. The single cell suspension was spun at
600 g for 5 minutes, and the resulting cell suspension was reconstituted and
seeded at a density of 4000 cells/cm
The company that manufactured the domes in previous literature is now out of business and there is no current manufacturer [31, 32]. As such, domes were 3D printed as a substitute. Domes were designed using Blender (Blender Foundation, Amsterdam, Netherlands) version 2.79. Ultimaker software version 3.6.0 was used for slicing (Cura, Geldermalsen, Netherlands). eSUN eLastic TPE 85A 1.75 mm Flexible 3D printer filament in natural white was used as the material (INTSERVO, Cary, NC, USA). Domes were 3D printed using a Qidi (Ruian, China) X-one 3D printer with a 400 µm nozzle at 212 °C on a heated print bed at 95 °C. The diameter of the hole in the flange was 1 cm. A 2 mm biopsy punch was used to create a hole in the top of the dome to allow for evaporation of cell media. Domes were sterilized in the autoclave at 105 °C, just below the melting temperature of the material.
NCr-Foxn1nu mice (Charles River Laboratories, Wilmington, MA, USA) at an age of 5–8 weeks were used. All animal procedures were conducted in a laminar flow hood with sterile materials. On the day of cell seeding (day 0), anesthesia was induced by 5% isoflurane in an induction box. After an appropriate plane of anesthesia was reached, the animal was transferred to a nose cone and maintained on 3.5% isoflurane for the remainder of the experiment. The skin was surgically prepped using Chloraprep (Becton Dickinson, Franklin Lakes, NJ, USA). A 1 cm in diameter full thickness circular excisional wound was created using curved scissors (Fig. 2A). The bottom flange of the dome was then placed under the skin (Fig. 2B). Buprenorphine was administered intraperitoneally at 0.05 mg/kg for pain relief. This administration was done prior to cell seeding so that the animal would not have to be turned supine after cell seeding. On the day of cell seeding, cells were thawed in a 37 °C water bath and were transferred to 10 mL of complete media to rinse off the DMSO. The cells were then re-suspended in complete media and counted. 8e6 fibroblast cells were re-suspended in 50 µL of a mixture of 50% Matrigel (Corning) and 50% Cnt-40 media (CellnTec). An iteration of the procedure where the cells were reconstituted in 500 µL of media was also performed and deemed to be far inferior to 50 µL. Additionally, normal cell media without Matrigel was attempted, however there were problems with cell leakage, and thus that method was discontinued. Likewise, the problem of leakage was attempted to be controlled with medical grade glue (Dermabond, Ethicon, Raritan, Franklin Township, NJ, USA) and with commercially available superglue, but these methods were toxic to cells and did not result in the creation of a robust xenograft.
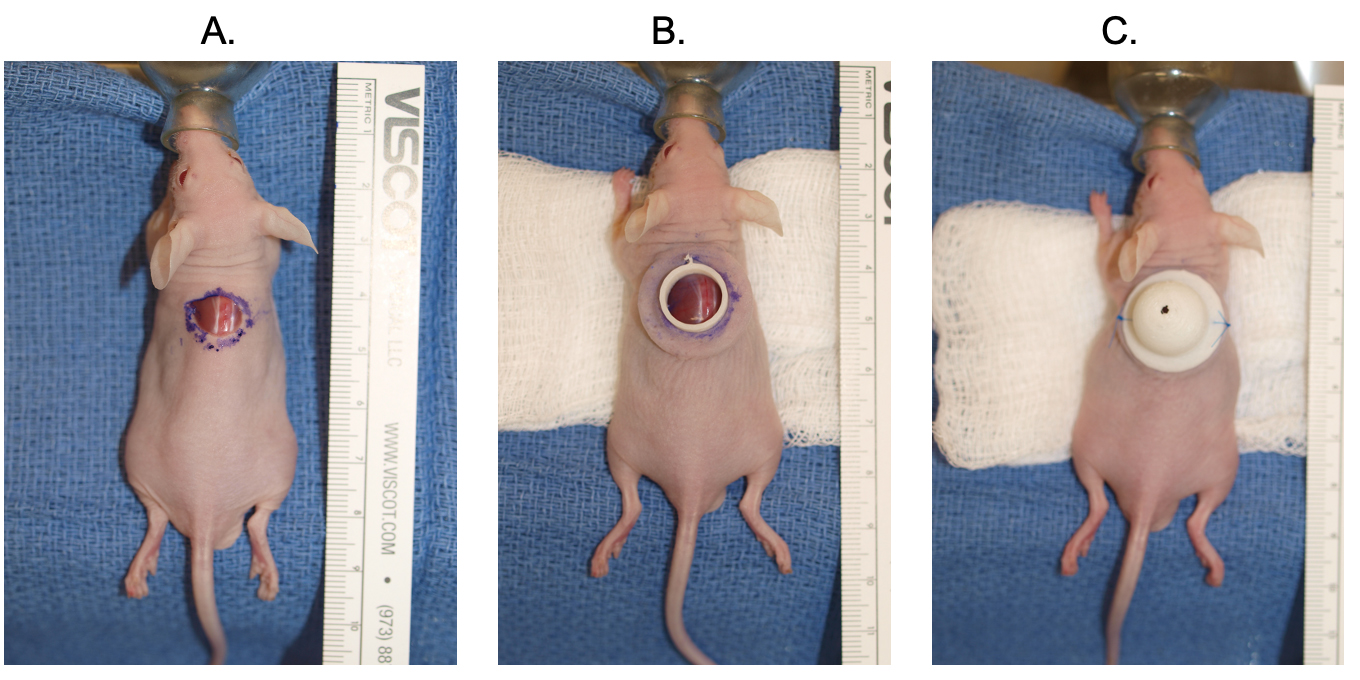
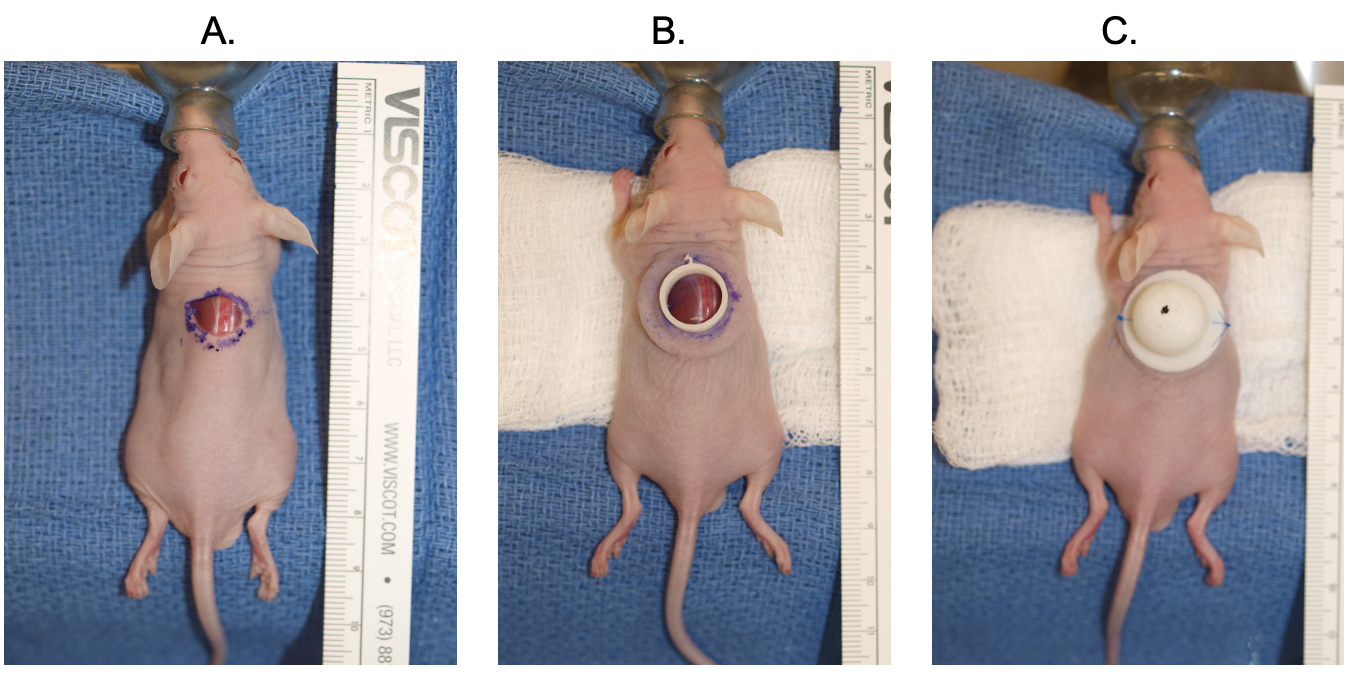 Fig. 2.
Fig. 2.The dome placement and cell seeding xenografting procedure is detailed. A full thickness excision is performed on the dorsum of the mouse (A). The bottom flange is placed (B). The cells are seeded, and the top dome is secured with suture on either side (C).
The suspension was aliquoted onto the open wound area with care taken to hold the flange in place to prevent leakage while the gel formed (Matrigel forms a gel after heating to 37 °C). After 60 seconds, the top portion of the dome was put into place over the flange such that the mouse skin was sandwiched between the top of the dome and the flange. Two 5-O proline sutures were then tied onto opposite sides of the dome to prevent it from moving (Fig. 2C). The animal was allowed to recover on heat and returned to normal housing once normal movement was observed.
On day 3, animals were anesthetized as described above and maintained on
isoflurane via nose cone. The right suture was removed, and the top portion of
the dome was moved to the side. On the day of cell seeding, cells were thawed in
a 37 °C water bath and were transferred to 10 mL of complete media to rinse off
the DMSO. The cells were then re-suspended in complete media and counted. 1-2e6
epidermal cells were re-suspended and seeded in 50 µL of a mixture of 50%
Matrigel and 50% Cnt-40 media as described above. The dome was then re-sutured
in place and the animal was allowed to recover. An iteration of the model where
fibroblasts and epidermal cells were seeded on the same day was attempted and
resulted in primarily dermis generation with minimal epidermis, and hence was not
pursued. This ratio of cells was determined empirically from our other published
grafting studies. Assuming a human basal keratinocyte ranges from 20 to
40 µm
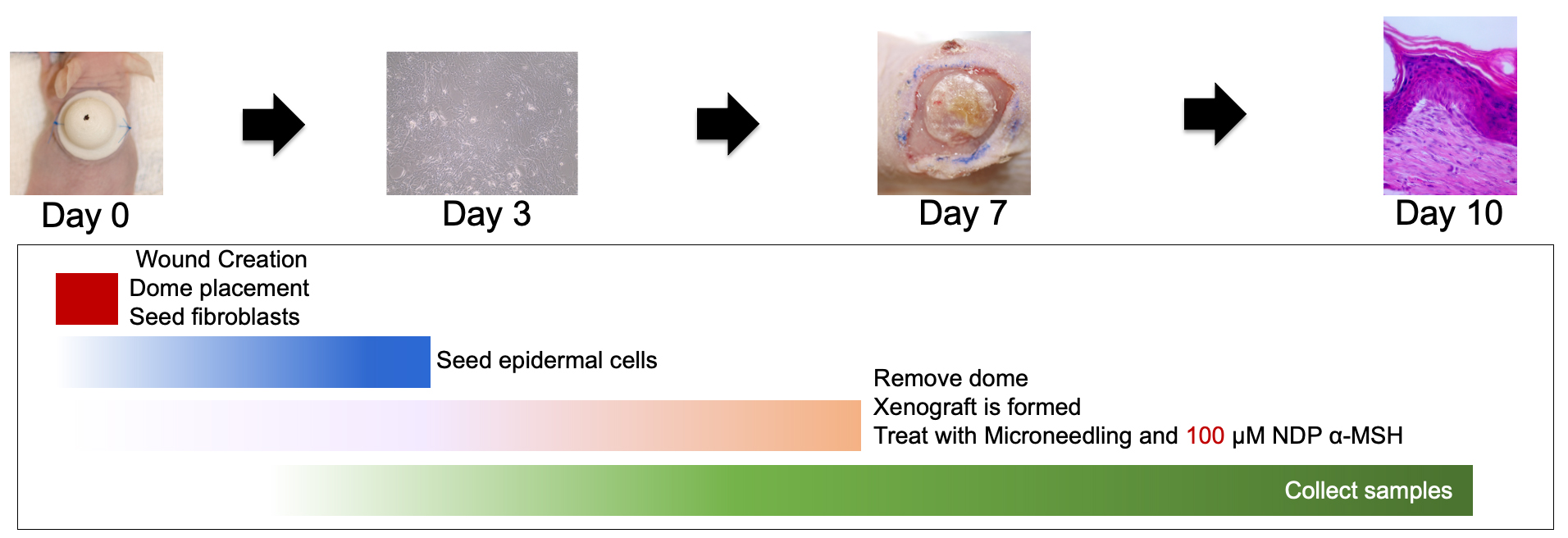
On day 7, the animals were anesthetized as described above and maintained on isoflurane via nose cone. The top of the dome was removed and discarded. The bottom flange was carefully removed, using a scalpel is necessary, with care taken not to disturb the fragile xenograft layer. The xenograft was photographed and covered with an occlusive Tegaderm dressing (Cardinal Health, Dublin, OH, USA) and the dressing was sutured in place at multiple contact points to prevent the animal from tampering with the xenograft (Fig. 3).
 Fig. 3.
Fig. 3.The xenografting procedure including dome removal, treatment, and sample
collection is detailed. NDP
On day 14, the animals were anesthetized as described above and maintained on isoflurane via nose cone. A necropsy was carried out and exsanguination was performed via cardiac stick. Once death was confirmed, the xenograft was excised and tied into a histology cassette to maintain the xenograft orientation. The cassette was then put into 10% neutral buffered formalin for fixation. Following fixation, the xenograft was paraffin-embedded, sectioned, and stained with H&E.
A number of different cell types (hyper-, hypo-, and normally pigmented scar and
skin) and treatment parameters were tested throughout the course of the model
development. Xenograft were successfully created using normally-pigmented
epidermal cells and normal porcine dermal fibroblasts (NPDF) (Table 1, Group 1).
Subsequently, hyper-pigmented (Table 1, Group 2) and hypo-pigmented (Table 1,
Group 3) xenografts were also successfully created. Once xenografts were
successfully created, normal porcine epidermal cells were used to test
treatments. A dosage of 10 µM NDP
| Group | Dermal Cell Type | Epidermal Cell Type |
| 1 | NPDF | NPE |
| 2 | HTSDF (P) | Hyper-PE |
| 3 | HTSDF (P) | Hypo-PE |
| 4 | NPDF | NPE |
| 5 | NPDF | NPE |
| 6 | HTSDF (P) | Hypo-PE |
| 7 | HTSDF (P) | Hypo-PE |
| 8 | HTSDF (P) | Hypo-PE |
Abbreviations: NPDF, Normal porcine dermal fibroblasts; NPE, normal porcine epidermal cells; HTSDF (P), Hypertrophic scar dermal fibroblasts-porcine; Hyper-PE, Hyper-pigmented porcine epidermal cells; Hypo-PE, Hypo-pigmented porcine epidermal cells.
| Group | Treatment | # of animals (sham/treatment) | Type of Sample Collected | Variable optimized or experiment performed |
| 1 | Day 7: 100 µM NDP |
3 sham/3 treatment | Day 10: | Treatment effect on normal cells? |
| Molecular preservation | ||||
| Punch bx for FFPE | ||||
| 2 | Day 7: 100 µM NDP |
3 sham/3 treatment | Day 10: | Treatment effect on hypo-pigmented cells? |
| Molecular preservation | ||||
| Punch bx for FFPE | Type of sample collected. | |||
| 3 | Day 7: 100 µM NDP |
3 sham/3 treatment | Day 10: | Treatment effect on hypo-pigmented cells? |
| Molecular preservation | ||||
| Punch bx for FFPE | Type of sample collected. | |||
| 4 | Day 7: 100 µM NDP |
3 sham/3 treatment | Day 10: | Treatment effect on hypo-pigmented cells? |
| Molecular preservation | ||||
| Punch bx for FFPE | Type of sample collected. |
FFPE, formalin fixed paraffin embedded.
A subset of animals with either normally-pigmented (n = 3) or hypo-pigmented (n
= 9) epidermal cells and HTSDFs were treated with NDP
From the formalin fixed paraffin embedded (FFPE) samples, sections were created and stained using H&E. Images were obtained to qualitatively characterize skin and scar xenograft structure and epidermal thickness.
From the samples preserved for molecular work, the RNeasy fibrous tissue kit was used to isolate and quantify mRNA from the xenografts. Melanin was also isolated from a group of tissues as previously described [41]. In brief, the pellet was dissolved in 1 N NaOH, incubated for 30 minutes at 100 °C, and melanin was measured by absorbance at 405 nm normalized to a standard curve with synthetic melanin (Sigma Aldrich, Burlington, MA, USA).
100 ng of RNA at a concentration of 10 ng/µL of mRNA was incubated with
primers, reverse transcriptase, and SYBR green. An increased amount of template
mRNA had to be used due to the low percentage of the xenograft that makes up the
epidermis, and the relatively low proportion of the epidermis that consists of
melanocytes. An iteration of this protocol was run with 10 ng of total mRNA and
the C
Protein was isolated from the molecularly-preserved xenografts using RIPA buffer with 10 µL/mL of protease inhibitor cocktail (Sigma Aldrich). A mortar and pestle was used to grind tissue in 1mL of RIPA buffer. The subsequent homogenate was sonicated (ThermoFisher Scientific, Waltham, MA, USA) with 5 pulses (1 sec pulse, 6 second pulse delay), then spun at 10,000 g for 10 minutes. The supernatant was saved and protein was quantified using a Bradford assay. Eighty mg of heated-denatured total protein per sample was subjected to Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 10% precast (Biorad) gels, along with precision plus protein western C ladder (Biorad). The proteins were transferred to a nitrocellulose membrane (Biorad, Hercules, CA, USA) and blocked for 3 hours with 3% non-fat dry milk. The primary antibody for TYRP1 (ab83774 at 1 µg/mL) was applied for overnight incubation. The membrane was rinsed with TBST, then the secondary antibody which was a goat-anti rabbit HRP-cinjugated antibody was applied at 1:10,000. The blot was then developed with SuperSignal West Dura substrate (ThermoFisher Scientific) and imaged with chemiluminescent imaging (Cytiva Life Sciences, Marlborough, MA, USA). Bands were quantified by densitometry analysis. Protein expression levels are divided by GAPDH to normalized to varying levels determined by densitometry, then plotted.
After the optimization of printing of domes, seeding volume, and cell leakage
(Table 1), normally pigmented porcine epidermal cells and normal porcine dermal
fibroblasts were used to create xenografts. The xenografts were imaged at day 10
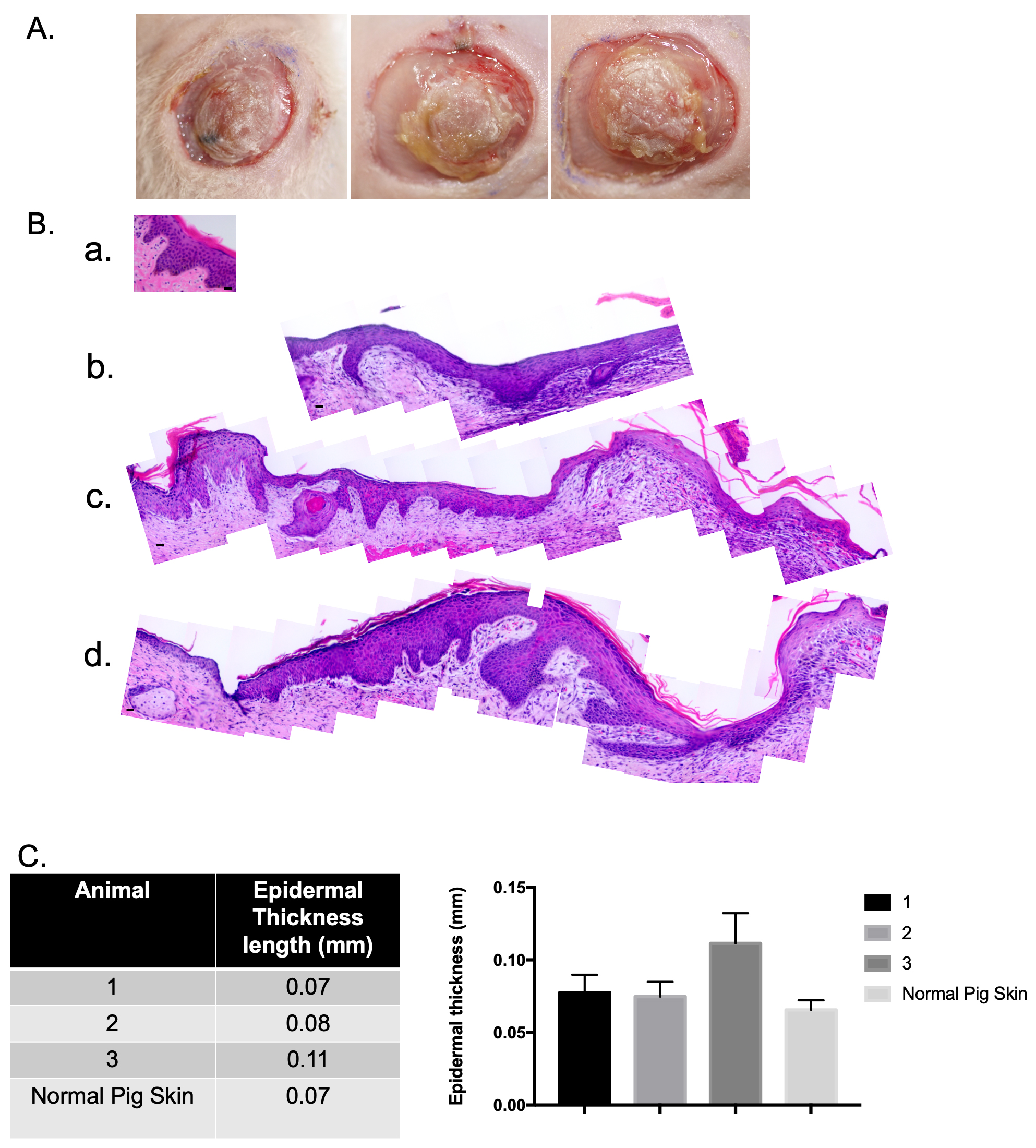
(Fig. 4A). Samples were collected on day 14 and were FFPE and stained with H&E.
The xenografts contain an epidermis and a dermis, and the epidermis looked
similar to uninjured pig skin (Fig. 4B) with the presence of epidermal rete
ridges and associated dermal papillae. When these different samples were measured
using ImageJ (Version 1.53a, National Institutes of Health, Bethesda, MD, USA),
it was concluded that a stratified epidermis in similar thickness to uninjured
pig skin was created (Fig. 4C). The xenograft was 0.11
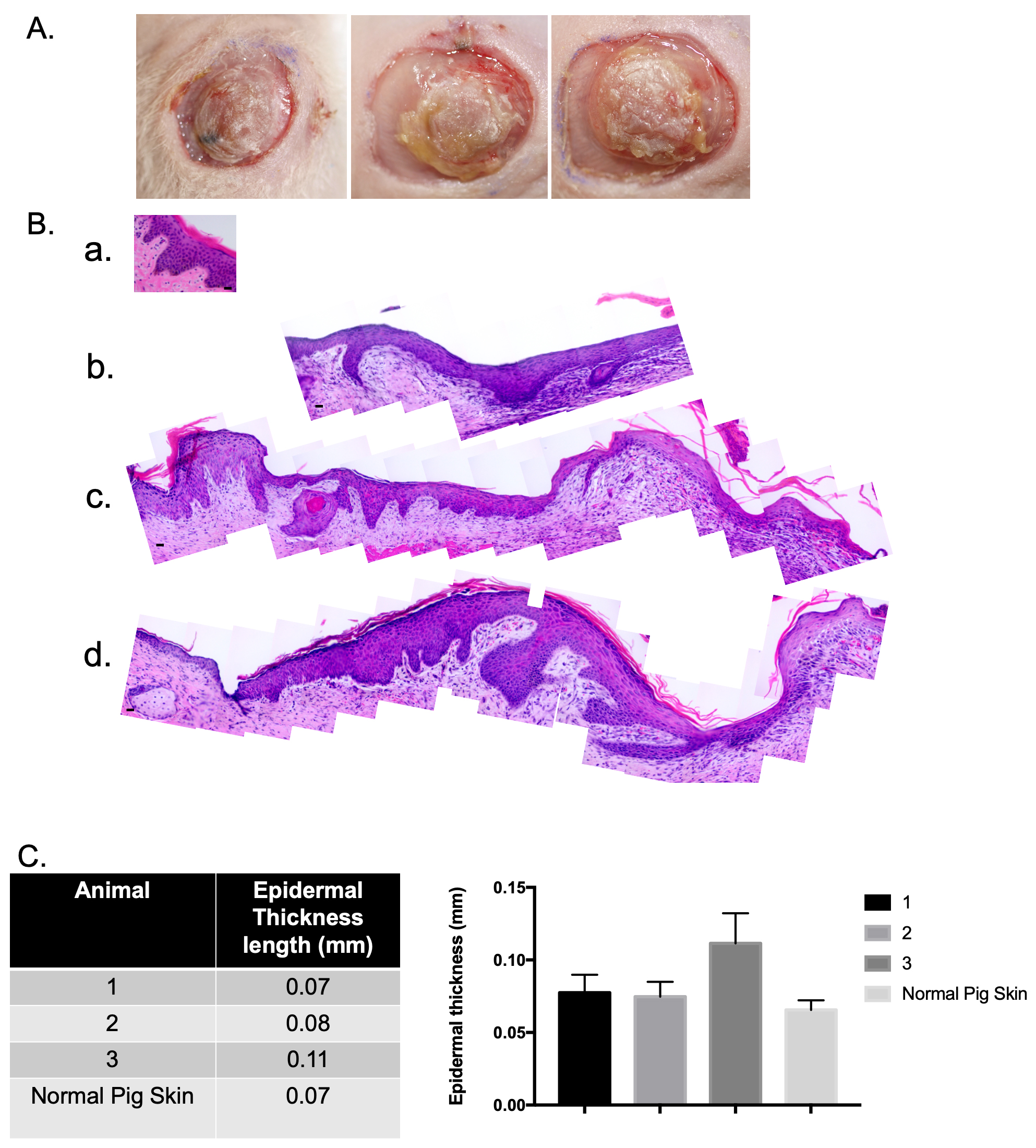 Fig. 4.
Fig. 4.Normally pigmented skin epidermal cells and normal porcine dermal
fibroblasts were used to create a xenograft that is histologically similar to
uninjured pig skin. Macroscopic images of xenografts at day 10 (A). Punch
biopsies of the xenografts were taken at day 14 and were FFPE and stained with
hematoxylin and eosin (H&E). Microscopic images of H&E-stained xenograft
sections (B). Un-injured porcine skin (a). Animal 1 (b). Animal 2 (c). Animal 3
(d). N = 3. Epidermal thickness was measured using ImageJ (C). (Scale bar = 20
µm) (mean
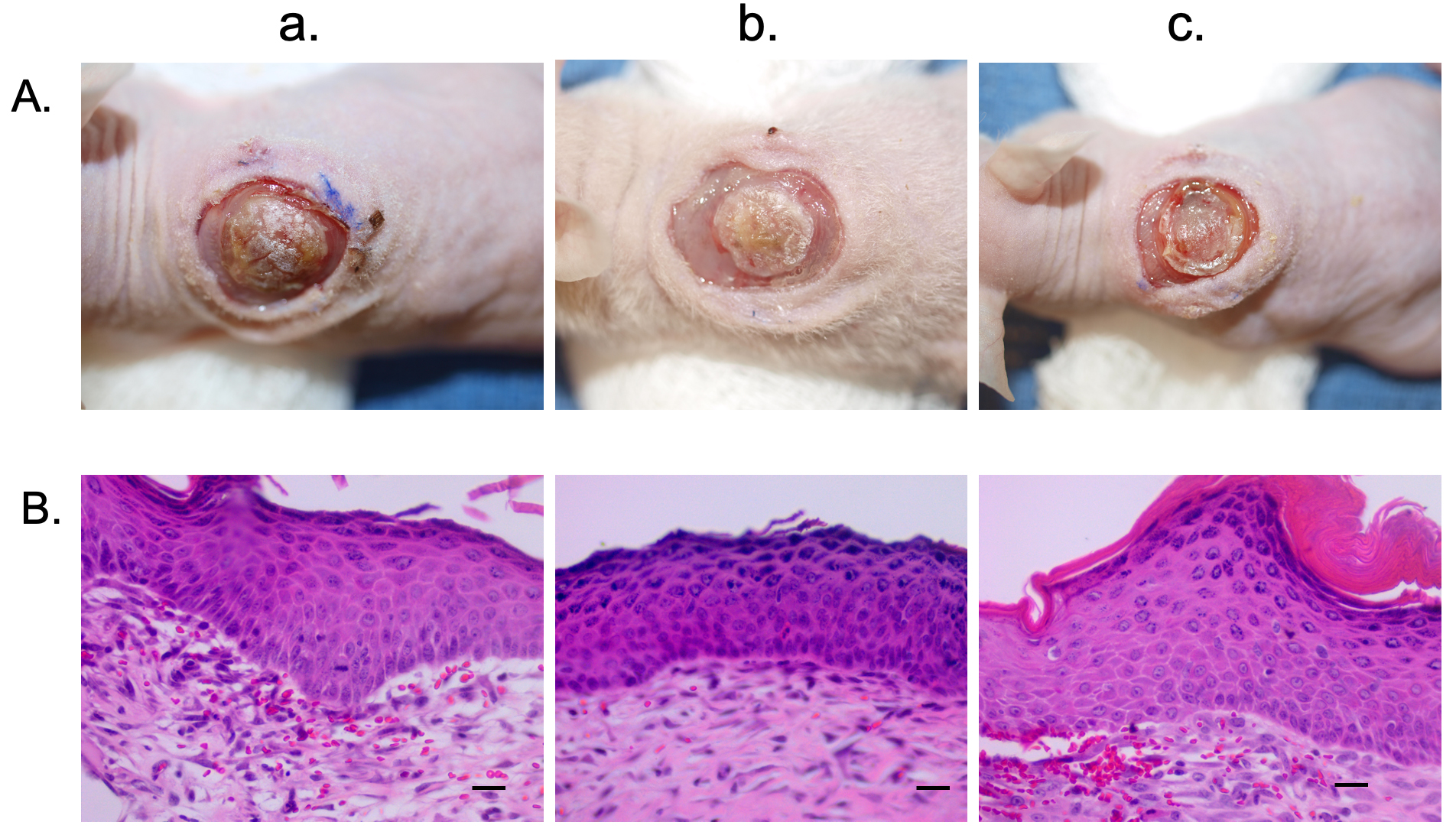
This model was also tested using hyper-pigmented scar epidermal cells. These cells resulted in similar xenografts to the normally pigmented xenografts (Fig. 5A). The histo-architecture of these xenografts revealed a xenograft similar in structure to hyper-pigmented scar in the pig model. There were few rete ridges, and the epidermis was thicker compared to the normally pigmented skin xenograft (Fig. 5B).
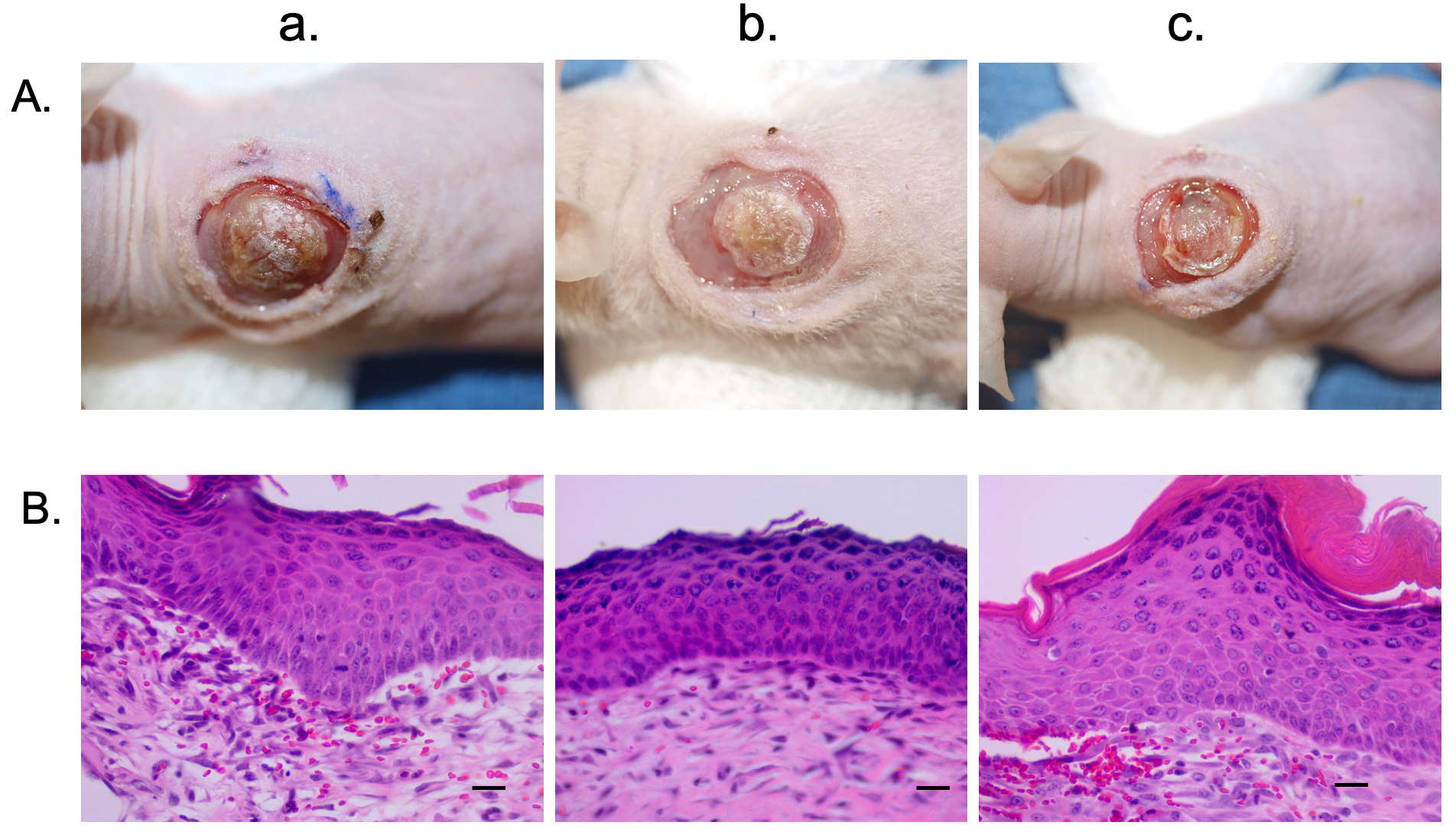 Fig. 5.
Fig. 5.Hyper-pigmented scar epidermal cells and HTS dermal fibroblasts were used to create a xenograft that is histologically similar to hyper-pigmented pig scar. Macroscopic images of xenografts at day 10 (A). Punch biopsies of the xenografts were taken at day 14 and were FFPE and stained with H&E. Microscopic images of H&E-stained xenograft sections (B). Animal 1 (a). Animal 2 (b). Animal 3 (c). N = 3 (bottom) (Scale bar = 20 µm).
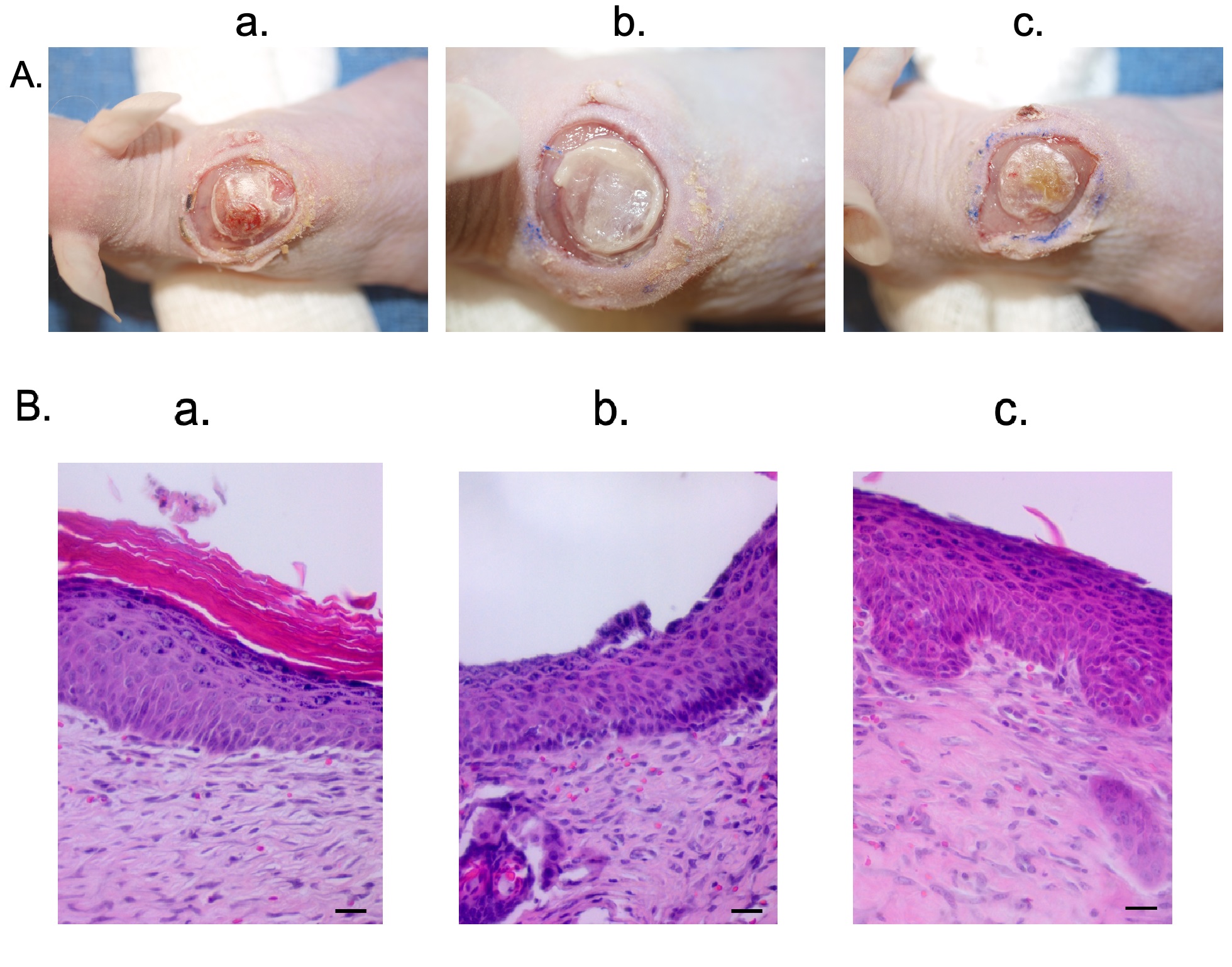
Last, the model was tested using hypo-pigmented scar epidermal cells. These cells resulted in very similar xenografts at the gross (Fig. 6A) and histologic level (Fig. 6B).
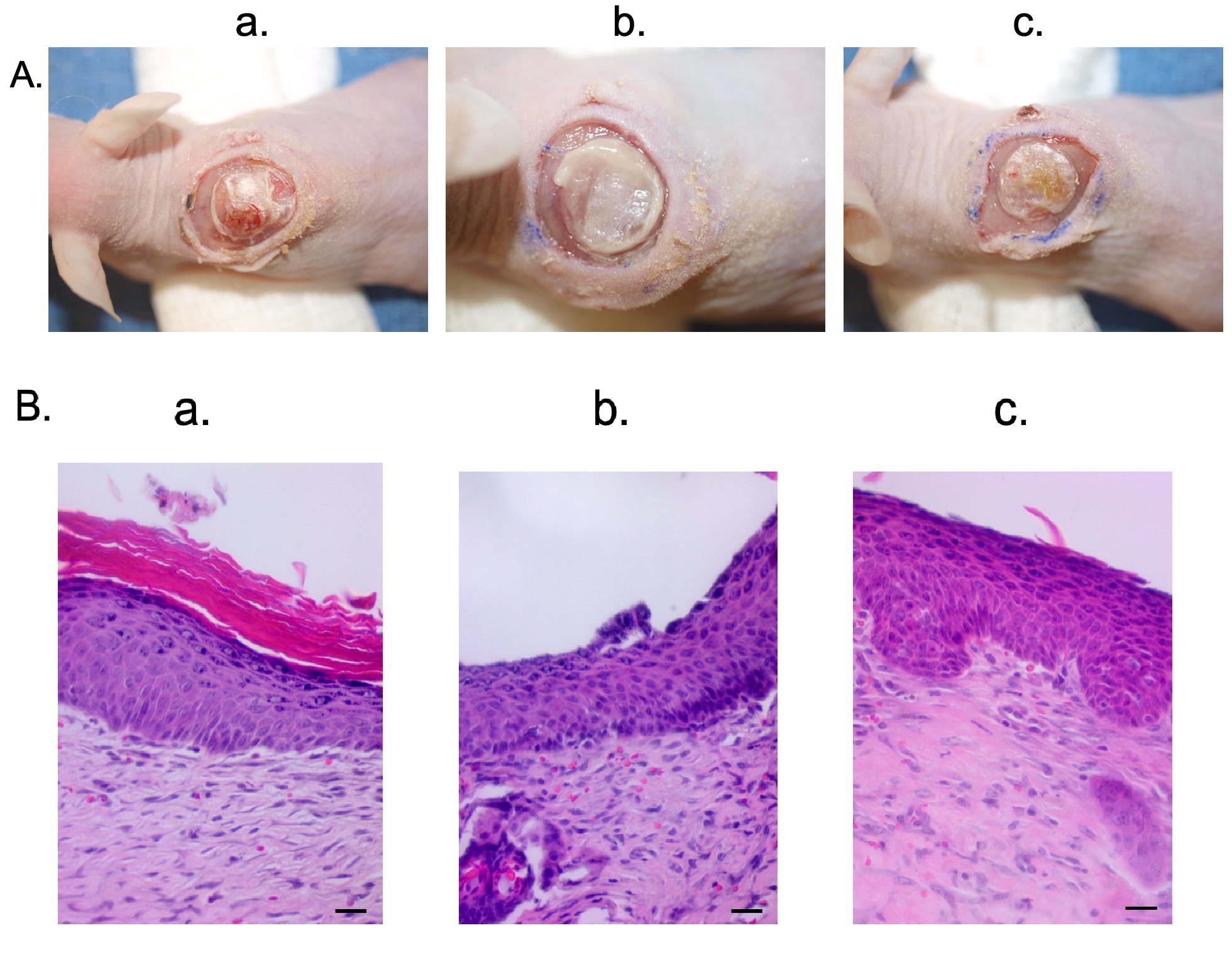 Fig. 6.
Fig. 6.Hypo-pigmented scar epidermal cells and HTS dermal fibroblasts were used to create a xenograft that is histologically similar to hypo-pigmented pig scar. Macroscopic images of xenografts at day 10 (A). Punch biopsies of the xenografts were taken at day 14 and were FFPE and stained with H&E. Microscopic images of H&E-stained xenograft sections (B). Animal 1 (a). Animal 2 (b). Animal 3 (c). N = 3 (bottom) (Scale bar = 20 µm).
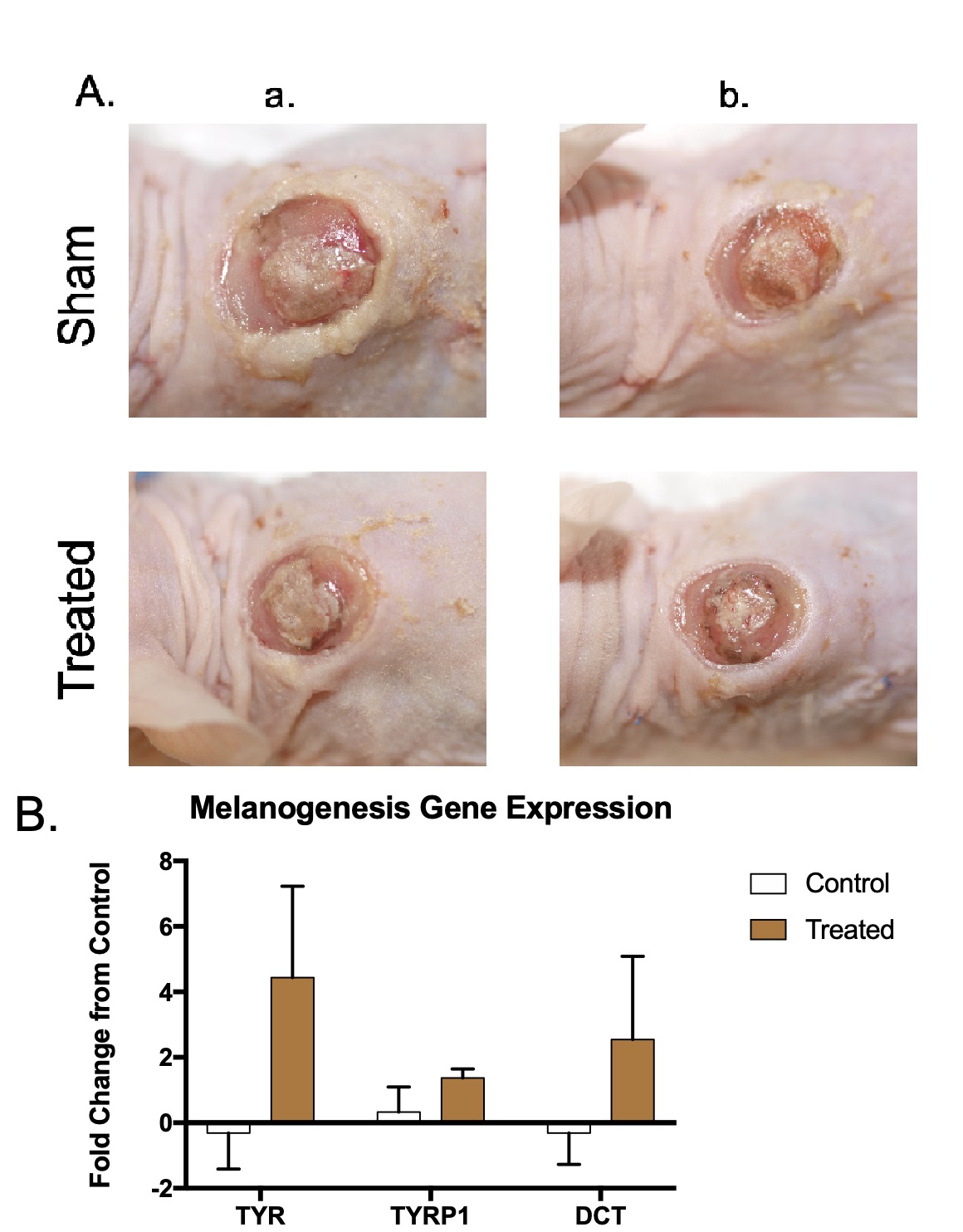
Normally pigmented xenografts were treated with microneedling and a 100 µM
topical application of NDP
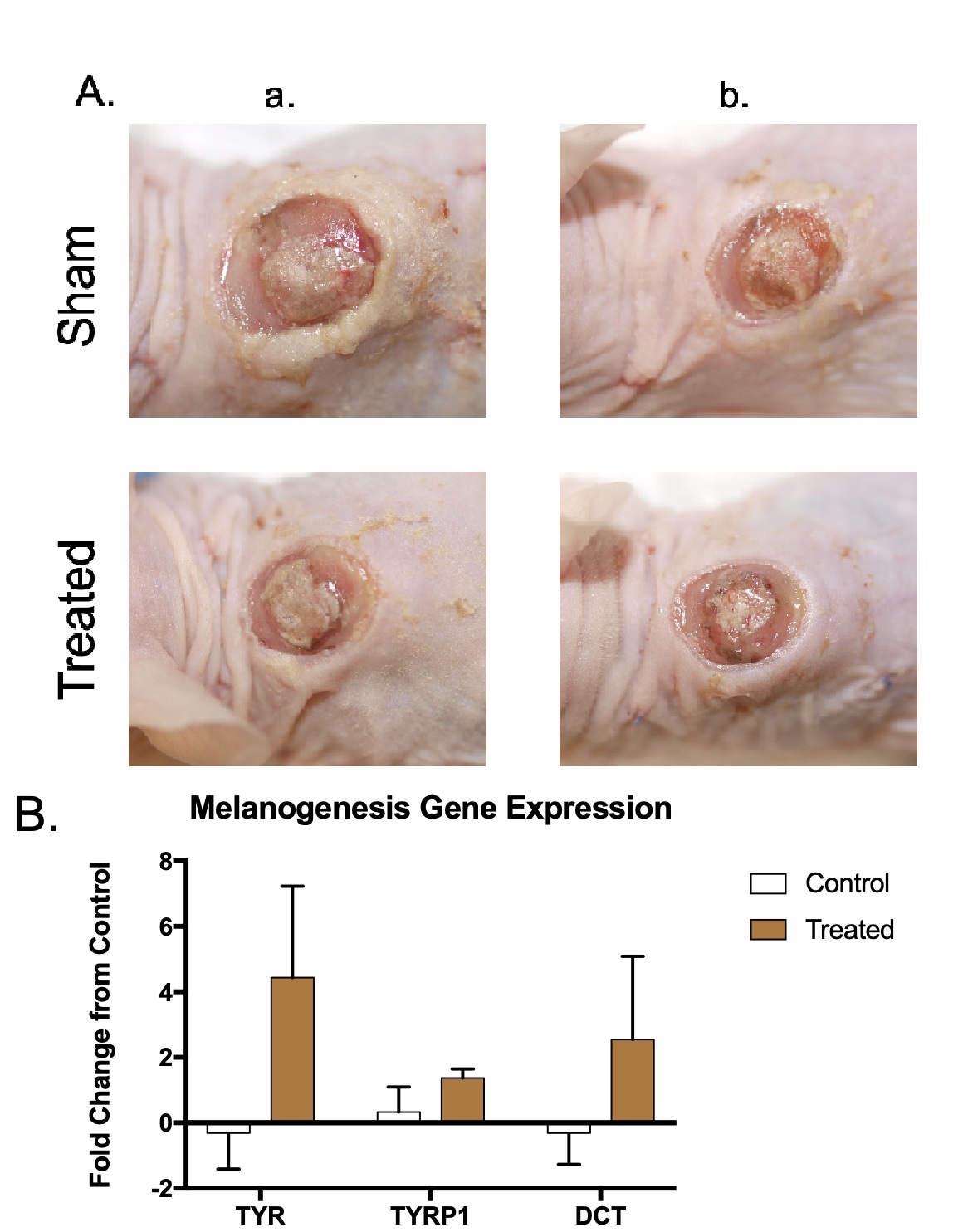 Fig. 7.
Fig. 7.Normally pigmented skin xenografts treated with microneedling and NDP
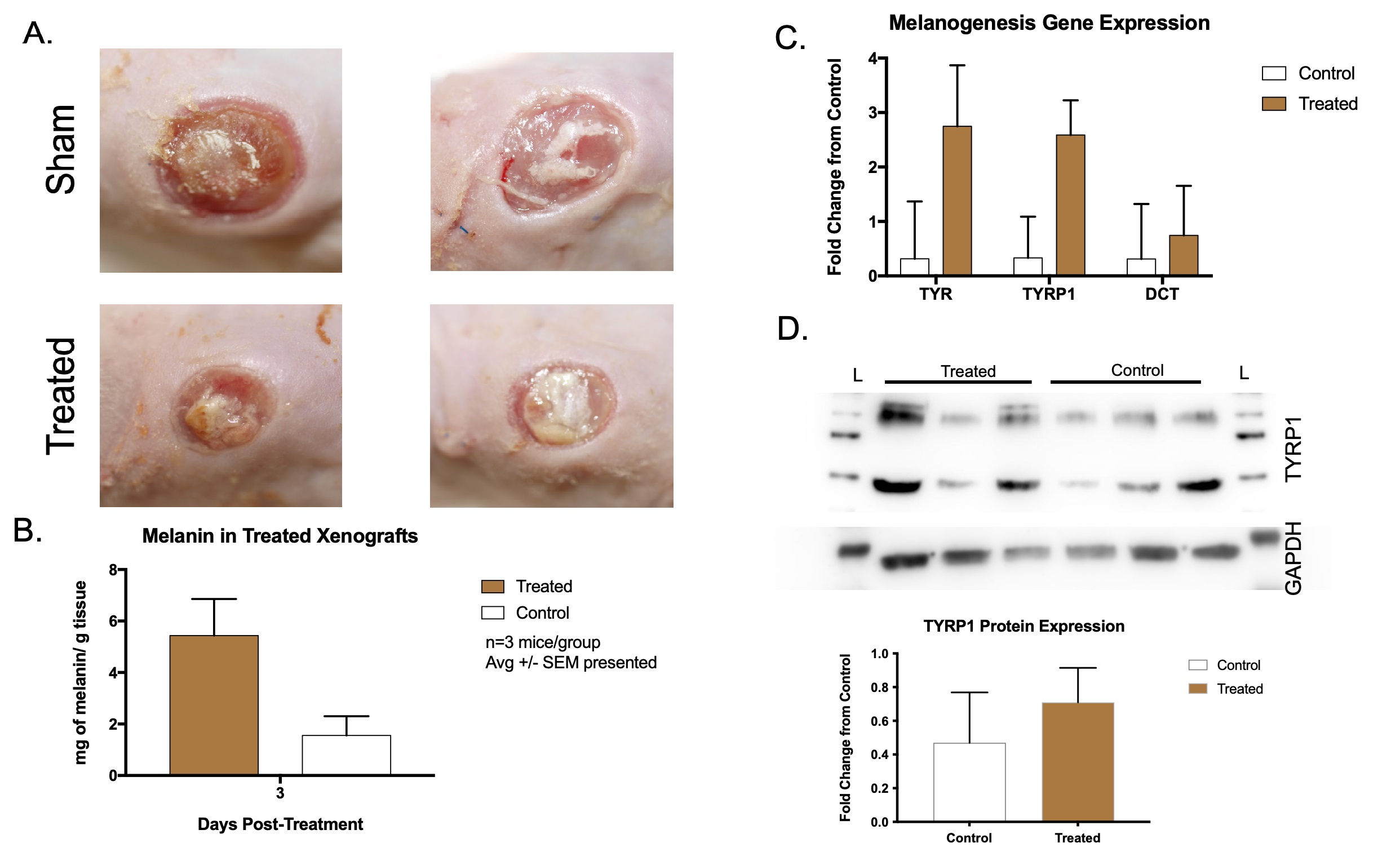
Hypo-pigmented xenografts were treated as above, and samples were collected at
day 10 for molecular analysis of melanin levels (Fig. 8A). Treated xenografts had
5.43
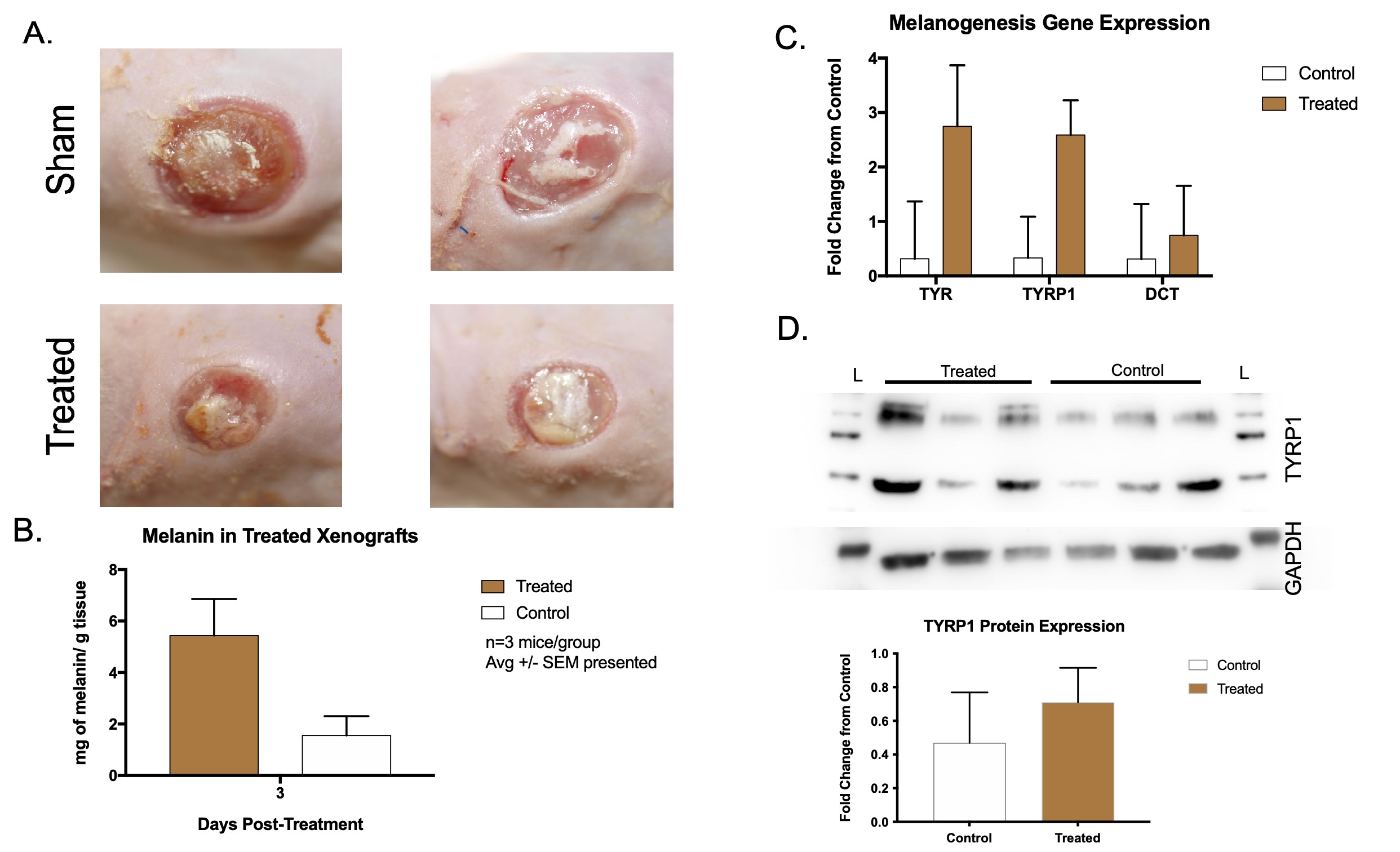 Fig. 8.
Fig. 8.Hypo-pigmented scar xenografts treated with microneedling and NDP
A nude mouse xenograft model is suitable for studying HTS in vivo. The
seeding of normal fibroblasts and normally-pigmented epidermal cells created a
full thickness skin equivalent that contained an epidermis and dermis. This
structure was similar to normal skin in its approximation of rete ridges. The
presence of melanocytes in this skin structure was confirmed when the
normally-pigmented xenografts were treated with synthetic NDP
HTS fibroblasts and hyper- and hypo-pigmented epidermal cells also created full
thickness skin with a stratified epithelium. These skin structures likewise
contained melanocytes of pig origin for the reasons described above. The use of
microneedling to create channels in the skin through which the topical
application of the drug could be delivered was a success in this model. One of
the reasons why it needed to be tested in vivo was to test if this
treatment could work to penetrate the epidermis and cause a response in
melanocytes. This work confirms that hypo-pigmented melanocytes do not lose the
ability to make melanin. They are simply in a state of non-pigment production,
and can be signaled to synthesize melanin with synthetic NDP
While we have previously xenografted primary human skin cells, patient-derived dyschromic HTS can only be derived in smaller amounts, within a limited time-frame, and only after healing. Additionally, patient HTS are most likely at the severe end of the spectrum because samples large enough to graft can most often only be collected from severe HTS that requires excision. Human wound studies are also limited by heterogeneity of the patients, wound types, and cells within the wound. Finally, HTS-prone patients are generally not included as a resource for preclinical studies because of the potential for worsening of scars.
There are several limitations to this model. One limitation is that, although the xenografts resemble skin upon histological examination, the xenografts do not entirely resemble normal porcine skin upon visual inspection. Although this is true, skin was deduced to be present when, 5–10 minutes after dome removal on day 7, the cells dried and the cornified layer typical of keratinocytes could be easily viewed (Supplementary Fig. 1). Another limitation is that the melanin that was made through transcription of TYR, TYPR1, and DCT could not be readily seen upon gross examination of the xenografts. This could be due to a loss of melanin after melanocyte production. Another possibility is that melanin levels did not reach those that could be detected by visual inspection. The advantage to this model is that it allowed for the confirmation of the hypothesis that hypo-pigmented cells could be stimulated to produce melanin related genes. Melanin was also detected using biochemical methods. However, future work will be aimed at using this treatment in the Duroc pig model. Another limitation of the xenograft model is the absence of professional immune cells, although human T cells have been used in xenografts in the study of psoriasis and other skin disorders. Clearly, the immune system plays an important role in hemostasis, wound healing, and scar formation. While the pathophysiological role of the immune system in HTS dyschromia has not been fully elucidated [42], pro-inflammatory cytokines, as well as reactive oxygen species, secreted in response to burns and other wounds may alter melanocyte proliferation and differentiation [43, 44, 45, 46].
This xenograft animal model can be used to study topical treatments for any skin condition. Cells from adult patients can be isolated, grown in culture, and seeded to make xenografts. Possible topic areas for study are systemic sclerosis, psoriasis, discoid lupus, or post-inflammatory hyper- or hypo-pigmentation. These xenografts could also be useful for topical drug delivery studies. In addition, different cell phenotypes can be mixed and matched using this model to study paracrine cell signaling. For example, normal fibroblasts could be mixed with pathogenic epidermal cells or vice versa. Finally, this model may have some advantages over direct testing of topical therapeutics in pig models because once promising compounds/techniques are studied in the xenograft mouse model, we can then go back from mouse xenograft to pigs with fewer animals yielding lower costs and more streamlined and time-efficient drug development processes.
We have also employed 3D cell culture models [47], derived either commercially (EpiDerm, MatTek) or within our lab, and found them to be invaluable for some applications [48]. In our hands, we found the xenograft model provides more reliable graft takes, especially with a 3-cell model (keratinocytes, fibroblasts, and melanocytes) than those from 3D cell culture models. The xenografts demonstrate longer survival of all three cell types, robust proliferation, and recapitulate epidermal biochemical and morphological differentiation. Further, the xenograft model works with or without cell immortalization or reprogramming, and works with adult and neonatally-derived skin cells. For these reasons, we have used it as a model for testing non-murine skin cells for its response to DNA alkylation [49], jet fuel toxicity [50], melanoma-melanocyte differentiation [32], sulfur mustard [29] and UVB-induced apoptosis. We find that the technique is easily learned, and completion time is similar to that of 3D histiotypic or organotypic culture models.
Adult skin cells can be xenografted onto nude mice to create a skin construct with epidermal and dermal layers that is responsive to pigmentation agonists. The skin constructs contain stratified epithelia and can be treated topically and through microneedling-assisted drug delivery.
Datasets are available upon reasonable request of the corresponding author.
BCC, CMSR, DSR, JWS designed the research study. BCC performed the research. BCC analyzed the data. BCC wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal work was approved by the MedStar Health Research Institute’s Institutional Animal Care and Use Committee (No. 2001000190 or 2018-001).
The authors acknowledge Nicholas J. Prindeze, MD for 3D printing the domes used in this work.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.