

 , Eryse Amira Seth 1, Mohd. Farooq Shaikh 1,2,*
, Eryse Amira Seth 1, Mohd. Farooq Shaikh 1,2,*
1 Neuropharmacology Research Laboratory, Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Bandar Sunway, SGR 47500, Malaysia
2 School of Dentistry and Medical Sciences, Charles Sturt University, Orange, NSW 2800, Australia
Abstract
Neuroinflammation has emerged as a shared molecular mechanism in epilepsy and cognitive impairment, offering new insights into the complex interplay between immune responses and brain function. Evidence reveals involvement of High mobility group box 1 (HMGB1) in blood-brain barrier disruption and correlations with epilepsy severity and drug resistance. While anti-inflammatory treatments show promise, translating these discoveries faces challenges in elucidating mechanisms and developing reliable biomarkers. However, strategically targeting neuroinflammation and HMGB1-mediated inflammation holds therapeutic potential. This review synthesises knowledge on HMGB1 and related biomarkers in epilepsy and cognitive impairment to shape future research and treatments targeting these intricate inflammatory processes.
Keywords
- neuroinflammatory biomarker
- epilepsy
- cognitive comorbidity
- HMGB1
Epilepsy and cognitive impairment are debilitating neurological conditions that often co-occur, and recent research has unveiled a common thread connecting them: neuroinflammation. Inflammation within the central nervous system, previously underappreciated in its significance, has now taken the spotlight as a key contributor to both the onset and progression of epilepsy and cognitive decline. With approximately 65 million individuals worldwide affected by epilepsy [1] and up to 80% experiencing cognitive dysfunction [2], the need for effective treatments is urgent. Unfortunately, current medications primarily manage symptoms [3], leaving one-third of epilepsy patients drug-resistant [4]. In many trials, anticonvulsant benefits of steroids and other anti-inflammatory drugs have been shown in drug-resistant epileptic patients [5, 6]. Understanding the mechanisms linking neuroinflammation, epilepsy, and cognitive decline is crucial, as it holds the potential to inform the development of more targeted and efficacious treatments and interventions [7].
Within the array of inflammatory mediators implicated in neuroinflammation, the protein High mobility group box 1 (HMGB1) emerges as a key molecule due to its pivotal roles in critical processes such as blood-brain barrier (BBB) disruption and its correlations with epilepsy severity, drug resistance, and symptomatic aetiology [8, 9]. This protein’s significance in the context of epilepsy and cognitive impairment stems from its role in exacerbating the neuroinflammatory processes that are central to the pathophysiology of these conditions. Elevated HMGB1 levels, particularly noted in patients with drug-resistant epilepsy, correlate with increased disease severity and a lack of response to conventional treatments, highlighting its importance [10]. This review aims to delve into the role of neuroinflammation, with a focus on HMGB1, in the pathogenesis of epilepsy and cognitive impairment. By synthesizing current knowledge surrounding HMGB1 and related inflammatory biomarkers, we aim to underscore their intricate involvement in the neuroinflammatory pathways of these conditions. Additionally, the challenges in translating these findings into clinical practice will be discussed, with emphasis placed on the potential for novel anti-inflammatory therapeutic strategies to shape future research and treatments targeting these intricate inflammatory processes.
The human body’s biological system responds to dangerous stimuli by activating the immune cascade and releasing inflammatory mediators as a defence mechanism. This process is called inflammation. The primary contributors to neuroinflammation are innate immune cells of the central nervous system (CNS), like microglia and astrocytes [11]. Neuroinflammation is also caused by adaptive immune cells like Tregs, Th, and B cells. The adaptive immune system affects neuroinflammation, and peripheral T cells initiate immune responses by interacting with other immune cells [12]. Though it is an immune response to protect the CNS, neuroinflammation can be dangerous when overexerted. This brain’s natural defence mechanism is triggered early in response to insults to stop neuronal damage and slow down neurodegeneration.
Various neural and non-neural cells interact in the molecular mechanisms underlying neuroinflammation to preserve cellular homeostasis, neuronal integrity, and tissue repair. In neurodegenerative diseases and neuroinflammation, microglia and astrocytes are important players. Their cytokine secretion and toll-like receptor (TLR) activation preserve synaptic plasticity and improve neurogenesis. Simultaneously, certain parallel mechanisms are triggered to regulate and improve neuroinflammation. Chronic inflammation can negatively impact the CNS, causing multiple system atrophy, Parkinson’s, Alzheimer’s and epilepsy [13]. Addressing these issues promptly and effectively is crucial to avoid long-lasting consequences.
A substantial body of research supports the key role of neuroinflammation in the pathogenesis of epilepsy, particularly brain injuries, infections, and certain immune-related conditions. Animal and human studies have demonstrated that brain inflammation contributes to cell loss and seizures [14, 15]. In both surgically removed brain tissue from epileptic patients and experimental models, particular inflammatory molecules and pathways have been found [16]. Many anti-inflammatory medications have demonstrated anticonvulsant properties in animal models and clinical epileptic patients [5]. However, there is no comprehensive body of evidence to support the direct correlation between neuroinflammation and epileptogenesis. The precise mechanisms behind neuroinflammatory signalling in epileptogenesis remain poorly comprehended [17, 18]. Several pathways may mediate how inflammatory mediators affect neuronal excitability and epilepsy. The anti-inflammatory therapies that benefit the susceptibility of epilepsy models are compiled in Table 1 (Ref. [15, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31]), along with their proposed mechanisms.
| Therapeutic intervention | Experimental model and Convulsant stimulus | Species, strain, age | Outcome | Effect and potential mechanism | Reference |
| Selective |
Fluid-percussion injury (FPI), Pentylenetetrazol (PTZ)-induced | Male Sprague-Dawley rats, 12 weeks old | - |
-ATI influences the production of inflammatory cytokines. | [19] |
| - |
|||||
| IL-1 receptor antagonist (100 mg/kg, s.c.) | Pediatric brain injury, PTZ-induced seizure | C57BL/6J mice, toddler-aged child | - |
-Blocking IL-1 |
[20] |
| - | |||||
| White rose petal extract (r. Hybrida) oral 50–200 mg/kg |
Kainic acid (KA) induced brain injury and epileptogenesis | Male Institute of Cancer research (ICR) mice, 7-weeks old | - |
-Downregulation of GPx, PHGPx, SOD1, and SOD2 prevents lipid peroxidation. | [23] |
| - |
-Downregulation of inflammatory markers, including COX2, and GFAP, HMGB1, IL-1 |
||||
| - |
|||||
| Monophosphoryl lipid A (MPL) and tri-palmitoyl-S-glyceryl-cysteine (Pam3Cys) toll-like receptor agonists, single dose (1 g/rat) i.c.v. | Controlled cortical impact device-induced trauma, then underwent amygdala kindling post24H trauma. | Adult male Wistar rats, 9 weeks old | -Return traumatic rats’ enhanced rate of epileptogenesis to normal. | -Suppression of overexpression of TNF- |
[25] |
| -MPL outperforms Pam3Cys in anti-inflammatory effects, restoring traumatised rats’ brain TNF- |
|||||
| Rapamycin 6 mg/kg i.p. after PTE induction. | FeCl2-induced PTE | Male Sprague-Dawley rats | - |
-Reduced levels of pmTOR and p-P70S6K levels indicate active mTOR signalling. | [26] |
| Isoliquiritigenin (ISL) | Kainic acid-induced epileptic | Young male Wistar rats | - |
-Attenuation of KA-induced expression of IB |
[27] |
| -Upregulation of superoxide dismutase and glutathione peroxidase activities. | |||||
| -Attenuation of the neuroinflammatory process through the TLR4/MYD88 signalling pathway. | |||||
| NAC (5 mg/kg, twice daily) + SFN (5 mg/kg, once daily) | Pilocarpine-induced epileptogenesis | Adult male Sprague-Dawley rats | - |
-Reduced oxidative stress inhibits the production of High mobility group box 1 (HMGB1). | [15] |
| - | |||||
| - |
|||||
| - |
|||||
| Retigabine (RTG), a prototype M-channel “opener”, 1.2 mg/kg i.v | Pilocarpine-induced status epilepticus | Adult C57BL/6 J mice, 10-weeks old | -Decrease in the expression of CD40L, a marker of inflammation, in the ipsilateral hemisphere. | [28] | |
| -Prevention of metabolic depletion of brain hemisphere cells ipsilateral to the damage site reduces neuroinflammation. | |||||
| Brivaracetam i.p, orally, s.c osmotic pump, or drinking water (combination or alone) | Rostral parasagittal fluid percussion injury (rpFPI) | Male Sprague- Dawley rats, 5-weeks old | - |
-Brivaracetam bound to the Synaptic Vesicle Glycoprotein prevents the cells in the ipsilateral brain hemisphere from reaching metabolic fatigue, suggesting a possible decrease in neuroinflammation 2A (SV2A) induced anti-inflammatory effect. | [29] |
| - | |||||
| - | |||||
| - | |||||
| Levetiracetam, atorvastatin and ceftriaxone (low doses) |
Intra-hippocampal KA-induced acquired temporal lobe epilepsy. | Outbred male NMRI mice | - |
-Levetiracetam modulates neurotransmitter release by binding to SV2A. | [30] |
| - |
-Atorvastatin is anti-inflammatory, antioxidant, and neuroprotective. | ||||
| -Ceftriaxone increases glutamate uptake and reduces excitotoxicity by upregulating GLT-1. | |||||
| Monophosphoryl lipid A (MPL) 1, or 0.1 µg/1 µL i.c.v. | TBI induced by a Controlled Cortical Impact (CCI) device, PTZ-induced seizure | Adult male Wistar rats, 9 weeks old | - |
-Decrease in TNF- |
[22] |
| -Upregulation of TGF- |
|||||
| -TBI-accelerated epileptogenesis returns to sham-operated levels. | |||||
| -Attenuating number of dead neurons. | |||||
| Jujuboside A 0.02 mg/kg/day |
FeCl3-induced model of PTE | Male Sprague-Dawley rats, 6-weeks old | - |
-Repression of p38 and ERK1/2 activation. | [31] |
| - | |||||
| Oxytocin, 1 µg/side/mouse, intra-mPFC infusion | Weight drops model, PTZ-induced seizure | Male C57/BL6J mice, 8–9 weeks old | - |
-Normalisation of BBB integrity. | [21] |
| -Inhibition of IL-1 | |||||
| EP2 receptor antagonist TG8260 (25 mg/kg, i.p.) | Rostral−parasagittal fluid percussion injury (rpFPI) in PTE | Sprague−Dawley rats | - |
-Reduced expression of pro- inflammatory mediators (COX-1, COX-2, EP2, IL-1 |
[24] |
| -Reduce rat cortex and hippocampus inflammation (COX-2, NOX2). |
s.c, subcutaneous; i.v, intravenous; i.p, intraperitoneal; i.c.v,
intracerebrovascular;
CD11b, Cluster of Differentiation 11b; COX, Cyclooxygenase; EP2, Prostaglandin
E2 receptor 2; FeCl3, Iron(III) Chloride; ERK, Extracellular Signal-Regulated
Kinase; GLT1, Glutamate Transporter 1; GFAP, Glial Fibrillary Acidic Protein;
GPx, Glutathione peroxidase; iNOS, Inducible Nitric Oxide Synthase; IBA1, Ionized
calcium-binding adapter molecule 1; mpges1, Microsomal Prostaglandin E
Synthase-1; NOX2, NADPH oxidase 2; NF-
Clinical immune activation data supports the theory linking neuroinflammation and seizure activity. The breakdown of the BBB which permits inflammatory mediators and peripheral immune cells to enter the brain parenchyma and intensify the inflammatory response, is a defining feature of neuroinflammation in epilepsy. This interference with brain communication routes impacts all CNS cells, including glial and neuronal cells. Pro-inflammatory cytokines, chemokines, and reactive oxygen species are released due to increased glial activation pathways. These factors contribute to neuronal hyperexcitability and synaptic dysfunction, which in turn cause seizures [19, 20, 21].
Pro-inflammatory mediators such as Interleukin (IL)-8, IL-12, IL-18, transforming growth factor (TGF), and pro-inflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor (TNF), are crucial in the neuropathology of epilepsy. These mediators cause glia, neurons, and BBB constituents to activate their intracellular signalling pathways. The TLR pathway starts innate immune responses by recognising pathogens-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). In epilepsy, TLR signalling dysregulation releases pro-inflammatory cytokines, worsens neuroinflammation, and affects cell function, turning a normal person into an epileptic one [22, 23, 24]. Understanding this event cascade is crucial to identifying potential therapeutic targets and interventions.
Epileptic individuals exhibit a significant prevalence of cognitive and psychiatric comorbidities, which impair their overall quality of life [32]. These complications can often be more incapacitating for pharmacoresistant patients than the seizures themselves. There appears to be a correlation between the type of epilepsy syndrome and cognitive impairment in epilepsy patients. People with generalised tonic-clonic epilepsy, for example, are significantly more likely than healthy subjects to have impairments in memory, attention, visuospatial, and language functions [33]. Status epilepticus (SE) causes memory loss that lasts weeks to years [34]. In fact, cognitive deficits are common at diagnosis, even for non-SE epilepsies [35]. In posttraumatic epilepsy (PTE), up to 80% of patients experience some form of cognitive dysfunction following TBI [36], but the mechanism remains poorly understood. Neuroinflammation has emerged as a key contributor to cognitive dysfunction in epilepsy patients. The disruption of neural networks and the loss of synapses following the brain injury insult may be the primary initial cause, which is followed by a cascade of neuroinflammatory processes.
Damaged neurons and glial cells release DAMPs such as calcium-binding protein B
(S100B) and high mobility group box 1 protein (HMGB1). They will then be
recognised by pattern recognition receptors (PRR) on immune cells, such as
microglia and astrocytes, activating these cells and causing the release of
pro-inflammatory cytokines [37]. The function of TLR in the innate immune
system’s reaction to DAMPs and PAMPs is crucial [38]. Through the activation of
receptors, including toll-like receptor 4 (TLR4) and receptor for advanced
glycation end products (RAGE), interleukin-1
Additionally, pro-inflammatory cytokines like tumour necrosis factor-
| Intervention | Preclinical model | Outcome | Neuroinflammatory pathology involved | Mechanism suggested | Reference |
| Astaxanthin (AST), 30 mg/kg, i.p | Lithium chloride and pilocarpine-induced SE on rats | - |
-ATP-P2X7R signal pathway. | - |
[41] |
| - |
-Inflammatory cytokine genes (TNF- |
||||
| N. incisum root extract (NRE) 1 g/kg/d oral for 7 days, then i.p on 8th day | PTZ-induced epilepsy in mice | - |
Inhibition of microglia and astrocytes in the hippocampus and cortex. | - |
[42] |
| - |
|||||
| - |
|||||
| Low thiamine (25 mg/kg), or high thiamine (50 mg/kg). | PTZ-induced kindling rat model | - |
Pro-inflammatory cytokines and mediators. | - |
[43] |
| - |
- |
||||
| Prostaglandin E2 (EP2) antagonist, TG11-77, 8.8 mg/kg. i.p. | Pilocarpine-induced SE on rats | - |
Cyclooxygenase-2 (COX-2)- related pathways. | -Following COX-2-induced brain seizures, EP2 receptor activation causes BBB leakage, inflammatory response, neuronal damage, and cognitive impairment. | [44] |
| -Microgliosis and delayed mortality. | -TG11-77 reduces COX-2 induction, delayed mortality, and SE memory deficit. | ||||
| Captopril (50 mg/kg/day) i.p. | Kainic acid (KA) induced SE in rats | - |
Immune complement signalling through C3 protein. | -Inhibited phagocytosis mediated by the complement system, reversal of glial stimulation, and production of inflammatory factors. | [45] |
| -Suppression of epilepsy-induced astrocyte-microglia activation and abnormal contact. | |||||
| - |
High Mobility Group Box 1 (HMGB1), initially recognised for its role in gene transcription regulation, has emerged as a pivotal player in epileptogenesis. Its duality as a chromatin-associated protein and a DAMP molecule highlight its intricate involvement in neuroinflammation, immune responses, and, more recently, cognitive impairment [46, 47]. Cumulative evidence consistently reports heightened HMGB1 levels in epilepsy patients, particularly those grappling with drug-resistant epilepsy (DRE). The concomitant correlation with disease severity, drug resistance, and symptomatic aetiology posits HMGB1 as a promising prognostic biomarker [45]. A summary of studies on the role of HMGB1 in epilepsy and cognition is given in Table 3 (Ref. [10, 48, 49, 50, 51, 52]).
| Study | Sample size, Age | Source of sample, Method | Study design, duration. | HMGB1 level | Predictive/diagnostic value | Effect on epilepsy/cognition |
| [10] | 105 epilepsy (29.65 |
Serum, ELISA | Case-control | Epilepsy: 6.3, 4.4–7.8 ng/mL | -HMGB1 predictive value of epilepsy risk: Area Uunder Curve (AUC) of 0.905 (95% CI, 0.864–0.946). | -HMGB1 levels are higher in epilepsy than HC (p |
| 100 HC (29.83 |
HC: 1.9, 2.4–3.3 ng/mL (p |
-HMGB1 expressions were linked to higher epilepsy risk and severity (p | ||||
| -Patients resistant to anti-epilepsy medications have higher HMGB1 (p = 0.002). | ||||||
| [49] | 28 severe epilepsy, 29 mild epilepsy, 27 HC (Children 4-17 y.o) | Serum, ELISA | Prospective, cross-sectional | HC: 721.40 |
- | -The group with severe epilepsy had a higher mean HMGB1 value than mild epilepsy and HC group (p = 0.001). |
| Mild: 859.40 |
||||||
| Severe: 1056.73 |
||||||
| [50] | 27 DRE, (15–67 y.o) | CSF and serum, ELISA | Prospective, cross-sectional | CSF | - | -DRE and NDE have higher CSF HMGB1 levels than ONND (p |
| 56 NDE, (14–79 y.o.) | DRE: 5.08 |
-Higher serum HMGB1 in DRE than NDE (p = 0.002) and ONND (p | ||||
| 22 ONNDs, (17–80 y.o) | NDE: 3.03 |
-Epilepsy severity, resistance cases, and symptomatic aetiology were linked to CSF HMGB1 levels (p | ||||
| ONND: 0.83 |
||||||
| Serum | ||||||
| DRE: 4.53 |
||||||
| NDE: 2.32 |
||||||
| ONND: 1.56 |
||||||
| [51] | 65 DRE (17– 65 y.o.) | Serum, ELISA | Prospective, cross-sectional | DRE: 8.70 |
-HMGB1 predictive value of DRE: AUC of 0.99; cut-off value 2.3 ng/mL | -DRE has higher HMGB1 levels than non-DRE and HC (p |
| 74 HC (19–66 y.o.) | Non-DRE: 1.25 |
-Higher HMGB1 levels in the DRE with abnormal MRI than without abnormalities (p | ||||
| 26 non-DRE (17–60 y.o) | DRE with abnormal brain MRI = 9.8 |
|||||
| DRE without abnormalities: 7.4 |
||||||
| [52] | 180 NDE, 40 HC (Children 1 month-13 y.o) | Serum, ELISA | Prospective, longitudinal (18 months) | Not mentioned. | -HMGB1 predictive value of seizure frequency: AUC of 0.947; cut-off value 9.625 ng/mL (sensitivity 80.6%, specificity 92.5%) | -Epilepsy patients had significantly higher levels of HMGB1, S-100B, IL-1 |
| -IL-1 |
-HMGB1, IL-1 | |||||
| -The AUC of HMGB1 was significantly higher than IL-1 |
-Serum HMGB1 predicted seizure frequency better than IL-1 | |||||
| [48] | 127 epilepsy, 120 HC, (18–79 y.o) | ELISA, serum | Prospective, Cross-sectional | Epilepsy type: | -HMGB1 diagnostic value for epilepsy: AUC of 0.870; cut-off value 4.260 ng/mL (sensitivity 71.65%, specificity 92.5%) | -Higher serum HMGB1 in epilepsy than HC (p |
| Primary 5.42 |
-MMP-9 and HMGB1 combined detection in diagnosis for epilepsy: AUC of 0.903; cut off value 0.531 pg/mL (sensitivity 79.17%, specificity 89.17%) | -HMGB1 level negatively correlates with MMSE scores in epilepsy patients (r = –0.732, p | ||||
| Secondary 5.39 |
-HMGB1 linked to epilepsy duration, seizures, and treatment (p | |||||
| Seizure activity: | ||||||
| Grand mal seizure 5.92 |
||||||
| Petit mal seizure 4.76 |
||||||
| Psychomotor seizure 5.88 |
||||||
| Focal seizure 5.22 |
||||||
| Complex partial seizure | ||||||
| 5.60 |
CSF, cerebrospinal fluid; HC, Healthy control; DRE, Drug-refractory epilepsy; NDE, newly diagnosed epilepsy; ONND, other non-inflammatory neurological disorders; ELISA, enzyme-linked immunosorbent assay. The underscores differentiate values for two different fluids CSF and Serum.
Recent investigations delve into the nuanced signalling pathways mediated by
HMGB1, notably the HMGB1/TLR4 axis and IL-1
In addition to its role in signalling cascades, HMGB1 contributes to disrupting the BBB, a pivotal event in epileptic pathology. Physiologically, HMGB1 exists at basal levels in the brain. However, under inflammatory conditions such as traumatic brain injury (TBI) events or stimulation by hypoxia or seizures, its release is induced from neurons, glial cells, and endothelial cells. The therapeutic potential of targeting HMGB1 and its downstream pathways with specific antagonists emerges as a promising avenue for epilepsy treatment, warranting further exploration [48].
However, HMGB1 is not the sole biomarker of interest. DAMPs, cytokines and
chemokines are also pivotal in the neuroinflammatory pathway. Notably,
IL-1
Fig. 1 illustrates the neuroinflammatory pathway linking a brain injury event of
TBI to the release of HMGB1, which triggers increased expression of
pro-inflammatory cytokines such as IL-1
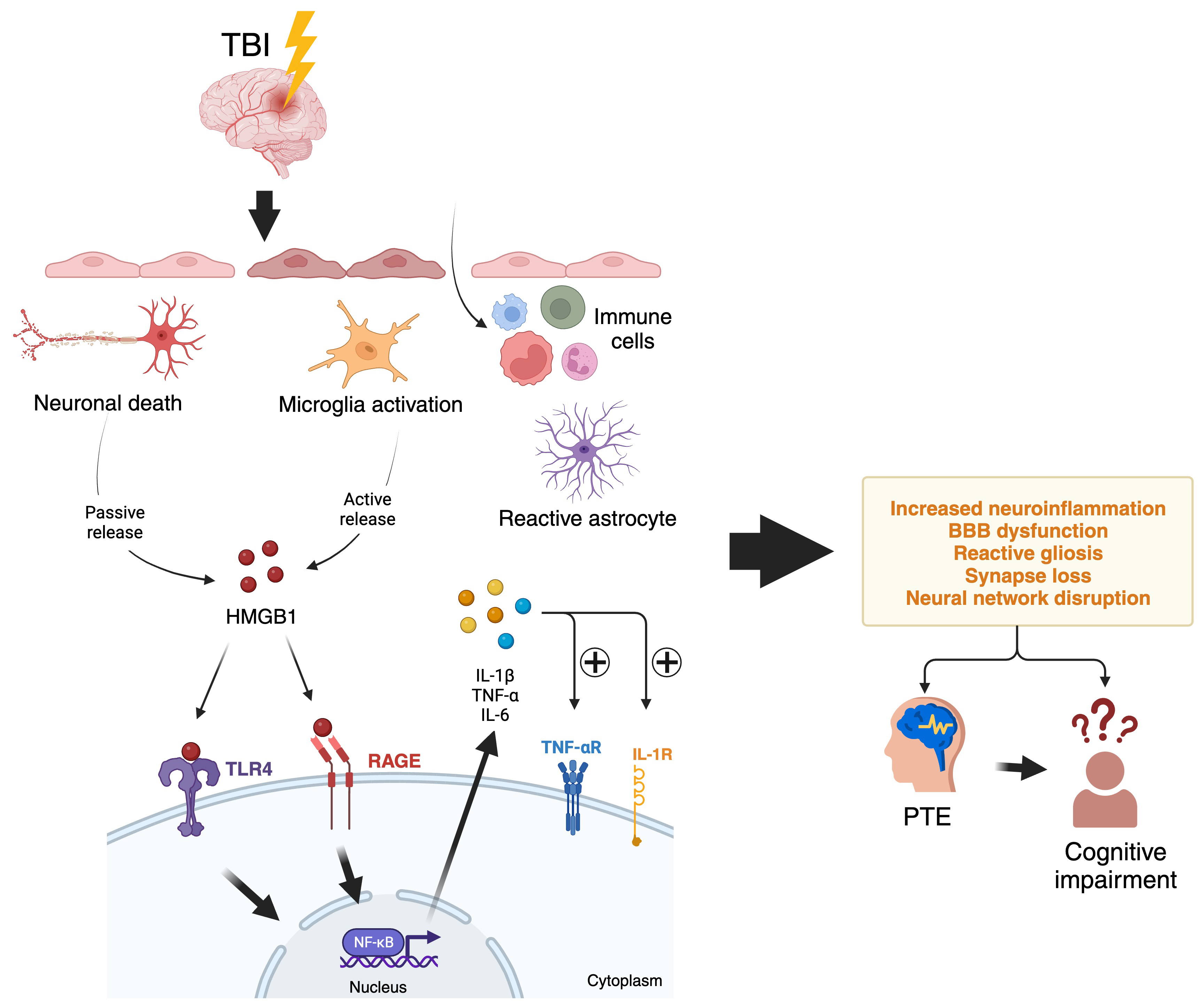 Fig. 1.
Fig. 1.Neuroinflammatory pathways from a TBI event that
triggers HMGB1 release and leads to epilepsy and cognitive impairment. A TBI
injury will cause acute cell damage and death in the brain. Cells undergoing
necrosis release HMGB1 passively due to loss of membrane integrity, while
stressed neurons also actively secrete HMGB1 in response to cell changes from
TBI. The extracellular HMGB1 binds to TLR4 and the RAGE receptors on microglia
and other immune cells, initiating a signalling cascade. This cascade activates
the transcription factor NF-
While there is increasing evidence of HMGB1’s role in neuroinflammation, there are significant challenges in translating this understanding into effective clinical interventions. One of the main hurdles lies in unraveling the complex molecular mechanisms underlying HMGB1’s involvement in neuroinflammation-associated pathologies. Although research has demonstrated that HMGB1 can activate pro-inflammatory pathways, disrupt the BBB, and worsen neurodegeneration, a comprehensive understanding of the precise spatiotemporal dynamics and context-dependent roles of HMGB1 in different disease states is still lacking [58]. The exact molecular mechanisms through which HMGB1 influences neurological disorders are subject to ongoing research, making it difficult to determine whether HMGB1 is a primary driver or a downstream consequence of neuronal damage.
The existence of different isoforms of HMGB1 in the extracellular matrix adds another layer of complexity to understanding the specific function of individual isoforms [58]. Consequently, utilising HMGB1 as a biomarker becomes increasingly challenging. Although HMGB1 has potential as a biomarker candidate, there are difficulties in standardizing detection methods, addressing isoform-specific variations, and establishing correlations between HMGB1 levels and disease severity or progression [15]. Significant knowledge gaps exist regarding the intricate nature of the neuroinflammatory pathway and the roles played by various biomarkers such as HMGB1. These gaps include our limited understanding of the causal relationships between neuroinflammation, epilepsy, cognitive impairment, and the temporal dynamics of biomarker expression [59]. Conflicting evidence exists regarding these causal relationships, hence addressing these gaps is necessary to advance research and develop therapeutic strategies effectively.
Ethical considerations also arise when exploring anti-inflammatory or immunomodulatory therapies, especially in vulnerable populations. The translation of HMGB1-targeted therapies from the laboratory to clinical practice requires robust preclinical safety assessments, rigorous clinical trial protocols, and transparent communication of risks and benefits. These aspects are crucial for ensuring ethical clinical translation in neuroinflammation research, particularly regarding vulnerable populations and the possibility of off-target effects or unintended immunomodulatory consequences [59].
In terms of therapeutic development, preclinical studies have shown promising results for HMGB1-targeted interventions, including small molecule inhibitors, monoclonal antibodies, gene silencing techniques, and cell-based therapies [15, 16]. However, translating these preclinical successes into clinical trials presents challenges such as optimizing drug delivery methods, selecting appropriate outcome measures, and accounting for individual variability in treatment response. Additionally, the multifunctional nature of HMGB1, with both pro-inflammatory and immunomodulatory properties, underscores the need for tailored therapeutic approaches specific to the disease context and patient population [8]. To effectively translate HMGB1-targeted therapies into clinical use, extensive research is required to verify their safety and effectiveness. Establishing the causal relationship between neuroinflammation, epilepsy, and cognitive impairment is like navigating a complex maze. Challenges in this effort include addressing heterogeneity, considering bidirectional influences, accounting for comorbidities, incorporating age-related factors, accommodating individual variability, utilising suitable experimental models, and adhering to ethical considerations.
The development of reliable biomarkers for neuroinflammation in epilepsy and cognitive impairment is critical for early diagnosis and monitoring. However, identifying such biomarkers remains challenging, as current diagnostic tools often lack sensitivity and specificity [60]. Further investigation using advanced imaging techniques, single-cell omics analyses, and disease-specific animal models is vital for fully comprehending the complexities of HMGB1’s involvement in neuroinflammation-related disorders, such as epilepsy and cognitive impairment [61].
The increasing recognition of HMGB1 and inflammatory biomarkers in
neuroinflammation not only enhances our understanding but also reveals promising
therapeutic opportunities. In epilepsy, studies have implicated inflammatory
molecules such as IL-1
In preclinical studies, HMGB1-targeted interventions have demonstrated the efficacy of various strategies, such as HMGB1-neutralizing antibodies, receptor antagonists, and inhibitors of HMGB1 release. These interventions hold potential for clinical applications, including the attenuation of neuroinflammation, reduction of seizure frequency, and improvement of cognitive function in epilepsy and cognitive impairment [63]. Importantly, the concept of biomarker-guided therapeutics, with HMGB1 as a potential candidate biomarker, offers real-time insights into disease progression and enables personalized interventions. For example, monitoring HMGB1 levels in cerebrospinal fluid or serum may provide valuable information on treatment response and disease prognosis in neuroinflammatory disorders.
The prospects of HMGB1 as a therapeutic target are promising, with specific evidence highlighting its relevance in neuroinflammatory pathways and potential clinical applications. By elucidating the roles of inflammatory molecules, TLR, and HMGB1-targeted interventions, we pave the way for more tailored and effective treatments for individuals with neurological disorders.
Neuroinflammation is emerging as a shared molecular mechanism in epilepsy and cognitive impairment, offering hope for novel therapeutic strategies. HMGB1, a versatile protein with dual roles, is a central player in this intricate pathway alongside other inflammatory biomarkers. However, there is much to uncover about the precise mechanisms and temporal relationships in these complex conditions. Overcoming the translational challenges, from biomarker development to personalised treatment strategies, is essential to harness the therapeutic potential of these inflammatory markers in these neurological disorders.
IWN and MFS designed the review concept. IWN analysed the data and wrote the original manuscript. EAS provided help with illustration. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was funded by the Jeffrey Cheah School of Medicine and Health Science Research Strategic Grant 2021.
The authors declare no conflict of interest. Given his role as Guest Editor, Mohd. Farooq Shaikh had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Lin-Hua Jiang and Igor Lavrov.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.