






 , Wiwit Ananda Wahyu Setyaningsih 1,2
, Wiwit Ananda Wahyu Setyaningsih 1,21 The Wellcome-Wolfson Institute for Experimental Medicine, Queen’s University Belfast, BT9 7BL Belfast, UK
2 Department of Anatomy, Faculty of Medicine, Public Health, and Nursing, Universitas Gadjah Mada, 55281 Yogyakarta, Indonesia
Abstract
Endothelial cell dysfunction is a complex process involving various causes, early and late events, and subsequent consequences. This review provides an overview of each aspect and outlines therapeutic interventions targeting these stages. Causes of endothelial dysfunction encompass a spectrum of risk factors including hypertension, diabetes, smoking, obesity, inflammation, oxidative stress, and genetic predispositions. Early events such as endothelial activation, inflammatory response, and dysregulated vasomotor tone precede late events like oxidative stress, endothelial apoptosis, and microvascular rarefaction. The consequences include endothelial remodelling, neovascularization, organ dysfunction, and clinical manifestations, highlighting the diverse impacts across multiple systems. While depicted linearly, the progression of endothelial dysfunction is dynamic, influenced by various factors such as the underlying cause and affected vascular bed. Understanding these dynamics is crucial for tailoring therapeutic interventions, ranging from lifestyle modifications to targeted therapies, to address the underlying causes and effects effectively. Here we provide comprehensive understanding of endothelial cell dysfunction that is essential for developing strategies to mitigate the impact of this dysregulation on health and cardiovascular diseases progression.
Keywords
- cardiovascular diseases
- metabolic disorders
- blood vessels
- disease modelling
- vascular dysfunction
- therapeutic interventions
Endothelial cells, the delicate inner lining of blood vessels, play a pivotal role in maintaining vascular health and regulating essential physiological processes [1]. Endothelial cell dysfunction, a pivotal factor in the pathogenesis of various cardiovascular diseases [2, 3, 4, 5] and metabolic disorders [6, 7, 8, 9, 10], represents a complex and dynamic process with multifaceted causes, events, and consequences. The causes of endothelial cell dysfunction are diverse, encompassing a spectrum of risk factors that range from traditional elements such as hypertension, diabetes, and hyperlipidaemia to lifestyle-related factors like smoking and obesity [11, 12]. Additionally, the influence of aging, inflammation, oxidative stress, shear stress disturbances, and genetic factors underscores the multifactorial nature of this phenomenon [12, 13].
Understanding the temporal sequence of events in endothelial cell dysfunction is indeed crucial. The early events involve endothelial activation, heightened expression of adhesion molecules, and increased vascular permeability [14, 15]. Additionally, impaired nitric oxide production can lead to abnormal vasodilation and vasoconstriction, resulting in narrowed blood vessels and contribute to high blood pressure [16, 17]. These early changes are often triggered by various cardiovascular risk factors such as hyperglycaemia, hyperlipidaemia, hyperinsulinemia, insulin resistance, and hypertension [18].
As the dysfunction progresses, late events begin to manifest. These include the induction of a pro-thrombotic state that can increase platelet production, causing blood clots [19, 20, 21]. Additionally, heightened oxidative stress can lead to inflammation in the artery walls, potentially leading to atherosclerosis [22, 23]. Furthermore, the potential initiation of endothelial-to-mesenchymal transition can occur, which is a critical process in the pathological progression of various diseases [15, 24, 25, 26, 27]. Lastly, endothelial cells (ECs) may undergo senescence and death [2, 28, 29]. As endothelial dysfunction advances, microvascular rarefaction becomes a significant late event [30, 31, 32]. This entire sequence of events, from early activation to late cell death, contributes to the overall process of blood vessels malfunction [14, 15, 18].
In Fig. 1, we provide a generalized chronological order of events associated with endothelial cell dysfunction. While these events are depicted in a linear fashion, it is important to note that the progression of endothelial dysfunction can be dynamic and influenced by various factors. Early events such as endothelial activation and inflammation often precede later events like oxidative stress and endothelial apoptosis, though the exact sequence may vary based on factors like the underlying cause of dysfunction and the specific vascular bed affected [33, 34]. Furthermore, certain events, such as inflammation and oxidative stress, may dynamically contribute to dysfunction, exhibiting varying degrees of involvement at different stages [9, 22, 35, 36, 37]. Moreover, the response of endothelial cells to various risk factors is diverse and multifaceted, which further influences the temporal progression of events and plays a crucial role in shaping the sequence of events in endothelial dysfunction [38, 39, 40]. For a deeper understanding of the multifaceted causes and responses to endothelial dysfunction, we delve into subsequent sections of this review.
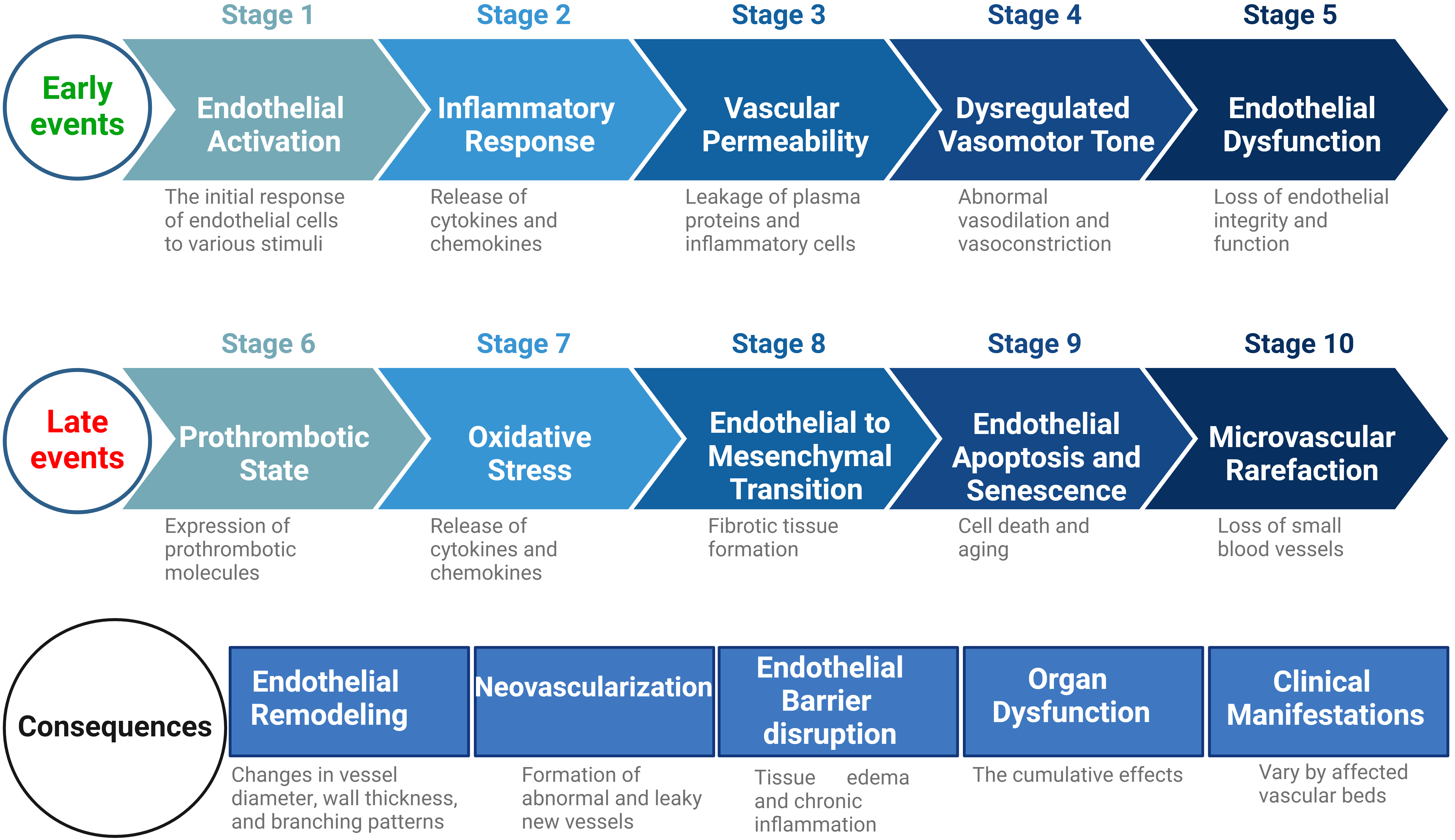 Fig. 1.
Fig. 1.General chronological order of adverse events in initiation and progression of the endothelial cell dysfunction. This figure illustrates the sequential progression of early and late events in endothelial cell dysfunction. Early events, including endothelial activation, inflammatory response, vascular permeability, dysregulated vasomotor tone, and endothelial dysfunction, precede late events such as the prothrombotic state, oxidative stress, endothelial-to-mesenchymal transition (EndoMT), endothelial apoptosis and senescence, and microvascular rarefaction. Subsequently, consequential stages include endothelial remodelling, neovascularization, endothelial barrier disruption, organ dysfunction, and clinical manifestations, emphasizing the wide-ranging impacts of endothelial dysfunction across multiple organ systems. While the events are shown in a linear order, the actual progression of endothelial dysfunction is dynamic and influenced by various factors. The exact sequence can vary depending on the underlying cause of dysfunction and the specific vascular bed affected. Additionally, some events like inflammation and oxidative stress may contribute to dysfunction in different degrees at different stages. Moreover, the diverse responses of endothelial cells to stimuli, further affecting the sequence of events. Created with BioRender.com.
This review not only delineates the intricate landscape of endothelial cell dysfunction from causes to consequences but also outlines therapeutic interventions such as antioxidants, anti-inflammatory therapies, and endothelial-specific interventions. Emphasizing the importance of tailoring treatments to the underlying causes and effects, this comprehensive understanding is essential for the development of effective strategies aimed at mitigating the impact of endothelial cell dysfunction on overall health and disease.
Understanding the causes of endothelial cell dysfunction is of paramount importance. Here, we delve into the origins of this dysfunction, where a constellation of risk factors emerges as its architects (Fig. 2). These risk factors, including hypertension [16], diabetes [41], smoking [11], obesity [42], hyperlipidaemia [43, 44], aging [45], inflammation [46], oxidative stress [47], shear stress disturbances [48], and genetic predispositions, underlie the initiation of endothelial cell dysfunction. These risk factors frequently exhibit interconnections, forming a mutually reinforcing cycle wherein the exacerbation of one factor contributes to the worsening of the others [22, 49, 50, 51]. A comprehensive analysis of these causative factors reveals their individual contributions and potential interactions in the initiation of endothelial dysfunction.
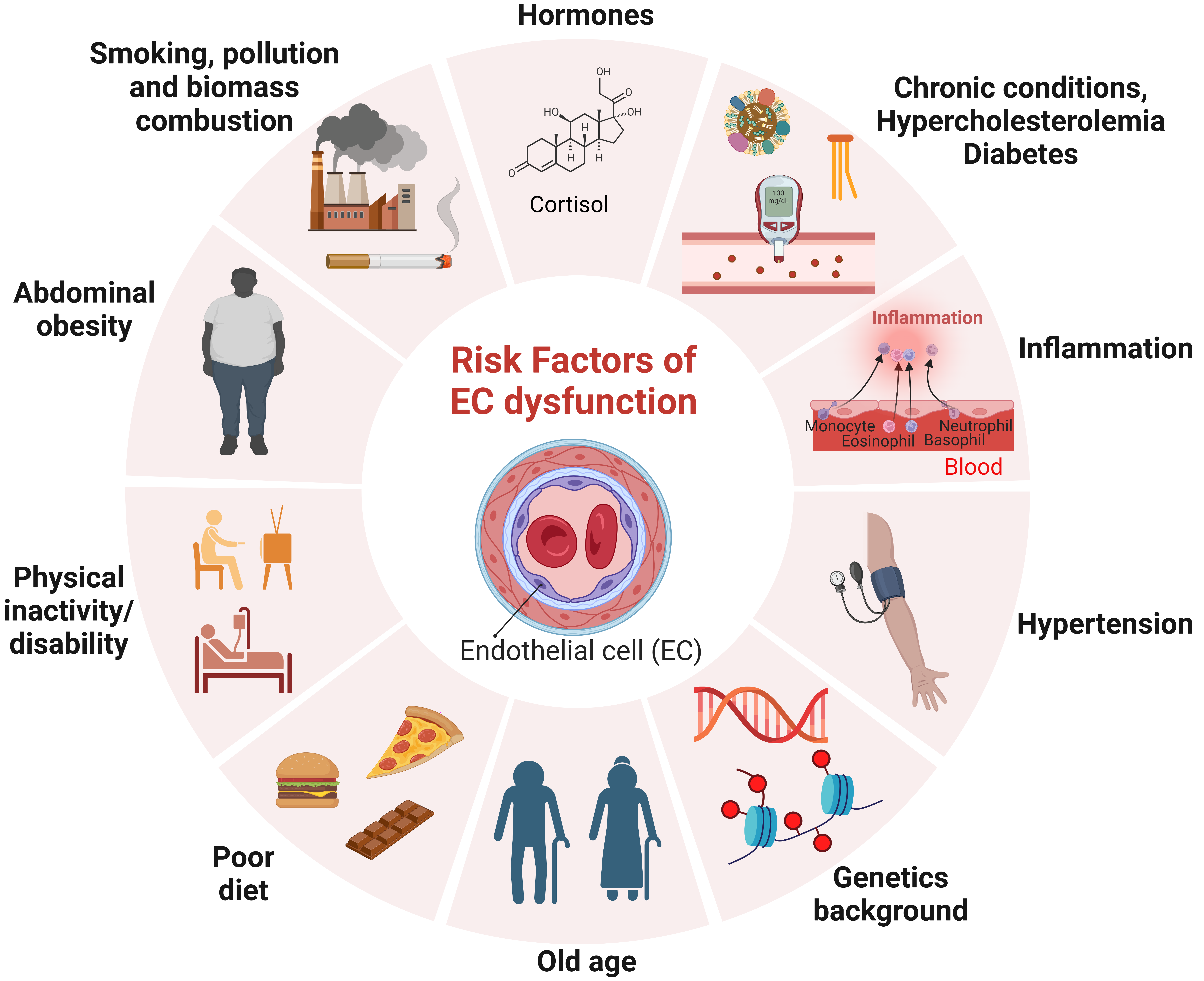 Fig. 2.
Fig. 2.Multiple risk factors contribute to endothelial dysfunction in a complex interplay. These factors, including hypertension, diabetes, smoking, obesity, hyperlipidaemia, aging, inflammation, oxidative stress, shear stress disturbances, and genetic predispositions, initiate endothelial dysfunction. This figure illustrates the intricate relationship among these risk factors, highlighting their interconnectedness and mutual reinforcement. Understanding the individual contributions and potential interactions of these causative factors is crucial for comprehending the initiation of endothelial dysfunction. Created with BioRender.com.
Excessive sugar consumption, a prevalent modern trend, directly threatens endothelial health by inducing stress, inflammation, and impaired vasodilation [52]. Diabetes exacerbates this condition, leading to chronic high blood glucose levels that adversely affect endothelial cells. Key mechanisms linking diabetes to endothelial dysfunction involve reduced nitric oxide production, increased oxidative stress, chronic inflammation, advanced glycation end products (AGEs), endoplasmic reticulum stress, and impaired angiogenesis [7]. These factors collectively contribute to compromised blood vessel function and increased risk of vascular complications, such as atherosclerosis, hypertension, and microvascular diseases. In the relentless battle for endothelial health, effective diabetes management takes centre stage, involving meticulous glycaemic control and lifestyle modifications [53, 54].
Four main pathways are involved in the mechanisms that underlie hyperglycaemia-induced diabetic vascular complications: the generation of AGEs, the activation of protein kinase C (PKC), an increase in the flux of the polyol pathway, and an increase in the flux of the hexosamine route [55, 56].
AGE Production: Nonenzymatic interactions between sugars and proteins,
lipids, or nucleic acids occur under hyperglycaemic circumstances and result in
the creation of AGEs. The activation of AGE in endothelial cells promotes
activation of several signalling pathways such as mitogen-activated protein
kinases (MAPK), p38, and ERK1/2, nuclear factor kappa B (NF-
Increase in the Flux of the Polyol Pathway: High glucose levels saturate hexokinase, which directs surplus glucose into the polyol pathway. Oxidative stress results from this pathway’s depletion of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) and NAD+. Aldose reductase (AR) produces ROS that causes endothelial cell death and dysfunction and accelerates thrombus production [55, 56, 57].
PKC Activation: The production of diacylglycerol is stimulated by
hyperglycaemia, which activates PKC. Through a variety of pathways, such as the
disruption of the IL-18 pathway, the stimulation of NF-
Hexosamine Pathway Flux Increase: Hyperglycaemia causes an increase in superoxide generation in the mitochondria, which in turn triggers the hexosamine pathway (HBP). Increased production of VEGF-A and ICAM-1, impaired tube formation and migration of endothelial cells, and vascular problems are all caused by elevated O-GlcNAc levels [55, 56, 57].
Hormones play a crucial role in regulating endothelial function, and endothelial dysfunction is associated with various hormonal imbalances and disturbances, including insulin, sex hormones (oestrogen and testosterone), thyroid hormones, cortisol (stress hormone), growth hormone, and adipokines. Insulin, a key hormone involved in glucose metabolism, is essential for maintaining endothelial health. Insulin resistance, a condition where cells become less responsive to insulin, is linked to endothelial dysfunction, and can contribute to the development of cardiovascular complications. Oestrogen [58] and testosterone [59], among other sex hormones, have vasodilatory effects that help regulate blood vessel function. Imbalances in sex hormones, such as those occurring during menopause or andropause, can impact endothelial health and contribute to vascular dysfunction [60]. Thyroid hormones influence various cardiovascular parameters, including heart rate and blood vessel responsiveness. Thyroid disorders, such as hypothyroidism or hyperthyroidism, can affect endothelial function and cardiovascular health [61]. Chronic stress can lead to elevated levels of cortisol, a stress hormone. Prolonged exposure to high cortisol levels is associated with endothelial dysfunction and may contribute to the development of cardiovascular diseases [3]. Growth hormone plays a role in vascular homeostasis and can influence endothelial function. Imbalances in growth hormone levels may contribute to vascular complications [62]. Adipose tissue acts as an endocrine organ producing adipokines and have been implicated in endothelial dysfunction [63]. Perivascular adipose tissue (PVAT) is the adipose tissue surrounding blood vessels composed of mature adipocytes, adipocytes, pre-adipocytes, and stem cells adipocytes. The PVAT is similar to other adipose tissues, which actively secretes adipokines and contributes to endothelial dysfunction [64].
Adipokines such as adiponectin and leptin play roles in inflammation and
vascular health [5]. Upon hyperglycaemia state, the level of adiponectin
decrease, resulting in a significant increase of the adhesion molecules (VCAM1
and ICAM1) and inflammatory molecules such as TNF-
Genetics plays a pivotal role in influencing endothelial function, and genetic factors are recognized contributors to endothelial dysfunction. Understanding the genetic determinants associated with endothelial dysfunction is crucial for unravelling the molecular mechanisms involved and developing targeted interventions. Numerous genetic factors have been implicated in endothelial dysfunction, including variations in genes related to nitric oxide production and vascular tone regulation [69, 70], oxidative stress response [71], and inflammation [72]. Polymorphisms in these genes can influence susceptibility to endothelial dysfunction and contribute to the risk of developing cardiovascular diseases. The identification of specific genetic markers associated with endothelial dysfunction enables a more personalized approach to cardiovascular risk assessment and management [69, 70, 71]. Advances in genetic research continue to shed light on the intricate interplay between genetics and endothelial function, providing valuable insights for the development of precision medicine strategies to mitigate the impact of endothelial dysfunction on cardiovascular health.
Age is a significant determinant of endothelial function, with distinctions between normal aging and abnormal aging conditions. In normal aging, there is a natural, gradual decline in endothelial function, marked by changes such as reduced nitric oxide production and increased oxidative stress. Abnormal aging conditions, on the other hand, may accelerate these processes, leading to more pronounced endothelial dysfunction and an elevated risk of cardiovascular complications [2, 46]. The question of whether age effects can be postponed is a subject of considerable interest. Lifestyle modifications, including a healthy diet [73], regular exercise [74], and proper management of risk factors like hypertension and diabetes, have been shown to positively influence endothelial function and potentially attenuate age-related decline [46, 74, 75]. Ongoing research endeavours would unravel the underlying mechanisms, seeking interventions that could potentially delay or alleviate the effects of aging on endothelial function, contributing to holistic cardiovascular health.
Smoking, a pervasive habit with far-reaching health implications, emerges as a significant contributor to endothelial dysfunction. Endothelial cells, the guardians of vascular health, face a barrage of detrimental effects induced by the toxic components of cigarette smoke. Smoking hampers the delicate balance of endothelial function, impairing the production of nitric oxide (NO), a vital vasodilator. Reduced NO availability leads to compromised blood vessel dilation and heightened susceptibility to vasoconstriction [76]. The intricate equilibrium within endothelial cells is disrupted by the onslaught of oxidative stress induced by smoking. Reactive oxygen species (ROS) generated from cigarette smoke inflict damage on endothelial cells, triggering inflammation and setting the stage for dysfunction [77]. The harmful constituents of cigarette smoke accelerate the aging of endothelial cells, diminishing their reparative and regenerative capacities. This premature aging process contributes to the progression of endothelial dysfunction [78].
Smoking acts as a catalyst for the upregulation of adhesion molecules on endothelial surfaces, fostering a pro-inflammatory environment. This heightened inflammatory state not only impairs cell-to-cell communication but also paves the way for the infiltration of harmful substances [79]. The risk of thrombosis escalates as smoking induces a pro-thrombotic state within the endothelium. Disruption of the delicate balance between pro-coagulant and anti-coagulant factors heightens the likelihood of clot formation [80]. Smoking has been implicated in triggering Endothelial-to-Mesenchymal Transition (EndMT), a phenomenon where endothelial cells undergo transformation into mesenchymal cells. This transition contributes to fibrosis and vascular remodelling, exacerbating the detrimental impact on overall vascular health [81]. In understanding the intricate interplay between smoking and endothelial dysfunction, it becomes evident that this habit transcends mere respiratory concerns. Smoking emerges as a potent catalyst for a cascade of events leading to compromised vascular health. Besides smoking, air pollutants have also been shown another serious risk factor giving rise to endothelial dysfunction [82], reviewed comprehensively elsewhere [83].
The impact of dietary choices on vascular health is profound, with a poor diet emerging as a significant instigator of endothelial dysfunction. The intricate interplay between nutrition and endothelial function sets the stage for a cascade of events that compromise the integrity of blood vessels. Diet consisting of high cholesterol and fat, especially trans-fat and saturated fat can cause endothelial dysfunction and significantly increase the risk of cardiovascular diseases. Excessive intake contributes to the accumulation of lipid deposits, fostering atherosclerosis and impeding the flexibility of blood vessels [84]. These poor dietary choices often contribute to obesity, a major risk factor for endothelial dysfunction. Adipose tissue dysfunction in obesity results in the release of pro-inflammatory cytokines, perpetuating a chronic inflammatory state within blood vessels [8].
The prevalence of processed foods in modern diets introduces an array of
additives and preservatives. Chronic exposure to these substances triggers a
sustained inflammatory response within the endothelium, fostering dysfunction
over time. For example, Excessive salt intake disrupts the delicate balance of
sodium-potassium within endothelial cells, leading to increased blood pressure.
Hypertension, in turn, contributes significantly to endothelial dysfunction by
inducing structural and functional alterations in blood vessels [16].
Additionally, excess dietary sodium negatively influences nitric oxide
(NO)-mediated endothelial function by inducing increases in oxidative stress,
promoting an increase in EC stiffness, and causing damage to the endothelial
glycocalyx [85]. Additionally, a diet deficient in fibre compromises gut health
and, subsequently, impacts the composition of the microbiota. Disruptions in the
delicate balance of gut bacteria have been linked to systemic inflammation and
endothelial dysfunction [10, 86]. Firmicutes, Proteobacteria, and Actinobacteria
are a few of the gut bacteria that produce trimethylamine (TMA) from dietary precursors like
betaine, L-carnitine, or choline. Trimethylamine N-oxide TMAO, a product of TMA
oxidation in the liver, associated with cardiovascular disease (CVD) [87]. Trimethylamine N-oxide (TMAO)
decreases nitric oxide (NO) synthesis and increases pro-inflammatory cytokines,
adhesion molecules, and ROS thereby exacerbating endothelial dysfunction.
Inducing the production of adhesion molecules facilitates monocyte adherence by
means of NF-
Oral microbiota and gut dysbiosis may affect cardiovascular disease onset and/or development [89, 90]. Dysbiosis in the oral cavity has emerged as a significant focus of current research. In fact, following third molar surgery, endodontic therapy, subgingival cleaning, tooth extraction, or tonsillectomy, low-grade bacteraemia has been reported. Periodontitis, an inflammatory disease of the periodontium, often begins with gingivitis due to inadequate plaque management. Chronic periodontitis has been associated with cardiovascular disease (CVD). Porphyromonas gingivalis (Pg) is known to oxidise lipoproteins, especially high-density lipoproteins (HDL), which reduces their ability to prevent atherosclerosis. When oxidising low-density lipoprotein (LDL), Pg is more capable of doing so than other pathogens such as C. pneumoniae and M. tuberculosis. This bacterium generates two types of proteases known as gingipain, which are able to degrade antioxidants and produce reactive oxygen species (ROS). ICAM-1 promotes Pg invasion and damage to endothelial cells; additional pathogens, such as F. nucleatum and T. forsythia, may also contribute to this invasion. Pg infection directly damages gingival epithelium and endothelial cells, reducing adenosine triphosphate concentration, an increase in ROS, and mitochondrial fragmentation [89]. On the other hand, the human microbiome, Helicobacter pylori, which is found in the stomachs of about half of the world’s population. It has been associated with changes in serum lipid levels resulting in dyslipidaemia, a significant contributor to atherosclerosis. A persistent infection can alter the metabolism of lipids, raising total cholesterol, triglycerides, and LDL while decreasing HDL. H. pylori infection causes oxidative stress and DNA damage by upregulating pro-inflammatory cytokines and adhesion molecules, which in turn causes systemic inflammation. This inflammation aggravates atherosclerosis by promoting the production of foam cells from oxidised LDL particles and accelerating plaque build-up [89]. These studies revealed that understanding the nexus between a suboptimal diet and endothelial dysfunction underscores the importance of dietary interventions in preserving vascular health.
Here, we will elucidate the sequence of early and late events in the context of diabetic vasculopathy. Diabetic vasculopathy is marked by endothelial cell dysfunction, activation, inflammation, and the formation of atherosclerotic plaques (Fig. 3). Exposing into risk factors such as hyperglycaemia, oxidative stress, FFAs, and AGEs manifest the early events as forerunners of endothelial cell dysfunction. These initial events encompass increased adhesion molecule expression [91, 92], changes in vascular permeability [93, 94], endothelial activation [92, 95, 96], and inflammation [97, 98]. The intricate molecular pathways involved in these early events are dissected in Fig. 3, offering insight into the subtle shifts that precede overt dysfunction.
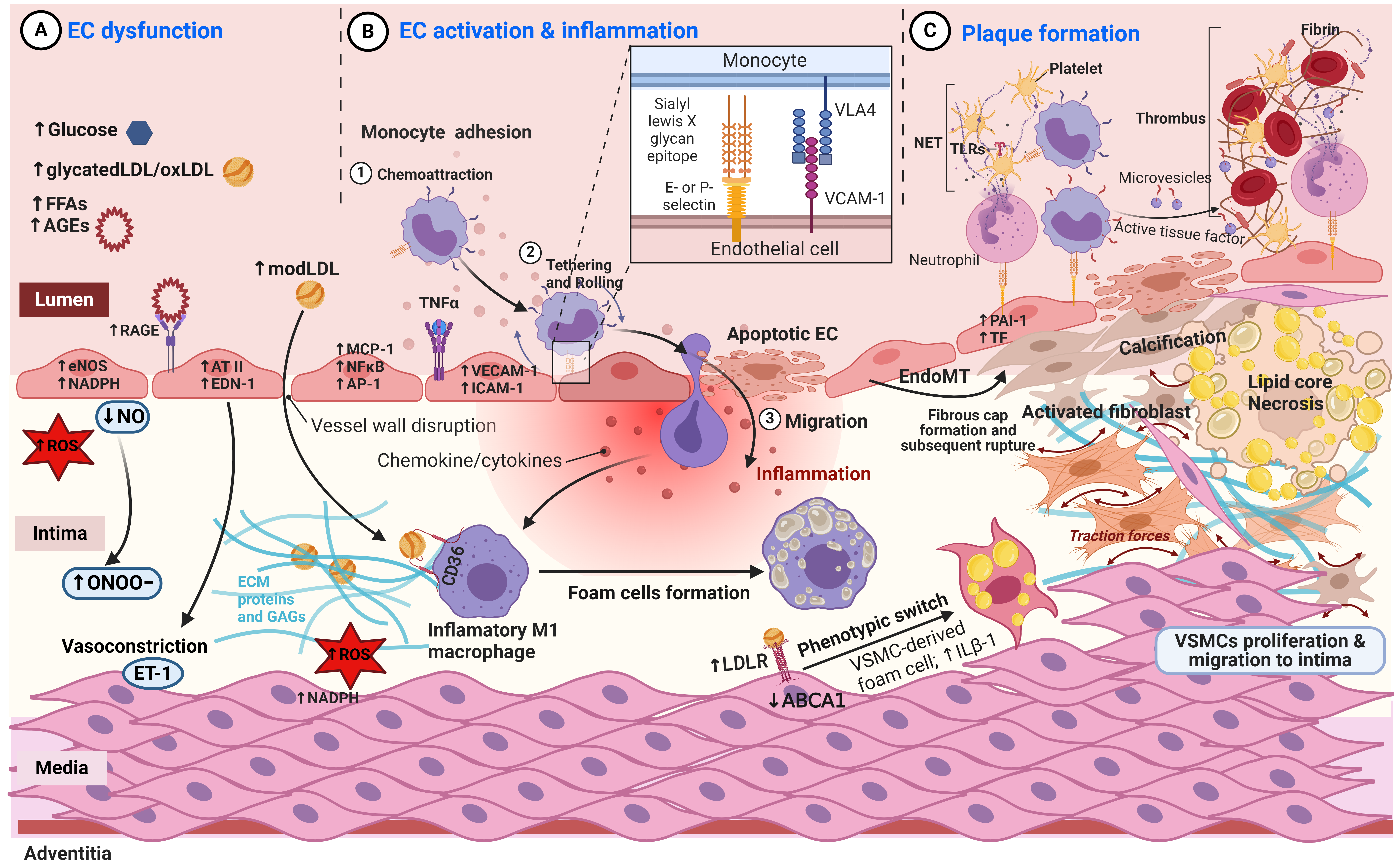 Fig. 3.
Fig. 3.Sequence of early and late events in diabetic vasculopathy. (A) Endothelial Cell (EC) Dysfunction: Endothelial cells initially become dysfunctional due to hyperglycemia, AGEs, FFAs, modified LDLs, and oxidative stress, disrupting mediator balances and creating a progressively proinflammatory and prothrombotic microenvironment. (B) EC Activation and Inflammation: Endothelial cells are activated, leading to the recruitment of immune cells to the inflammatory ECs. Lipids and immune cells accumulate in the tunica intima. Macrophages and vascular smooth muscle cells (VSMCs) uptake modified LDL, forming foam cells, and subsequently build an atheromatous plaque. (C) Plaque Formation: Local platelet activation and the expression of prothrombotic factors result in intravascular thrombus formation. Modified LDLs contribute to calcification by inhibiting osteoclastic macrophage activity and promoting the osteogenic differentiation of smooth muscle cells. Activated fibroblasts produce fibrotic tissue, and the core of the plaque undergoes necrosis. Abbreviations: FFAs, Free Fatty Acids; LDL, Low-Density Lipoproteins; AGEs, Advanced glycation end products; RAGE, receptor for AGEs; NO, Nitric Oxide; eNOS, endothelial NO synthase; ROS, reactive oxygen species; ET-1, endothelin 1; AT-II, angiotensin II; EDN-1, Endothelin 1 gene; MCP-1, Monocyte Chemoattractant. Created with BioRender.com.
Chronic disruption of the physiological balance between vasoactive,
inflammatory, and thrombotic mediators results in pathologic vasodilation, as
well as a proinflammatory and prothrombotic microenvironment [99]. Nitric oxide
(NO) is a potent vasodilator and key regulator in healthy blood vessels,
providing vascular protection through the prevention of cell proliferation,
inflammation, and platelet aggregation [100]. Because of risk factors such as
diabetes, however, endothelium-derived NO interacts with reactive oxygen species
(ROS) to form peroxynitrite – a cytotoxic oxidant that disrupts endothelial
function and contributes to the formation of oxidated low-density lipoproteins
(oxLDLs) [101]. In addition to oxidative modifications, the hyperglycaemic
diabetic microenvironment promotes nonenzymatic glycation of proteins and lipids,
such as LDL, resulting in the synthesis of advanced glycation end products
(AGEs). AGEs increase oxidative stress, stimulate vasoconstriction, and drive
local inflammation by binding to receptors for AGEs (RAGEs). AGE/RAGE binding
promotes oxidative stress by stimulating NADPH oxidase to generate ROS through
electron transfer from NADPH to oxygen and exacerbates vasoconstriction due to
the release of vasoactive factors such as endothelin-1 (ET-1) and angiotensin II
(AT-II) [99, 102]. In addition, AGE/RAGE signalling stimulates local inflammation
by inducing transcription factors NF-
Similar condition is observed in the heart failure (HF) in which there is a complex interplay between an alteration of the gut microbiota-heart axis, neurohormonal activation, and inflammation. Oxidative stress is another factor that can lead to the development of inflammation in any stage of the HF. It is produced via both the control of endothelial cells apoptosis and the stimulation of eNOS uncoupling [106]. There are different types of cell death in endothelial dysfunction such as apoptosis [106, 107], autophagy [107], necroptosis [107], pyroptosis [107], entosis [107], ferroptosis [107], ferroautophagy [107], and parthanatos [107]. Furthermore, the pathophysiology of pulmonary arterial hypertension (PAH) involves both dysregulation of the immune system and inflammation. Dysregulation of the immune system signalling results in apoptosis-resistant endothelial cells are selected by apoptosis and uncontrolled vascular smooth muscle cells proliferation leading to vascular remodelling [108].
The expression of adhesion molecules on the endothelial cell surface promotes immune cell binding and migration. Meanwhile, endothelial dysfunction impairs the endothelial barrier integrity, thereby facilitating the influx of immune cells and lipids into the tunica intima [109]. Several phosphorylation locations on vascular endothelial (VE)-cadherin, an adherent junction, have different impacts on the function of endothelial cells. In response to inflammatory stimuli, phosphorylation at tyrosine residue 685 enhances endothelial permeability, whereas dephosphorylation at tyrosine residue 371 enhances leukocyte extravasation during leukocyte-endothelial cell contact. Increase vascular permeability facilitates the migration of the blood cells and the extravasation of the macromolecules to the sites of infection which helps to resolve the infection. For example, conversion of the extravasated fibrinogen to fibrin forms an extracellular matrix which support the angiogenesis [94].
Over time, macrophages and modified lipids, such as oxLDL, accumulate within the intima. Increasing oxLDLs affect a wide range of local cell types by activating platelets, further increasing endothelial monocyte adhesion, and stimulating trans-differentiation of macrophages and vascular smooth muscle cells (VSMCs) [110]. Modified LDLs (models) are taken up by macrophages which acquire the inflammatory M1 phenotype and eventually become lipid-loaded foam cells [111]. Lipid ingestion impairs cell migration and traps the foam cells within the vessel wall, where they aggregate and form an atheromatous plaque [112]. Similarly, activated VSMCs within the tunica intima take up modified LDLs, acquire a macrophage-like phenotype, and eventually become foam cells that significantly contribute to plaque formation [113, 114]. Foam cell apoptosis results in the formation of necrotic lipid cores that cannot be sufficiently cleared due to impaired efferocytosis [115, 116]. Accumulation of necrotic cores increases local inflammation and destabilizes the atheromatous plaque [117]. Furthermore, modLDL contributes to the calcification of the vessel wall by inhibiting osteoclastic macrophage activity while simultaneously stimulating osteogenic differentiation of VSMCs within the plaque core [118, 119]. Besides plaque development and calcification, thrombus formation is an important aspect of diabetic vasculopathy (Fig. 3). The hyperglycaemic vascular microenvironment as well as the decreased activity of protective factors such as NO contribute to local platelet activation and adhesion [120]. AGE/RAGE signalling promotes hypercoagulation and thrombosis by increasing the expression of tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) [100]. The activation of platelets and the expression of prothrombotic factors facilitate fibrinogen platelet binding and thrombus formation—besides plaque formation and calcification a third key outcome of diabetic vasculopathy [109].
The path through endothelial cell dysfunction continues into later stages, where we confront more advanced changes. Here, we observe the consequences of prolonged dysfunction, including impaired nitric oxide production, the emergence of a pro-thrombotic state, escalated oxidative stress, and the enigmatic phenomenon of endothelial-to-mesenchymal transition (EndoMT) [27, 121]. These late-stage events have profound implications for vascular health and disease progression.
Endothelial dysfunction casts a pervasive influence, extending well beyond the endothelial layer and intricately impacting diverse facets of human health, depending on the initial cases (risk factor/s). The progression of endothelial cell dysfunction may culminate in the onset of symptomatic disease states, characterized by the manifestation of clinical signs and symptoms related to vascular dysfunction and organ damage. These clinical manifestations vary depending on the affected vascular beds and may include chest pain, dyspnea, cognitive impairment, renal insufficiency, visual disturbances, and other symptoms indicative of end-organ dysfunction [34, 122].
Chronic endothelial dysfunction may lead to structural alterations in the vasculature, including changes in vessel diameter, wall thickness, and branching patterns. Endothelial remodelling can contribute to alterations in blood flow dynamics and tissue perfusion, exacerbating tissue damage and organ dysfunction, particularly in high metabolic demand organs like the heart, brain, and kidneys [1, 123]. In response to chronic hypoxia and tissue ischemia resulting from endothelial dysfunction, there may be a compensatory increase in the formation of new blood vessels through processes such called neoangiogenesis. However, this neovascularization process is often dysregulated and inefficient, leading to the formation of abnormal and leaky vessels that exacerbate tissue injury and dysfunction, resulting in retinopathy of eyes [124]. Additionally, prolonged endothelial dysfunction can lead to significant disruption of the endothelial barrier function, resulting in increased vascular leakage and impaired regulation of fluid and solute exchange between the blood and surrounding tissues. This breakdown of the endothelial barrier particularly in brain contributes to tissue edema, chronic inflammation, and organ dysfunction [125].
Endothelial dysfunction’s influence extends to fostering a pro-thrombotic state, increasing the likelihood of blood clot formation, and raising the risk of thrombotic events such as deep vein thrombosis or pulmonary embolism [123, 126]. Additionally, impaired endothelial function contributes to a chronic inflammatory state within the vascular system, linked to various cardiovascular diseases and systemic inflammatory responses [6, 19, 127]. Further, dysfunction in the endothelium disrupts the normal regulation of blood vessel tone, contributing to vasoconstriction and elevating blood pressure [1, 4]. These, in turn, amplify the risk of cardiovascular events such as heart attacks and strokes. Ultimately, the cumulative effects of endothelial dysfunction, including impaired perfusion, inflammation, atherosclerosis, thrombosis, hypertension, and fibrosis, can manifest as functional deficits in target organs such as the heart, brain, kidneys, and eyes [4, 123]. Organ dysfunction may present clinically as cardiovascular disease, stroke, renal failure, retinopathy, or other end-organ damage (Fig. 4). These systemic consequences underscore the central role of endothelial dysfunction in the pathogenesis of diverse cardiovascular and metabolic disorders. Effectively addressing endothelial health becomes a pivotal strategy in preventing and managing a spectrum of health conditions that transcend the immediate endothelial layer [6, 19, 125, 127].
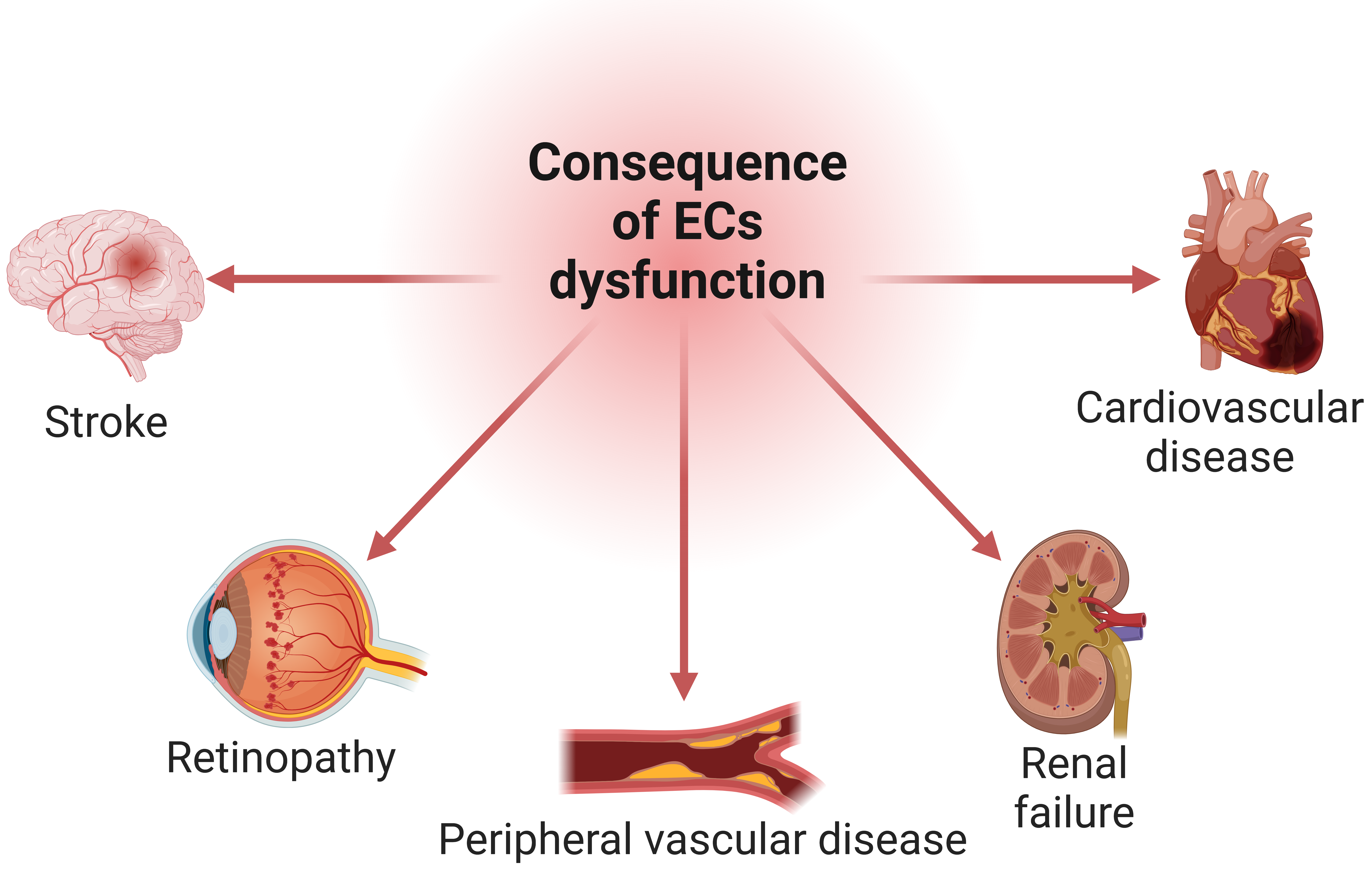 Fig. 4.
Fig. 4.The cascade of endothelial cells (ECs) dysfunction on organ health. Impaired vascular function adversely impacts organ perfusion, potentially leading to dysfunction and damage in vital organs such as the heart, brain, kidneys, eyes, and peripheral vessels. Created with BioRender.com.
The precise mechanisms behind endothelial dysfunction are still being investigated. Therefore, endothelium-specific interventions are still regarded as speculative. As depicted in Fig. 5, a spectrum of therapeutic interventions including antioxidants, anti-inflammatory therapies, cholesterol lowering drugs, and emerging endothelial-targeted strategies can be used to either prevent or alleviate dysfunctional endothelium. Pharmacological agents such as Hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitors and angiotensin-converting enzyme inhibitors, have been suggested to protect vessels in ways other than those of their main therapeutic effects (hypotension, hypocholesterolaemia, etc.). On the other hand, the application of epigenetic medications, referred to as epigenetic therapeutic agents, has increased recently [128, 129]. In the face of this complex phenomenon endothelial dysfunction, there is hope. The critical importance of tailoring treatment approaches to address the underlying causes and effects is emphasized, offering a glimpse into the future of personalized medicine in vascular health.
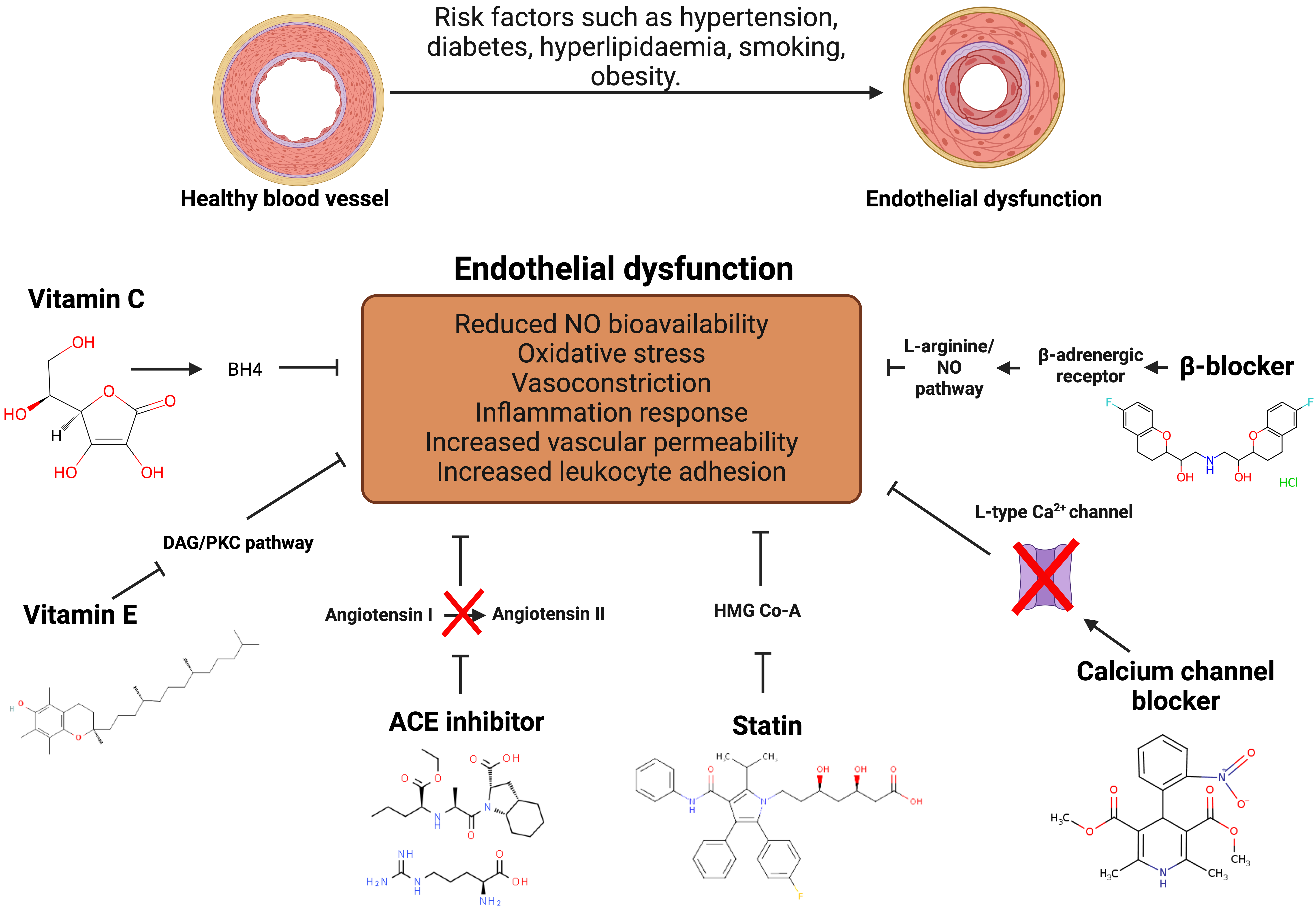 Fig. 5.
Fig. 5.Pharmacological treatments to alleviate endothelial
dysfunction. Antioxidant treatment is associated with improvement of the
endothelial function. Angiotensin-Converting Enzyme (ACE) medications reduce
blood pressure and enhance endothelial function by inhibiting the
angiotensin-converting enzyme, thereby lowering angiotensin II synthesis. Statins
reduce LDL cholesterol levels and the risk of atherosclerosis and cardiovascular
disease by blocking Hydroxymethylglutaryl-CoA (HMG-CoA) reductase which potentially inhibit endothelial
dysfunction. Calcium channel blockers work by lowering Ca
Antioxidant has been linked to an improved health condition such as diabetes and cancer and most likely found in plant-based food. Vitamin C and E showed an ability to restore endothelial function in diabetes mellitus, hypercholesterolaemia, and hypertension [130]. Vitamin C stimulates endothelial cell proliferation, prevents apoptosis, and increases NO production [130, 131] through recycling tetrahydrobiopterin (BH4) [131]. Active BH4 keeps the catalytic iron in its ferrous state and supports the correct operation of dioxygenase enzymes. Trihydrobiopterin radical (BH3) is produced when BH4 donates an electron to the active dimeric form of eNOS. However, vitamin C effectively reduces this radical back to BH4. If the BH3 is not reduced, it decomposes into inactive BH2, which then competes with BH4 for binding on eNOS but does not promote the production of NO•. When BH4 is absent, and BH2 is elevated, eNOS is decoupled, leading the enzyme to produce superoxide rather than NO. A combination of vitamin C (500 mg/day) and vitamin E (400 IU/day) for 6 weeks improves endothelial-dependent flow-mediated dilation (FMD) of the brachial artery in children with familial hypercholesterolaemia [131].
The administration of vitamin E functioned through donating hydrogen from the
chromanol head hydroxyl group to lipid radicals produced in lipid peroxidation
chain reactions, resulting in increased endothelial vasodilatation and
neutralises the circulating free radical that quenches NO before it reaches
vascular smooth muscle. Furthermore, vitamin E (
Beta-blockers are a class of medications that primarily block the effects of the hormone epinephrine (adrenaline). They belong to a larger group of drugs known as beta-adrenergic blocking agents. These medications target beta receptors in the body, which are typically found in the heart, lungs, blood vessels, and other tissues. By blocking the action of epinephrine on beta receptors, beta-blockers reduce the heart rate, lower blood pressure, and decrease the workload on the heart. This class of drugs is commonly used to treat various cardiovascular conditions, including hypertension and angina [134].
Some of the third generation of beta blockers drugs exhibited favourable effects on endothelial cells of hypertension patients [135] and smokers [136]. The third generation of beta blockers serve as a vasodilator by stimulating the L-arginine-NO pathway [135]. The meta-analysis data suggested that beta blockers treatment improved endothelial dysfunction. Investigations were conducted on two generations of beta-blockers: atenolol and metoprolol from the second generation, and carvedilol and nebivolol from the third generation. The third-generation beta-blockers significantly improve flow-mediated dilation (FMD) more than second-generation ones did. When compared to other antihypertensive medications, beta-blockers exhibit similar endothelial function effects comparable to angiotensin receptor blocker (ARB). Nevertheless, Angiotensin-Converting Enzyme (ACE) inhibitors are more effective than beta-blockers [137].
Angiotensin-Converting Enzyme inhibitors (ACE inhibitors) and Angiotensin Receptor Blockers (ARBs) are two classes of medications commonly used to treat conditions related to the renin-angiotensin-aldosterone system, a crucial system in regulating blood pressure and fluid balance in the body. ACE inhibitors and ARB are used for the management of hypertension [138], and atherosclerosis [139]. However, utilization of the combination of ACE inhibitors and ARB was linked to the side effects such as renal failure [140, 141]. ACE inhibitors work by blocking the action of the ACE, which is responsible for converting angiotensin I to angiotensin II. Angiotensin II is a potent vasoconstrictor that can raise blood pressure [142]. ACE inhibitors link to elevated bradykinin level that enhance the release of prostacyclin, NO, and endothelium-derived hyperpolarizing factors (EDHF) [143]. By inhibiting ACE, these medications reduce the production of angiotensin II, leading to vasodilation (widening of blood vessels), decreased blood pressure, and reduced workload on the heart [142]. ARBs, also known as angiotensin II receptor blockers, target the angiotensin II receptors directly. Instead of inhibiting the formation of angiotensin II like ACE inhibitors, ARBs prevent its binding to receptors [144]. By blocking the action of angiotensin II, ARBs result in vasodilation and reduced secretion of aldosterone, a hormone that regulates salt and water balance. This leads to lowered blood pressure and reduced strain on the heart. ARBs are prescribed for similar conditions as ACE inhibitors, including hypertension and heart failure. They are often an alternative for individuals who cannot tolerate ACE inhibitors due to side effects like cough [144, 145].
The choice between them depends on various factors, including the patient’s specific health conditions, tolerability, and any contraindications. Although they target different parts of the renin-angiotensin system (RAS), ACE inhibitors and ARBs both inhibit it. Specifically, ACE inhibitors prevent angiotensin I from becoming angiotensin II, while ARBs prevent angiotensin II from binding to AT1 receptors. It has been known that ACE inhibitors attenuate endothelial dysfunction in animal model of cardiovascular disease and diabetes [136]. The Perindopril-Function of the Endothelium in Coronary artery disease Trial (PERFECT) study showed that long-term perindopril use is consistent with a reduction in FMD, even though these results lacked statistical significance. The perindopril group exhibited a significant improvement in endothelial function over time, with improvements approaching statistical significance when compared to the placebo group [146]. The effect of ACE inhibitor to improve endothelial dysfunction by reducing the synthesis of angiotensin II and increase production of eNOS. Treatment of enalapril, perindopril, quinapril, ramipril, and trandolapril promoted upregulation of the eNOS protein expression and reduction of the systolic blood pressure (SBP) in hypertension rat model [147]. Bradykinin might mediate the escalation of eNOS expression through promoting the production of NO and endothelial-derived hyperpolarizing factor by binding to the bradykinin B2 receptor [146, 148].
Calcium channel blockers (CCBs) are a class of medications that interfere with the movement of calcium into heart and blood vessel cells. Calcium plays a crucial role in the contraction of heart muscles and the dilation of blood vessels. By blocking calcium entry, these medications reduce the workload of the heart and widen blood vessels, leading to various therapeutic effects. Calcium channel blockers are commonly prescribed to treat several cardiovascular conditions [149]. CCBs enhance endothelial function by shielding endothelial cells from free radicals through their antioxidant properties. These results are explained by CCBs’ strong lipophilicity and chemical structure, which support antioxidant processes. Consequently, this improves endothelial function through generating nitric oxide (NO) more available [136]. Research indicates that patients with coronary heart disease who receive nifedipine treatment have better endothelial function. By lowering oxidative stress markers, increasing NO bioavailability, and enhancing arterial structure and function, nifedipine treatment also helps patients with essential hypertension. Similar results, including a decrease in inflammatory markers, have also been shown by other CCBs, such as amlodipine, and valsartan in hypertensive patients [135]. Nifedipine administration reduced glucose-induced ROS release in both native endothelium and suspended endothelial cultures in a concentration-dependent manner. This transient effect was also noted when nifedipine was administered to endothelial cells for 48 hours without any nifedipine present at the time of the ROS measurement. Nifedipine pre-treatment at concentrations of 0.1 and 1 µmol/L significantly decreased the release of ROS induced by glucose in endothelial cells [150].
Statins are a class of medications commonly prescribed to lower cholesterol levels in the blood. They are widely used to manage and prevent cardiovascular diseases, particularly atherosclerosis, by reducing the production of cholesterol in the liver. The generic names of some common statins include atorvastatin, simvastatin, rosuvastatin, and pravastatin [151]. Statins primarily work by inhibiting an enzyme called HMG-CoA reductase, which is involved in the production of cholesterol in the liver [151, 152]. By blocking this enzyme, statins reduce the synthesis of low-density lipoprotein (LDL) cholesterol, often referred to as “bad” cholesterol. Elevated LDL cholesterol is a significant risk factor for atherosclerosis and coronary artery disease. Statins may also have secondary effects that contribute to their cardiovascular benefits. For example, they have anti-inflammatory and antioxidant properties that can help stabilize plaques in blood vessels [151].
Statins have been shown in meta-analyses of data from the Cardiovascular Treatment Trialists (CTT) to be effective in lowering the risk of cardiovascular disease (CVD) in individuals without a history of the condition [136, 151]. Numerous studies have revealed that the restoration of endothelial function is preceded by a significant decrease in serum cholesterol levels [153, 154, 155]. Statins impact endothelial cell function via a variety of signalling pathways, such as the AMPK, Rho/ROCK, and PI3K/Akt pathways, which produce NO and increase fibrinolytic capacity by lowering oxidised low-density lipoprotein (LDL) [155]. Statins could also ameliorate serum lipid profiles and CRP levels in coronary artery diseases (CAD) patients. Additionally, the endothelium dependent vasodilatation (EDV) valued was significantly increased after three months of atorvastatin administration resulted in improvement of the endothelial functions of coronary arteries and myocardial perfusion [156].
Epigenetic changes are thought to underlie diabetes complications such as vascular complications. Histone post-translational modification, DNA methylation, chromatin remodelling, and non-coding RNA regulate epigenetic mechanisms. Altering the epigenome has long been seen as a promising avenue for therapeutic intervention in a variety of disease areas, with a number of approved therapies currently on the market, mainly for cancer [157, 158]. Several drugs that inhibit epigenetic writers and erasers have been approved by Food and Drug Administration (FDA) which comprised as DNA methyltransferase (DNMT) inhibitors [159], histone deacetylases (HDACs) inhibitors [157, 160] and histone methyltransferase [157].
HDACs play an essential role in vascular biology. Several HDAC inhibitors are
designed to inhibit HDACs, but most are not specific to the HDAC subclass.
According to the chemical structure, HDACs inhibitors are classified into four
groups: short-chain fatty acids, benzamides, hydroxamic acids, and cyclic
peptides [157, 159, 160]. Treatment with first generation of histone deacetylase
inhibitors such as suberoylanilide hydroxamic acid (SAHA) [159], romidepsin
[157, 161], and sodium butyrate [159] attenuates inflammation and ROS production.
The aorta of ApoE-/-mice injected intraperitoneally with SAHA showed decreased
atherosclerosis plaque and macrophage infiltration; however, the lipid profile
remained unchanged [162]. Furthermore, administration of Romidepsin (40 nM) in
TNF
The journey through endothelial cell dysfunction is one marked by its complexity. In traversing the intricate landscape of endothelial cell dysfunction, we have revealed the multifaceted nature of this phenomenon, from its origins to its far-reaching consequences. Our journey through the causes, early events, late events, and therapeutic interventions has illuminated the complex interplay of factors that underlie this critical aspect of vascular health.
As we conclude this exploration, several key takeaways emerge:
• A holistic understanding: Our comprehensive examination underscores the need for a holistic understanding of endothelial dysfunction. Recognizing that it is not a singular event but rather a dynamic process influenced by diverse factors is pivotal.
• Personalized approaches: Tailoring therapeutic approaches to the specific causes and stages of endothelial dysfunction is imperative. One size does not fit all, and precision medicine holds promise in optimizing treatment outcomes.
• Promising interventions: The array of therapeutic interventions we have discussed offers hope for mitigating the impact of endothelial dysfunction. From lifestyle modifications to cutting-edge targeted therapies, our arsenal against this condition is expanding.
• Ongoing Research: This review underscores the ongoing research efforts to unravel the complexities of endothelial cell dysfunction. As we continue to delve into its molecular intricacies, new insights will undoubtedly emerge, further refining our strategies for prevention and intervention.
In closing, the significance of understanding the initiation and progression of endothelial cell dysfunction cannot be overstated. It is a critical piece of the puzzle in the broader context of cardiovascular diseases, metabolic disorders, and vascular dysfunction. By delving into this intricate narrative, we pave the way for innovative approaches to safeguard vascular health and, ultimately, improve the well-being of individuals affected by this multifaceted condition. Our journey continues, with the hope that the insights shared here will inspire further exploration and discovery in this vital field of research.
HNM contributed to the design of the work, collected literature for sections 1–5, and drafted Figs. 1,2,3,4 and the conclusion. WAWS collected literature for section 6 and created Fig. 5. Both authors wrote the manuscript, contributed to editorial revisions, and read and approved the final version. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
All the figures were created with BioRender.com.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.