







1 School of Biosciences and Technology, Vellore Institute of Technology, 632014 Vellore, India
2 Department of Sensor and Biomedical Technology, School of Electronics Engineering, Vellore Institute of Technology, 632014 Vellore, India
3 Department of Biomedical Sciences, College of Health Sciences, QU Health, Qatar University, 2713 Doha, Qatar
4 Bioinformatics Laboratory, College of Computing, Mohammed VI Polytechnic University, 43150 Ben Guerir, Morocco
Abstract
Background: The incidence rate of oropharyngeal squamous cell carcinoma
(OPSCC) worldwide is alarming. In the clinical community, there is a pressing
necessity to comprehend the etiology of the OPSCC to facilitate the
administration of effective treatments. Methods: This study confers an
integrative genomics approach for identifying key oncogenic drivers involved in
the OPSCC pathogenesis. The dataset contains RNA-Sequencing (RNA-Seq) samples of
46 Human papillomavirus-positive head and neck squamous cell carcinoma and 25
normal Uvulopalatopharyngoplasty cases. The differential marker selection is
performed between the groups with a log2FoldChange (FC) score of 2, adjusted
p-value
Keywords
- biomarker discovery
- explainable artificial intelligence
- human papillomavirus
- oropharyngeal squamous cell carcinoma
- RNA-sequencing
- shapley additive explanations
Head and neck squamous cell carcinoma (HNSCC) significantly impedes increases in global life expectancy, resulting in a substantial number of deaths worldwide. With over 562,328 annual occurrences, HNSCC ranks as the seventh most common cancer, posing a significant global burden and epidemiological risk [1]. The term HNSCC encompasses a range of diseases that manifest in the head and neck region, including cancers linked to the oral cavity, nasopharynx, and oropharynx [2]. Each subset under this category has a different etiology, epidemiological patterns, and medical regimen [3]. HNSCC has historically been linked to smoking and alcohol abuse. However, it has been increasingly evident in recent years that Human Papillomavirus (HPV) infection, primarily affecting the oropharynx, is critical in developing HNSCC [4]. Oropharyngeal squamous cell carcinoma (OPSCC) has drawn attention recently due to an incredible surge in cases, distinctly those pertinent to HPV [5, 6, 7]. On a global scale, HPV infection contributes to 20–60% of OPSCC cases, with HPV-16 being the predominant strain, accounting for almost 80% of HPV-related OPSCC cases [8, 9, 10].
HPV-associated OPSCC conditions are predominantly observed in individuals below 60 years. They tend to have less smoking and drinking habits than non-HPV-associated OPSCC conditions, with a more significant percentage of cases in men reportedly having more oral sex partners belonging to a higher fiscal community [11, 12, 13]. The disparities can explain the contrarieties in age in sexual habits between the older and younger consociates [10]. Additional risk factors include deep kissing, vaginal intercourse, having oral sex before the age of 18, marijuana abuse, and prior incidences of cervical HPV infection. Despite the efforts, very little is known about the carcinogenic pathway in OPSCC from initiation of HPV infection to cancer development; most disease hypotheses are adapted from cervical cancer studies. In addition to immune cells, pathogens such as HPV can infiltrate the mucosal lining of palatine tonsils [14]. Untreated HPV infections may progress, leading to premalignant lesions and potentially culminating in an aggressive variant of OPSCC.
HPV-positive OPSCC exhibits unique auguring traits and genetic patterns compared to HPV-negative conditions. With a greater survival rate and fewer reported side effects, HPV-positive OPSCC is linked to a better overall treatment outcome than HPV-negative OPSCC. Radiation therapy alone or combined with concurrent chemotherapy has demonstrated a more favorable prognosis for HPV-OPSCC [15, 16]. Despite the favorable prognosis, diagnosis of OPSCC at its initial stages remains challenging due to its multifaceted nature [17]. Early identification is crucial as it can increase the 5-year survival chances of OPSCC by up to 85% [18]. Current methods for diagnosing and monitoring OPSCC remain insufficient; the development of biomarkers can help in early patient diagnosis and monitoring of treatment response, thereby improving the survival rates and lowering the recurrence rates. The development of predictive biomarkers can predict a disease’s course and ascertain therapeutics’ success [19]. Differential expression analysis scrutinizes gene expression motifs and collocates the molecular processes underlying convoluted illnesses. Gene expression profiles delineate with high dimensional data and significant correlations between genes. Due to this, thousands of robustly linked genes are frequently included as outputs from the differential expression analysis. It is inefficient to use all the differential genes to develop a predictive model; here, dimensionality reduction comes into play.
Oncology has led the way in integrating AI into cancer management [20]. Researchers are actively implementing AI-based machine learning (ML) approaches to investigate the genetic differences among malignancies, which may be utilized to enhance the accuracy of cancer diagnosis, identification of novel biomarkers, and development of novel cancer therapeutics [21, 22, 23]. Although ML approaches help to identify putative biomarkers, their potentiality must be assessed by highly developed computational techniques. In addition, greater model complexity is typically used to attain higher performance, turning these systems into black-box methods that create ambiguity in their functionality and decision-making ability [24, 25, 26]. Relying on models whose findings cannot be comprehended efficiently is quite laborious. Building a pipeline combining efficient algorithms for identifying and characterizing biomarkers that incorporate expert knowledge with data-driven analysis is imperative. Using a particle optimization technique (PSO) and explainable A.I. (XAI) approach, in this study, we introduce a refined multi-objective approach for prioritizing marker genes as potential biomarkers instrumental in predicting the pathogenesis of OPSCC. Our methodology integrates sophisticated feature subset selection techniques within the biomarker identification pipeline, enhancing the precision and efficacy of predictive markers in OPSCC.
The study intends to find oncogenic biomarkers associated with OPSCC from the RNA-Seq data. The dataset consists of samples collected from 46 HPV-positive OPSCC tumor samples, and 25 normal tissue samples from uvulopharyngoplasty (UPPP) sequenced using the HiSeq 2500 sequencing approach at John Hopkins University (Accession ID: GSE112026) [27]. The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/.) was used to explore the OPSCC-related RNA-Seq data; the selection criteria follow the dataset size and group disparities matching the need of building a machine-learning model.
The disparities in gene expression within the diseased and healthy control
groups can be assessed by performing differential gene expression analysis. Using
the DESeq2 package (Version 1.44.0), the count matrix file was employed to scrutinize the
differentially expressed genes (DEGs) between OPSCC and the normal tissue samples
[28]. The count matrix file was subjected to preprocessing steps such as removing
genes with low expression counts, collapsing replicates, and data normalization.
The multidimensional scaling analysis (MSA) evaluated the expression pattern for
OPSCC tumors and normal tissues [29]. The DEGs were identified with the criteria
of Log2fold change (FC)
The PSO algorithm containing the multicollinearity issue tends to model overfitting when training with machine learning algorithms. Feature selection methods strategically identify the best subset by computing the relevancy of features with the target variable. Many techniques exist, such as filter, embedded, non-linear, wrapper, and meta-heuristic search optimization methods, each equipped with several ingenious algorithms. This investigation uses PSO, a robust stochastic optimization technique, to meticulously select salient candidate genes [31]. PSO mimics the collaborative dynamics and trajectories of swarm particles within a multidimensional search domain, aiming to pinpoint the most effective solution. Each particle symbolizes a prospective solution, and their collective, known as the swarm, systematically navigates the search terrain to identify the optimum solution. This process embodies both exploration and exploitation attributes. During initial phases, particles engage in an exploratory behavior by varying their positions extensively. As the optimization process advances, these particles gradually gravitate towards areas of high potential, marked by individual and collective optimal positions, thereby exploiting the search domain for superior solutions. The culmination of this procedure is the generation of a refined subset by the PSO algorithm, which serves as the foundational dataset for training various machine learning models.
Several algorithms have been developed to categorize cancer subtypes and potential biomarkers linked to a cancer etiology [32, 33, 34, 35, 36, 37, 38, 39]. However, the users of these models still need to discern how different characteristics contribute to the job at hand [40]. To overcome this problem, XAI has emerged to tackle the void in deep learning models [41]. The dexterity of XAI methods to expound the comportment of the model and establish confidence in it has been demonstrated in several applications [42, 43, 44]. One such application is the identification of cancer biomarkers; our study aims to use the XAI-based feature selection approach to identify a limited set of biomarkers associated with OPSCC. XAI framework aids in identifying the crucial genes that training models can use to categorize the disease state. SHapley Additive exPlanations (SHAP) is a methodology for interpreting machine learning model predictions. It offers a mechanism to assign the contribution of individual features to the concluding prediction made by the model. The mathematical representation of SHAP can be described as follows:
SHAP evaluates the impact of each feature in an input instance by assigning a specific value to it. The assigned value signifies the difference in the predicted outcome when the feature is incorporated versus when it is omitted. Denoted as:
- X: The input features of an instance (a vector of length n).
- f: The predictive model that anticipates the output value.
-
The SHAP value function
Where:
-
SHAP values satisfy certain properties, including local accuracy, consistency, and missingness. These attributes ensure that the summation of SHAP values across all features equals the disparity between the model’s prediction for a particular instance and the anticipated baseline outcome. SHAP values represent the contribution of each feature to confidence scoring in a local summary graphic. A SHAP summary graphic also displayed the global feature relevance generated from the training data. Further, a comprehensive literature study was carried out to understand the relevance of the genes with the highest average SHAP values in the pathogenesis of OPSCC.
The model validation process involves employing the 10-fold cross-validation technique. Bayes Net, Logistic Regression, Support Vector Machines, Random Forest, and Adaboost algorithms were utilized to train the model using the gene subset. The model’s performance is evaluated using accuracy, f-score, precision, recall, and Matthew’s correlation coefficient. The variation in the performance between three different feature subsets is benchmarked to find the potency of the biomarker selected in each process.
The clusterProfiler R package was used to evaluate the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways to comprehend the role of DEGs associated with OPSCC [45]. The clusterProfiler examines genes through GO and KEGG terms, aiming to identify enriched terms and pathways associated with the input DEGs. The GO analysis encompassed three separate domains: biological processes (BP), cellular components (CC), and molecular functions (MF). The cnetplot, dot plot, emapplot functions, and Enrich R package were employed to illustrate the enriched pathways. A p-value of less than 0.005 was used to evaluate whether genes and pathways have a higher enrichment ratio.
Predicting protein interactions for tracing behavior throughout the biological mechanism is crucial. Studying new protein interactions helps identify the association with a disease by unraveling complicated molecular pathways and novel cellular activities. A confidence threshold of 0.7 was employed to assess the Protein-Protein Interactions (PPI) of the DEGs using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (STRING v11.5, https://string-db.org/.) [46].
The Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia2.cancer-pku.cn/#survival) tool was further used to examine the associativity between the three DEGs (ECT2, LAMC2, DSG2) and the prognosis of OPSCC [47]. The OPSCC tumor and normal samples were contrasted with the The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and Genotype-Tissue Expression (GTEx) databases (https://gtexportal.org/home/). The tumor samples were grouped into high and low-expression categories to assess their relationship with survival. The results were considered significant if the p-values were below 0.05.
The Drug Gene Interaction Database (DGIdb, version 3.0.2, https://www.dgidb.org/.) examined potential interactions between drugs and genes related to the DEGs identified through PSO. The drug-gene interactions (DGI) for the genes were acquired by utilizing the ‘query DGIdb()’ function within the ‘rDGIdb’ R package [48].
The DESeq2 package was utilized to compare the gene expression profiles of OPSCC and normal tumor samples. We identified 714 genes DEGs, comprising 455 downregulated and 259 upregulated genes (Supplementary Table 1). Utilizing a Euclidean distance matrix, the DEVis package rendered a heat map, where each row signifies a distinct sample and its comparative distance from other dataset samples. A pale yellow shade highlighted the outlier samples, which were subsequently excluded to mitigate batch effects (Fig. 1A). Fig. 1B illustrates the volcano plot for the DEGs. To scrutinize the divergence and overlap between OPSCC tumor and normal tissue samples in the dataset, a multidimensional scaling analysis (MCA) was conducted, revealing a pronounced, distinct clustering of the two groups (Fig. 1C). A density plot was computed to compare the aggregation for the p-values and DEGs in both tissue types. Comparative analysis revealed a notable elevation in DEGs within OPSCC samples relative to normal tissues (Fig. 1D). The PSO algorithm effectively pinpointed key markers, condensing the initial gene subset from 714 to 73 (Supplementary Table 2).
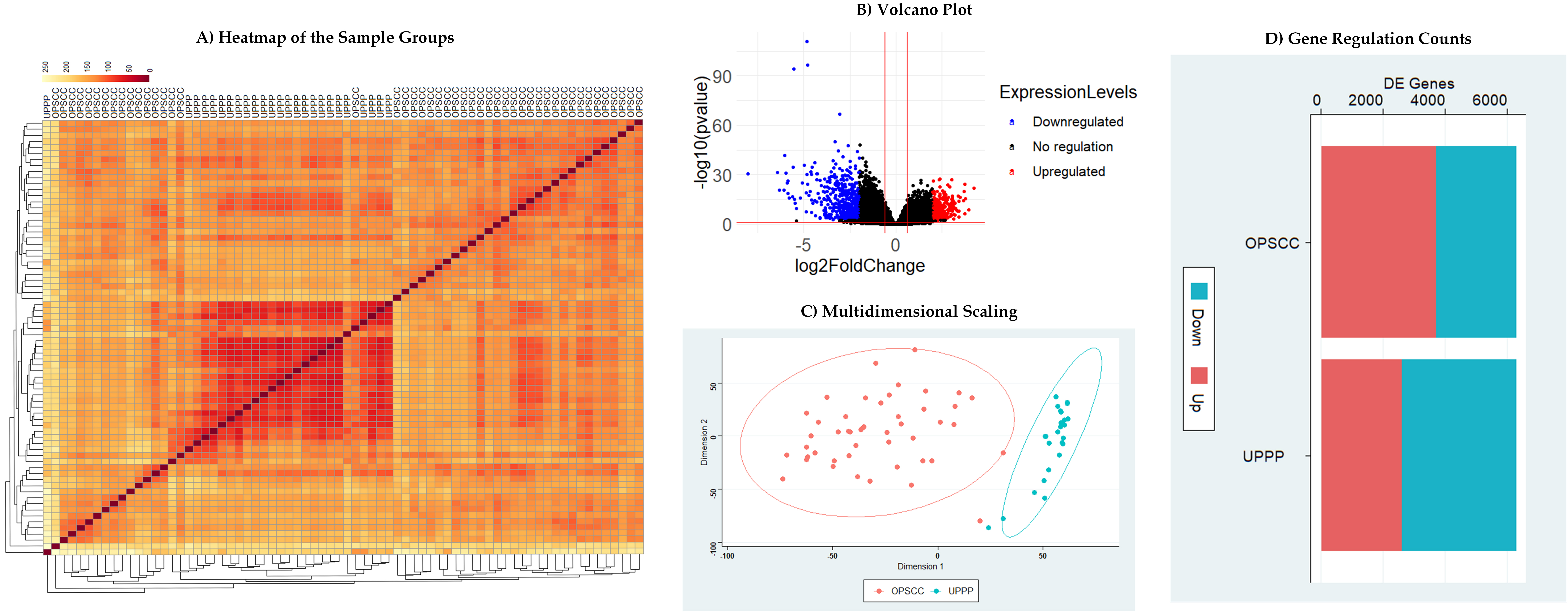 Fig. 1.
Fig. 1.Statistical analysis of Differentially Expressed Genes. (A) Heat map representation of oropharyngeal squamous cell carcinoma (OPSCC) and uvulopharyngoplasty (UPPP) sample groups based on Euclidean distance matrix. (B) Volcano plot illustrating genes with differential expression. A total of 714 genes exhibited statistically significant differences: depicted as up-regulated (in red) and down-regulated (in blue). (C) Multidimensional scaling plots for OPSCC and UPPP clusters. Each dot represents a sample. (D) Density plot comparing the aggregation of differentially expressed genes (DEGs) in OPSCC and UPPP tissue sample groups.
The random forest model is trained with the PSO-selected feature set for SHAP analysis. This algorithm interprets the results of the pre-trained model to understand the predictions. A bar plot function was utilized to construct a local feature importance plot by supplying a collection of SHAP values. Using this significant bar plot, the most relevant genes were depicted in a downward trend. The top genes have a higher predictive power in the ML model as they tend to contribute more. The bar plots in Fig. 2A,B illustrate the genes in order of their connotation, with the most significant genes placed at the top and vice versa for both OPSCC and UPPP tissue samples. ECT2, LAMC2, DSG2, FAT1, PLOD2, COL1A1, and PLAU were observed to have higher SHAP values. Waterfall plots were generated to provide explanations for diacritic predictions. In OPSCC, the waterfall plot for the genes mentioned above depicts contributions with ECT2 (–0.99), LAMC2 (–0.59), and DSG2 (–0.53) was found to have the higher negative score (Fig. 2C), indicating the prediction is favorable towards OPSCC.
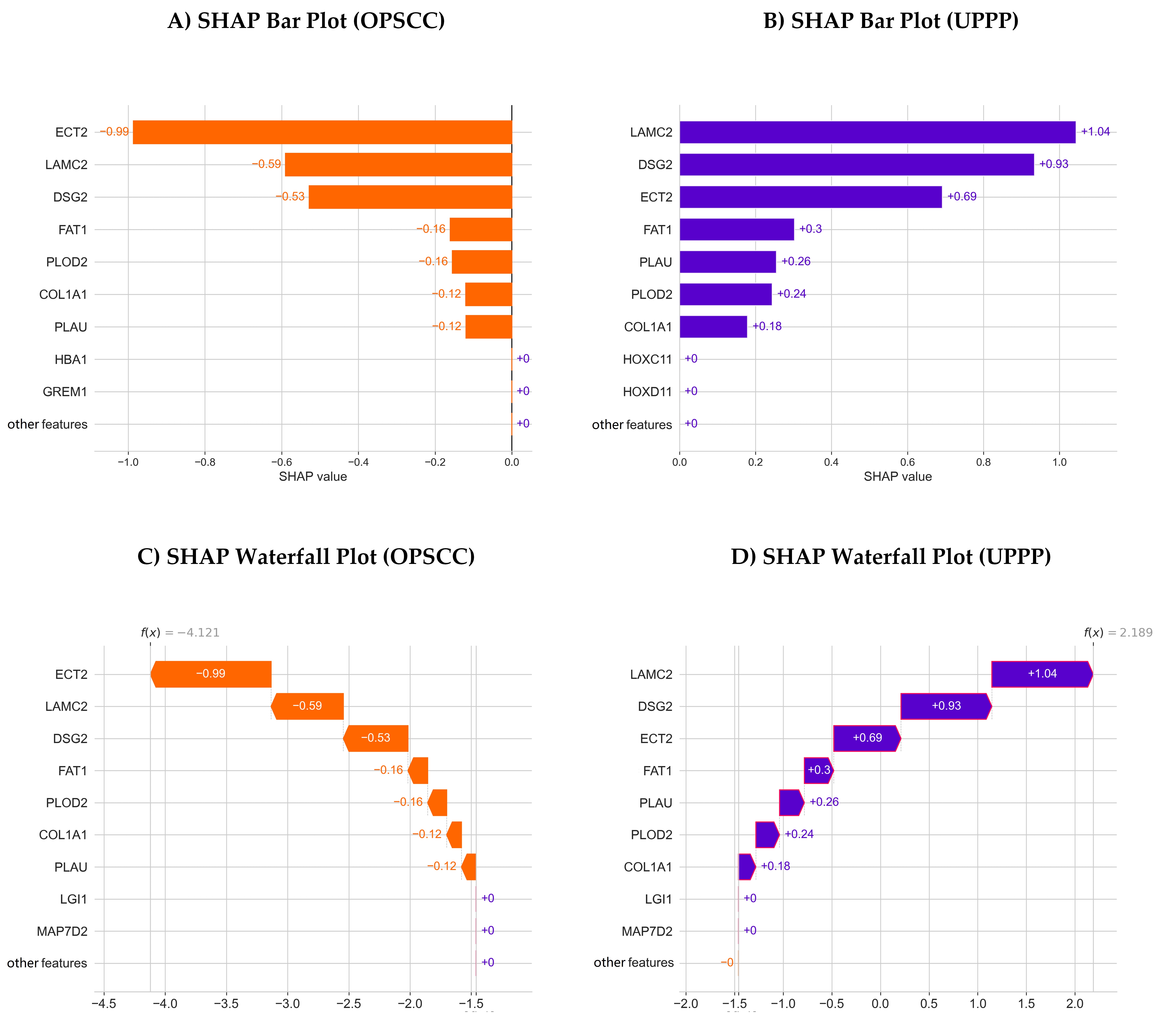 Fig. 2.
Fig. 2.Local bar plot and Waterfall plot representation of a randomly selected OPSCC and UPPP samples. (A) Local bar plot representation for OPSCC sample group generated using the Shapley Additive exPlanation (SHAP) values. (B) A local bar plot representation for the UPPP sample group was generated using the SHAP values. (C) Waterfall plot for OPSCC sample group. (D) Waterfall plot for the UPPP sample group. The OPSCC and UPPP sample groups are represented in orange and violet colors, respectively.
In contrast, for the UPPP sample, the waterfall plot displayed a positive contribution, with LAMC2 (+1.04), DSG2 (+0.93), and ECT2 (0.69) showing the highest contribution (Fig. 2D). The positive SHAP value evinces the prediction that the input sample is more to the class UPPP. The SHAP summary plot and SHAP beeswarm plot clearly distinguish the genes between OPSCC and UPPP; in both cases, the ECT2 gene shows higher feature importance, followed by LAMC2 and DSG2 based on the SHAP values (Fig. 3A,B). The low feature value with a positive SHAP score predicts the input as UPPP, whereas the sample with a higher feature value and negative SHAP score is predicted as OPSCC. Observing that, the higher feature value represents the over-expression of gene value on OPSCC samples. The cohort plot, depicted using a bar plot, represents the consequence of each gene contributing to the disease states (Fig. 3C). ECT2 gene was found to be of more global importance in the cohort plot, followed by LAMC2 and DSG2. A heatmap was generated to plot the instances based on the sample clustering (Fig. 3D). In the composite visualization, the bar plot positioned to the right side of the heat map distinctly illustrates elevated SHAP values for the trio of genes: ECT2, LAMC2, and DSG2. This elevation is marked compared to the SHAP values associated with other genes in this study. In summary, the SHAP interpretation of the trained random forest model reports that a sample with a positive SHAP value is predicted as UPPP and the negative to OPSCC, respectively (Supplementary Table 3).
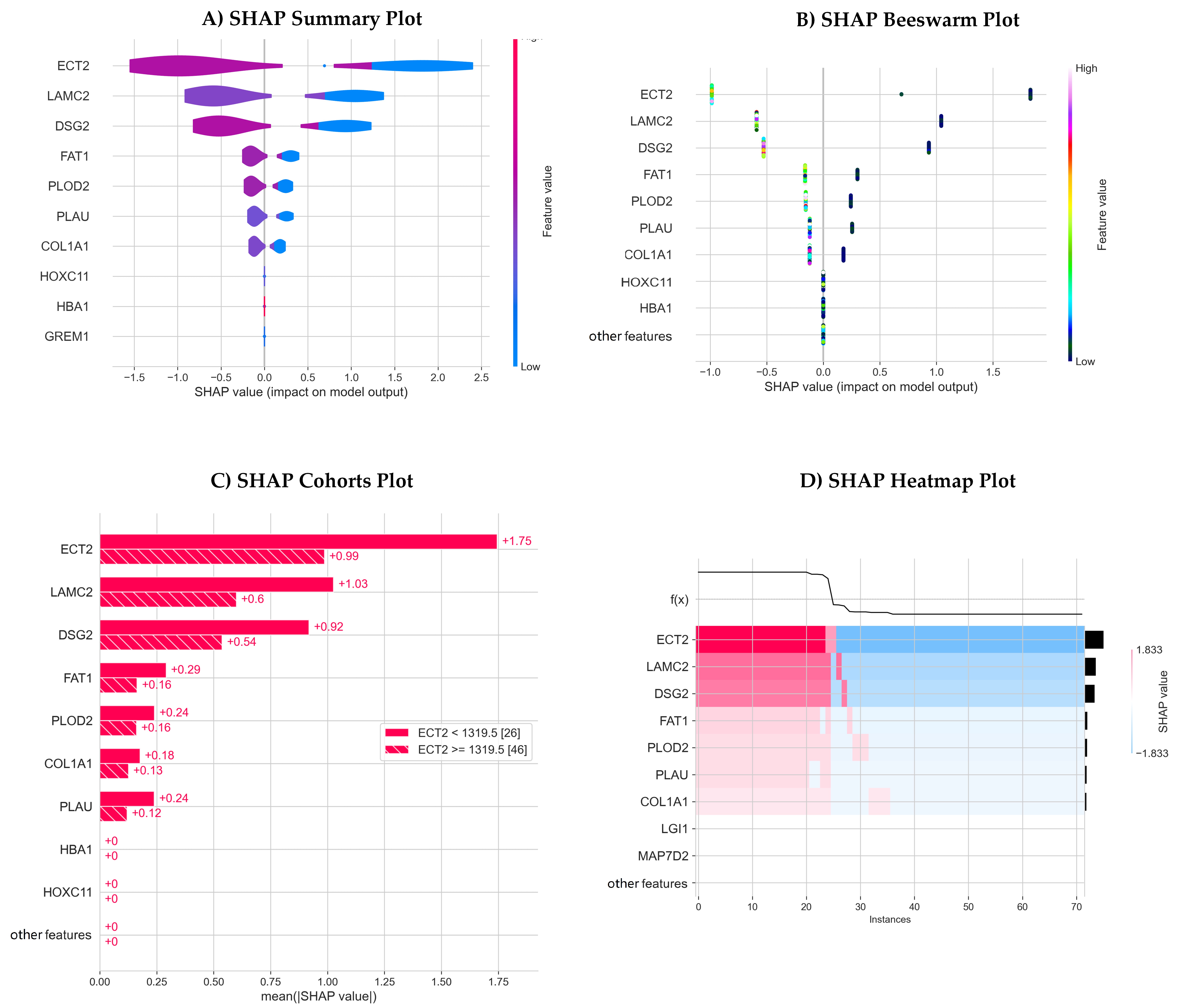 Fig. 3.
Fig. 3.Visualization of top genes identified with SHAP summary plot, Beeswarm plot, Cohorts plot and Heatmap plot using SHAP values. (A) SHAP summary plot illustrating the feature importance of the top 10 genes. The color intensity represents the value of gene features in increasing order. (B) Beeswarm plot representing the impact of top gene features on the SHAP model. (C) Cohort plot showing the global summary of gene features. The summary is shown separately for the top 9 genes belonging to UPPP and OPSCC sample groups, respectively. (D) A SHAP heatmap plot was generated based on the sample clustering. The bar plot on the right-hand side of the heatmap signifies the SHAP values.
The PSO subsets were trained with five ML classification algorithms to benchmark the performance between different models. The models achieved better results when the irrelevant features were eliminated during each process. The scores of the models are represented in Tables 1,2,3 for the log2FC adjusted set, PSO subset, and SHAP gene list.
| Classifier | Accuracy (%) | Precision (%) | Recall (%) | F-Score (%) | MCC (%) |
| BayesNet | 95.8 | 95.9 | 95.8 | 95.9 | 90.9 |
| Logistic Regression | 93.0 | 93.0 | 93.1 | 93.0 | 84.6 |
| Support Vector Machines | 95.8 | 95.8 | 95.8 | 95.8 | 90.8 |
| Random Forest | 94.4 | 94.4 | 94.4 | 94.4 | 87.7 |
| AdaBoost Classifier | 95.8 | 95.8 | 95.8 | 95.8 | 90.8 |
MCC, Matthew’s correlation coefficient.
| Classifier | Accuracy (%) | Precision (%) | Recall (%) | F-Score (%) | MCC (%) |
| BayesNet | 100 | 100 | 100 | 100 | 100 |
| Logistic Regression | 93.0 | 94.2 | 93.1 | 93.2 | 86.3 |
| Support Vector Machines | 97.2 | 97.4 | 97.2 | 97.2 | 94.2 |
| Random Forest | 100 | 100 | 100 | 100 | 100 |
| AdaBoost Classifier | 95.8 | 95.8 | 95.8 | 95.8 | 90.8 |
PSO, Particle Swarm Optimization.
| Classifier | Accuracy (%) | Precision (%) | Recall (%) | F-Score (%) | MCC (%) |
| BayesNet | 100 | 100 | 100 | 100 | 100 |
| Logistic Regression | 94.4 | 94.7 | 94.4 | 94.5 | 88.1 |
| Support Vector Machines | 94.4 | 95.2 | 94.4 | 94.5 | 88.8 |
| Random Forest | 97.2 | 97.2 | 97.2 | 97.2 | 93.9 |
| AdaBoost Classifier | 95.8 | 95.8 | 95.8 | 95.8 | 90.8 |
We utilized machine learning classifiers, including BayesNet, Logistic Regression, Support Vector Machines, Random Forest, and AdaBoost, to evaluate the effectiveness of predictive screening performed with PSO and SHAP algorithms. Seven genes were elected for model training: the initial gene set after the log2FC and adjusted p-value screening (714), candidate markers obtained from the PSO algorithm (73), and the final set of marker genes (7) from the SHAP model. Tables 1,2,3 represent the classifiers’ performance on log2FC-p-value adjusted, PSO, and SHAP gene subsets. The validation test showed a clear disparity for gene sets before and after the feature screening process. The F-score, a statistical assessment that combines accuracy and sensitivity parameters, was compared among the classifications for the seven genes. A higher F-score was observed in both PSO and SHAP-identified gene sets, indicating accuracy in algorithm predictions. Matthew’s correlation coefficient (MCC) score is higher in PSO and SHAP-subset values than the initial log2FC-p-value adjusted set, indicating the algorithms’ effectiveness in predicting the gene subsets. The BayesNet and Random Forest classifier attained 100% performances for PSO and SHAP genes compared to the log2FC-p-value adjusted gene set, outperforming the benchmark classifiers.
The 73 identified marker genes were further subjected to gene enrichment analysis using the clusterProfiler R package. Gene functions and associated disorders were rigorously analyzed through Disease Ontology (DO) via the DOSE R package (Version: 3.30.1). This comprehensive analysis aimed to establish a well-defined understanding of gene activities and their correlations with specific diseases. Employing the Enrichplot package, the most significant ten disorders linked to the identified genes were meticulously illustrated using barplot and dot plot methodologies. These visual depictions emphasized a noteworthy association between these genes and neoplasms related to head and neck cancer, especially within the DEGs framework (Fig. 4A,B). The Jaccard correlation coefficient linked the GO terms to the 73 identified PSO gene markers. The gene-related GO terms were systematized into five categories associated with adenocarcinoma, biliary head neck cancer, focal lipodermatosclerosis-glomerulosclerosis, ascending aorta aneurysm syndrome, and cerebral choroidal infarction artery (Fig. 4C). The line graphs in Fig. 4D generated by the PMC plot indicate an upward trend in publications on adenocarcinoma and head and neck cancer neoplasms. To further understand DEGs’ role in the pathogenesis of OPSCC pathway enrichment, analyses such as GO and KEGG were carried out. Predominantly, genes implicated in this study are integral to the extracellular matrix and its structural organization (Supplementary Fig. 1A). Based on the molecular function study, most genes were implicated in integrin binding, cytokine activity, and extracellular matrix structural components (Supplementary Fig. 1B). The top two cellular components of the identified genes were the collagen-comprising extracellular matrix and the endoplasmic reticulum (ER) lumen (Supplementary Fig. 1C).
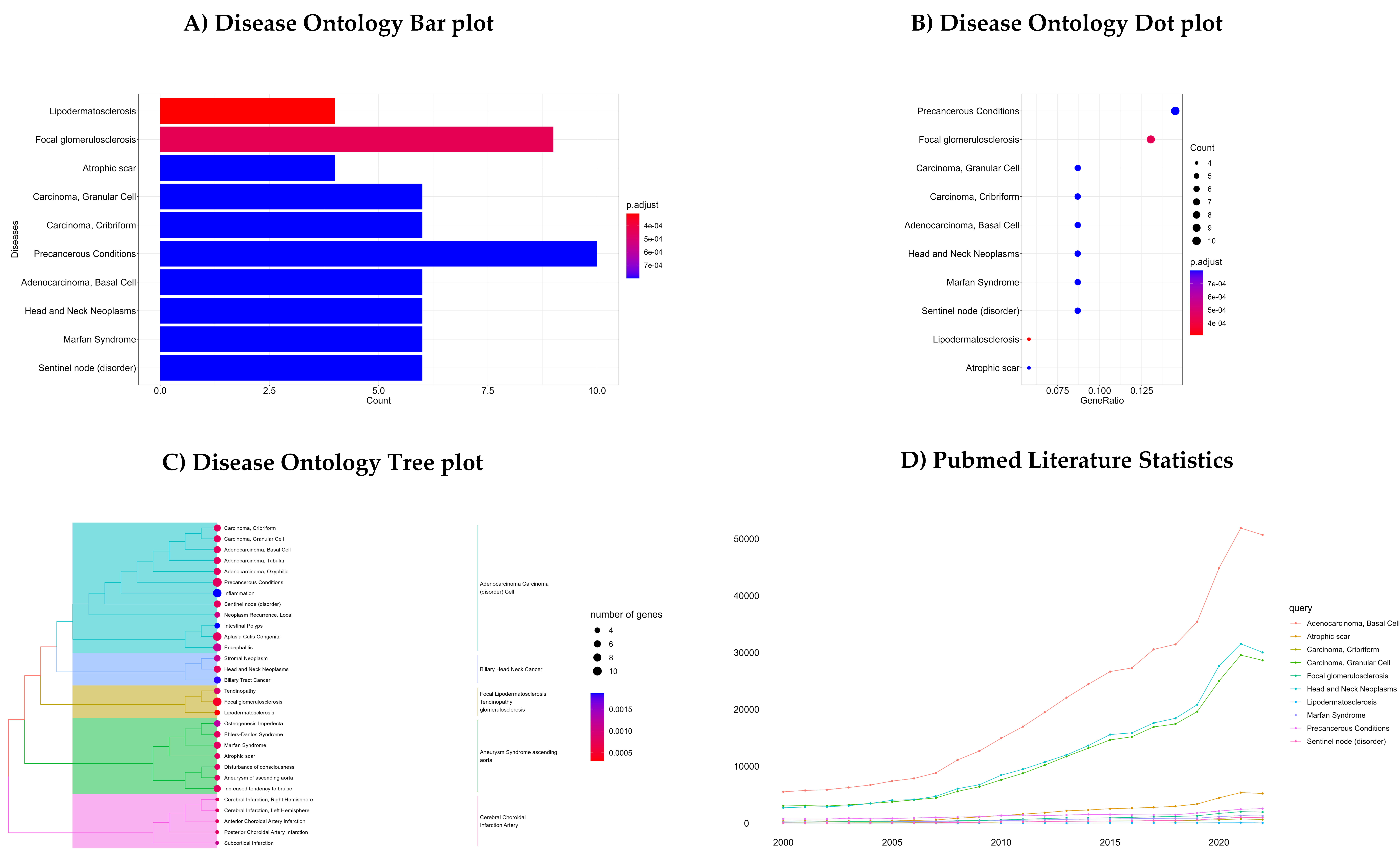 Fig. 4.
Fig. 4.Disease Ontology analysis of candidate biomarkers. (A) The bar plot illustrates the top diseases associated with the 79 candidate markers by count and Q-score. (B) Dot plot depiction of top diseases linked to 79 candidate markers based on gene ratio. The circle size increases, and the intensity of color deepens to dark red, signifying the increase in gene count. (C) A tree plot was generated to categorize marker genes to the Gene Ontology (GO) terms. (D) Line graphs made using pmcplot functions exhibit the trend in the number of publications from 2000–2020.
The seven biomarkers (ECT2, LAMC2, DSG2,
FAT1, PLOD2, COL1A1, and PLAU) identified
from the SHAP analysis were input into the STRING database. The PPI network, with
an average clustering coefficient of 0.768 and an enrichment p-value of
3.33
The survival analysis was carried out by employing the GEPIA platform. Kaplan-Meier (K-M) survival curves and log-rank tests were used to subtend the differences in survival between the high-risk and low-risk gene expression groups. The head and neck cancer samples in the TCGA cancer dataset were divided into high-risk (n = 519) and low-risk (n = 44) groups. The correlation between the tumor cell composition and patient outcomes were anticipated by assigning the risk categories to ECT2, LAMC2, and DSG2 genes.
The hazard ratio was computed for three genes, of which the LAMC2 gene showed a higher hazard ratio (1.4) and a Logrank p-value of 0.013, followed by DSG2 with a hazard ratio of 1.2 and Logrank p-value of 0.13, whereas ECT2 genes showed a hazard ratio of 1 with a Logrank p-value of 0.99 (Fig. 5A). Using whisker plots, the expression levels for the three biomarker genes were depicted for both high and low-risk groups. Fig. 5B shows that higher enrichment scores were observed for all three genes present in the higher-risk groups.
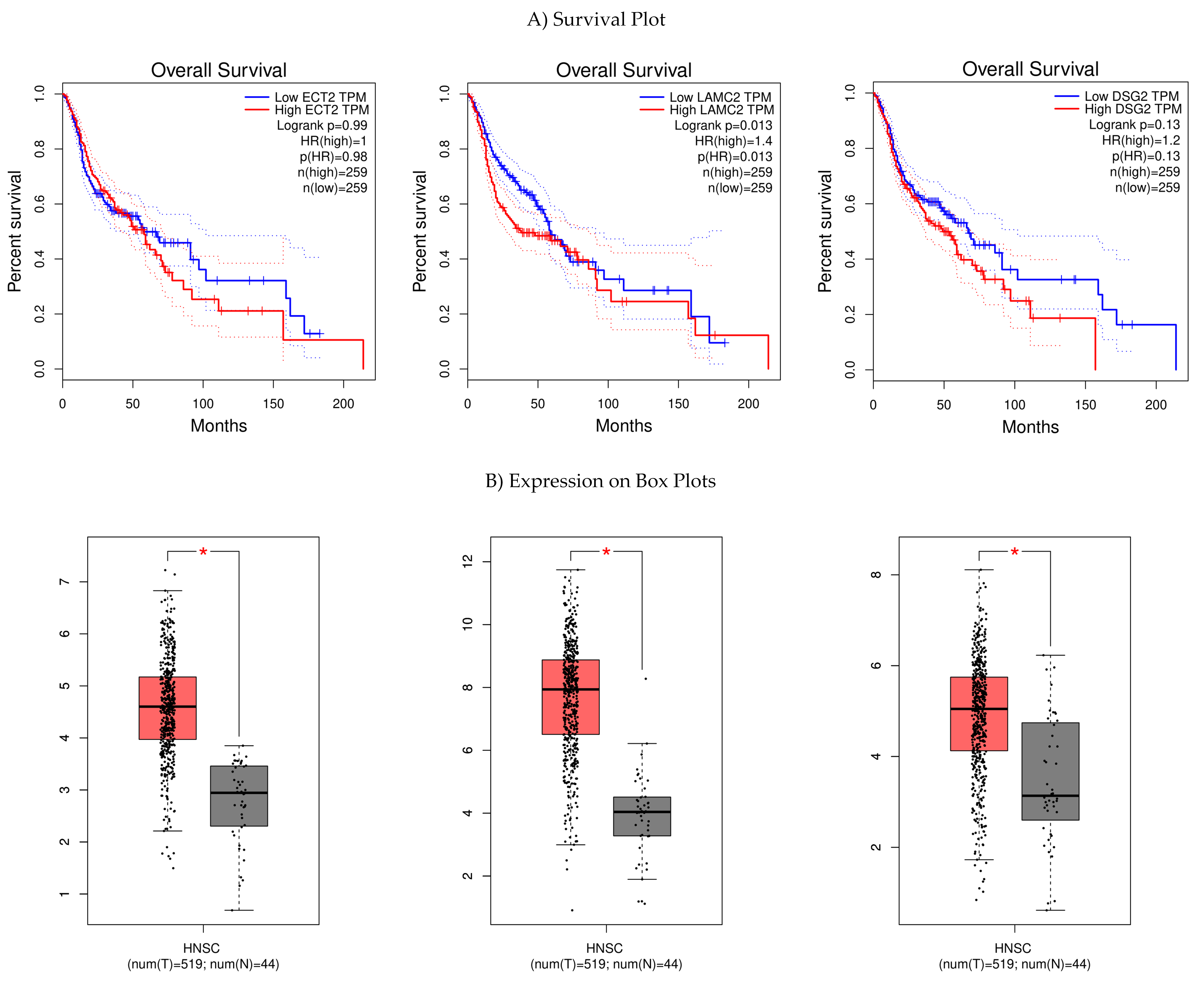 Fig. 5.
Fig. 5.Survival Analysis for the top-three genes (ECT2, LAMC2, and DSG2). (A) The K-M plots represent the survival curves from the log-rank test, comparing high-risk and low-risk groups for the top three genes (ECT2, LAMC2, and DSG2). (B) The box plot displays the enrichment scores for each risk group across the three genes. * represents the outlier.
To explore the potential therapies that may assist in disrupting cancer etiology, the 73 DEGs were analyzed by using the established DGI database. The DGI analysis revealed that 251 potential therapeutic targets were identified for DEGs. LAMC2 and PLAU genes have been identified to play imperative roles in tumor progression in OPSCC patients. Identifying potential inhibitors for the genes to promote the antitumor activity in OPSCC patients is crucial. Ocriplasmin was identified as a potential target for LAMC2, whereas 26 drug targets were identified for the PLAU gene (Supplementary Table 4).
The reproducibility and the effectiveness of the findings from the current study
are validated with similar datasets. The GEO database is queried to identify the
dataset with HPV active OPSCC and normal control samples. GSE55546 is the only
identified dataset relevant to this study, with 12 HPV active and 4 Uvula control
samples. The differential marker selection is performed between the groups with a
log2FC score of 2, adjusted p-value
The intricate molecular, environmental, and epigenetic coalition makes understanding the biological mechanism of OPSCC pathogenesis more laborious. Recently, several genes have been identified to play a significant role in the pathogenesis of OPSCC, including ECT2, LAMC2, and DSG2. ECT2 (epithelial cell transforming sequence 2) is a gene that codes for a protein functioning as a guanine nucleotide exchange factor (GEF). ECT2 regulates the Rho family of GTPases, playing a crucial role in cell migration, proliferation, and survival. In OPSCC, ECT2 downregulation has been reported in several studies [49, 50, 51, 52]. LAMC2 (laminin subunit gamma 2) encodes a component of the extracellular matrix (ECM) protein laminin-332. Laminin-332 is a heterotrimeric protein involved in cell adhesion and differentiation. LAMC2 downregulation is associated with OPSCC tumor progression [53, 54]. DSG2 (desmoglein 2) is a gene that encodes a desmosomal cadherin protein. Desmosomes are cell adhesion complexes critical in maintaining tissue integrity and strength. Research delineates that the DSG2 gene modulates pathways in individuals afflicted with OPSCC [55, 56]. The down-regulation of ECT2, LAMC2, and DSG2 increases the invasion and metastasis of OPSCC. Their knockdown has been shown to inhibit cell proliferation and decrease tumor growth. These genes could serve as potential targets for therapeutic interventions in OPSCC. This actuality is evidently proven by reproducing the same workflow with a similar dataset (Supporting Information: Supplementary Document 1).
The extant literature robustly correlates the genes delineated herein with diverse oncological phenotypes. High levels of ECT2 expression have been observed in clinical samples, such as colorectal, ovarian, and hepatocellular carcinoma [57]. Elevated levels of LAMC2 expression have been observed in several cancer types, such as gastric cancer, breast cancer, and ovarian cancer [58]. Aberrant expression of DSG2 has been documented in different cancer types, including breast cancer, prostate cancer, and oral squamous cell carcinoma [59]. The increased mortality in OPSCC cases is associated with a lack of understanding of the disease’s prognosis and dynamics. Comprehending the molecular mechanism is vital in deciding optimal treatment strategies to improve the condition. The clinical biomarkers identified in the present study give credence to developing potential therapeutics. PSO approaches help to impute the disparity between profoundly edifying and non-edifying features. Identifying such informative features tends to make a monumental contribution when subsumed within molecular signatures. The algorithm reduces the feature subset size to 73 by selecting the genes correlated with the target class type. By applying Shapley values and visual representations, we accentuate the significant features and interpretations of the ML model [60]. The Shapley values evaluate the conspicuousness of the output, considering all possible feature combinations, and deliver consistent and accurate assessments for each feature within the prediction model.
The results of the SHAP approach identified seven genes concurrent with the OPSCC, of which three genes were considered most vital for model decision. The genes unveiled by the SHAP model on the PSO-identified genes, such as ECT2, LAMC2, and DSG2, contribute directly to the OPSCC etiology. The vital information unraveled by the model reinforces that the overexpression of gene values is linked with OPSCC and is a contributing factor. Supervised machine learning classification algorithms were trained with the different subsets of the datasets (log2FC-p-value adjusted, PSO subset, and SHAP interpretations). The results exhibit significant improvement in the scores when eliminating the irrelevant features. BayesNet and Random Forest models performed best with the PSO subset compared to other benchmark algorithms and attained 100% scores against all evaluation metrics. These models displayed effective results with the seven marker genes of SHAP analysis. The experimental results further support the XAI models’ significance in accurately disclosing the interpretations.
This study has a few limitations, primarily the benchmarking with multiple datasets. The lack of similar datasets matching the phenotypic information turned the study validation with an independent dataset impractical. However, the current findings stand as evidence by supporting the relevance of existing literature. With the advent of explainable artificial intelligence methods, interpreting the black-box predictions of machine learning algorithms becomes facile. Future studies endeavor to integrate multi-omic biological information logically to untangle the pathophysiology behind complex diseases.
OPSCC remains an erratic illness with a dismal prognosis. The necessity for progressive diagnostic methodologies in OPSCC is underscored by the significant comorbidities and high recurrence rates associated with existing treatments. Exploring biomarkers, particularly those accessible through non-invasive collection methods, holds promise for transformative patient management strategies. The current study aimed to identify oncogenic drivers implicated in the pathogenesis of OPSCC by using a genomics approach. Integrating bioinformatics analysis and machine learning techniques with a thorough examination of RNA-seq datasets led to the identification of ECT2, LAMC2, and DSG2 as viable molecular markers for OPSCC. The findings from the study may be beneficial for improving the survival rates of OPSCC patients. Further research could use the findings and methodology of this work in clinical and experimental settings. In the future, we intend to increase the sample sizes to validate and closely monitor our findings.
All analyzed data during this study are included in this published article. The raw data can be found via https://github.com/karthiksekaran/OPSCC-RNASeq-XAI/.
The study’s design involved KS, RPV, SK, HZ, AEA, and GPCD. The data collection and experiment involved KS, RPV, and AEA. KS, RPV, SK, and AEA acquired, analyzed and interpreted the results. HZ and GPCD supervised the entire study. KS and RPV drafted the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the works.
Not applicable.
Not applicable.
The authors would like to thank the Vellore Institute of Technology, India authorities, for providing the necessary support in completing the manuscript. The authors acknowledge the Indian Council of Medical Research (ICMR), the Government of India agency, for the research grants (No. BMI/12(13)/2021, ID No: 2021-6359) and (No. VIR/COVID-19/31/2021/ECD-I, ID. NO: 2021-5570).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.