






1 Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, 530021 Nanning, Guangxi, China
2 Guangxi Key Laboratory of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention, Guangxi Medical University, 530021 Nanning, Guangxi, China
3 Guangxi Clinical Research Center for Cardio-Cerebrovascular Diseases, Guangxi Medical University, 530021 Nanning, Guangxi, China
Abstract
Background: Rheumatic heart disease (RHD) is caused by inflammatory
cells mistakenly attacking the heart valve due to Group A Streptococcus (GAS)
infection, but it is still unclear which cells or genes are involved in the
process of inflammatory cells infiltrating the valve. Inflammatory infiltration
into the target tissue requires an increase in the expression of phosphorylated
vascular endothelial-cadherin (p-VE-cad), p-VE-cad can increase the endothelial permeability and
promote the migration of inflammatory cells across the endothelium. P-VE-cad is
potentially regulated by RAS-related C3 botulinum substrate 1 (RAC1), together
with phosphorylated proline-rich tyrosine kinase 2 (p-PYK2). While
RAC1/p-PYK2/p-VE-cad is triggered by the activation of vascular cell adhesion
molecule-1 (VCAM-1). VCAM-1 is related to M1 macrophages adhering to the
endothelium via very late antigen 4 (VLA4). Inflammatory infiltration into the
valve is extremely important in the early pathogenesis of RHD. However, there is
no relevant research on whether M1/VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad is involved
in RHD; therefore, what we explored in this study was whether
M1/VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad is involved. Methods: We established
a rat model of RHD and a cell model of M1 macrophage and endothelial cell
cocultivation. Subsequently, we measured the degree of inflammatory cell
infiltration, the levels of IL-6/IL-17, the degree of fibrosis (COL3/1), and the
expression levels of fibrosis markers (FSP1, COL1A1 and COL3A1) in the heart
valves of RHD rats. Additionally, we detected the expression of M1/M2 macrophage
biomarkers in rat model and cell model, as well as the expression of
M1/VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad. We also tested the changes in endothelial
permeability after coculturing M1 macrophages and endothelial cells.
Results: Compared to those in the control group, the levels of
inflammatory cell infiltration and fibrotic factors in the heart valves of RHD
rats were significantly higher; the expression of M1 macrophage biomarkers (iNOS,
CD86 and TNF-
Keywords
- rheumatic heart disease
- M1 macrophages
- VLA4/VCAM-1
- inflammatory infiltration
Rheumatic heart disease (RHD) is associated with Group A Streptococcus (GAS) infection and causes approximately 300,000 deaths annually [1, 2]. It is one of the reasons that should be most responsible for the morbidity and mortality in individuals with cardiovascular disease (CVD) [3]. RHD affects mainly younger individuals and underdeveloped regions, where it is the most common reason for CVD and early death in young people [4]. However, RHD is also one of the most neglected diseases in terms of available treatment, with surgery being the main effective treatment strategy [5]. The implementation of preventive measures is not ideal, and there is a lack of low cost treatment methods that require little equipment [6]. The pathogenesis of RHD is still poorly understood, and is the main reason why alternative treatment methods have yet to be found [7]. Investigation of the pathogenesis of this disease is therefore crucial for the prevention and treatment of RHD.
A potential connection exists between pathological changes in heart valves and the pathogenesis of RHD, with valvular disease being the most common type of RHD [8]. The occurrence of valve lesions is related to the presence of cross antigens between heart tissue and the M protein of GAS, causing immune cells to mistakenly attack the heart valves [9, 10, 11, 12, 13]. Our previous research demonstrated significant infiltration of inflammatory cells into the heart valves of RHD model rats [14, 15]. However, the mechanism by which inflammatory cells infiltrate the valve during the pathogenesis of RHD has yet to be determined, together with identification of the cells and molecules that are involved. To date, there is a paucity of relevant research on the inflammatory cell infiltration of valves in RHD.
The process of inflammatory cells migrating and infiltrating target tissue across the endothelium is an important initiating process of autoimmunity [16], inflammatory development [17], and cellular attack [18] and has a potential connection with the autoimmune process of RHD. In autoimmune diseases, for inflammatory cells to infiltrate target tissue, the barrier function of the endothelium needs to be impaired, after which permeability increases, thereby promoting the infiltration of inflammatory cells across the endothelium into the target tissue [19].
A close relationship exists between macrophages and the changes in endothelial
permeability. In the early stages of inflammation, M1 macrophages often play a
role in promoting the inflammatory response [20]. The association between M1
macrophages and endothelial cells (ECs) occurs through the formation of strong
adhesions between very late antigen-4 (VLA4, also known as integrin
VLA4 is expressed mainly on macrophages and binds to VCAM-1 on ECs, thereby mediating leukocyte transport during inflammation [27]. VLA4 is crucial in the infiltration of inflammatory cells in autoimmune diseases. For example, inhibiting the expression of VLA4 can alleviate lesions in multiple sclerosis [28].
The VCAM-1 protein is typically involved in leukocyte adhesion and migration during inflammation. The expression of VCAM-1 is considered to be a biomarker of immunologic diseases, cancer and autoimmune myocarditis. It is also a predictor of incidence and mortality in chronic heart failure (CHF), and of endothelial damage in patients with coronary heart disease and arrhythmia [29]. The expression of VCAM-1 on ECs is considered to be a marker of inflammation and indicates cell infiltration [30].
The high expression of VCAM-1 promotes the expression of phosphorylated vascular endothelial-cadherin (p-VE-cad), resulting in loss of EC adhesion and dysfunction of the endothelial barrier, thus enhancing leukocyte transport during inflammation [31]. Previous studies have reported that p-VE-cad is potentially regulated by RAS-related C3 botulinum substrate 1 (RAC1) and proline-rich tyrosine kinase 2 (PYK2) [32]. RAC1 plays a major role in important cellular processes, particularly in the control of cell motility and dynamics, regulation of oxidative stress, and monitoring inflammation and immunity [33]. PYK2 belongs to the non-receptor tyrosine kinase family of focal adhesion kinase (FAK) proteins [34] and is a key factor in EC inflammation. Specific targeting of PYK2 is potentially effective for vascular inflammatory diseases associated with EC inflammation and intravascular coagulation [35]. RAC1/p-PYK2/p-VE-cad is potentially regulated by VLA4/VCAM-1 to form a cascade reaction chain, which is closely related to the adhesion of ECs and to vascular permeability [27]. This chain starts with M1 macrophages adhering to ECs and ends with increased VE-cad phosphorylation giving rise to weaker adhesion and increased permeability between ECs.
Based on current knowledge of the role of M1 macrophages and the VLA4/VCAM-1 pathway, we speculated that M1 macrophages activate downstream cascade reactions (RAC1/p-PYK2/p-VE-cad) via the VLA4/VCAM-1 pathway, thereby promoting inflammatory infiltration during RHD by increasing endothelial permeability. What we investigated in this study was whether M1 macrophages, the VLA4/VCAM-1 pathway, and the downstream cascade reaction RAC1/p-PYK2/p-VE-cad have potential involvement in RHD.
We established an RHD rat model and a coculture model of M1 macrophages and endothelial cells (ECs) to investigate whether M1 macrophages and the VLA4/VCAM-1 pathway are potentially involved in RHD. Inflammation and fibrosis of the heart valve are the core processes of RHD, and the infiltration of inflammatory cells into the valve to initiate false attacks is an important initiating step in the pathogenesis of RHD. This study focused on exploring the possible factors involved in the process of inflammatory infiltration in RHD patients, providing new references for updating the theory of pathogenesis and developing prevention and control measures for RHD.
Medium for Group A streptococci (GAS, cat. no. ATCC19615; American Type Culture
Collection, Mansas, VA, USA): brain heart infusion (Cat. no. HCM019; Guangdong Huankai Microbial
Sci. & Tech4. Co., Ltd., Guangdong, China), the condition for culturing: 24 h at
37 °C. After that, normal saline (NS) was used to wash the cultured GAS.
Following inactivation with 10% neutral formalin for 12 h and washing with NS,
the GAS were resuspended and the density adjusted to 4.0
We randomly divided 12 Lewis rats (Cat. no. 113; 150–180 g, eight-week-old,
female; Beijing Vital River Animal Technology Co., Ltd., Beijing, China) into 2
groups: the control group (n = 6) and the RHD group (n = 6). Animal experiments
conducted in a specific pathogen-free animal laboratory, which belongs to the
Animal Experiment Center of Guangxi Medical University. Comfortable living
conditions for the rats were guaranteed. The temperature was controlled at 23
°C, with a daily fluctuation of
The classical method was used to establish the RHD rat model [14, 37, 38, 39, 40] over a period of 9 weeks. At the beginning of the experiment, rats in the RHD group were treated with unilateral rear footpad injections of a 1:1 (v/v) mixed solution of 0.1 mL of antigen and CFA. After 4 weeks, a 0.5 mL mix of the antigen and CFA solution (1:1, v/v) was injected subcutaneously into the abdomen. After this, the injection solution was changed to 0.5 mL of antigen only, and subcutaneous injections in the abdomen were performed weekly for 4 weeks. The injection protocol for rats in the control group was the same as for those in the RHD group, except that the injection solution contained NS only (the flowchart is shown in Fig. 1A).
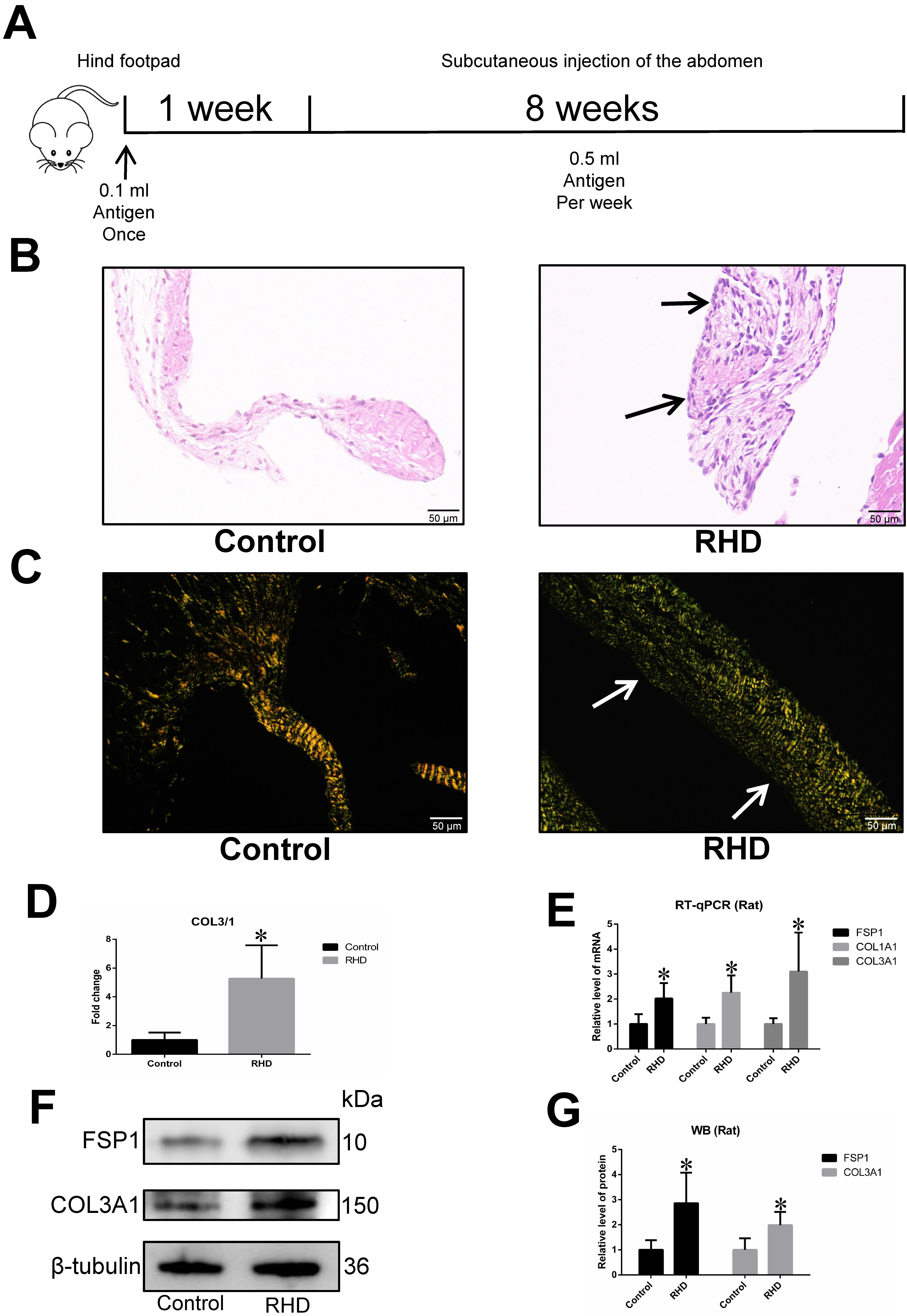 Fig. 1.
Fig. 1.Increased degree of inflammatory infiltration and fibrosis in
RHD group. (A) Flow chart for the establishment of a rat model of RHD. (B) H&E
staining of the heart valves of rats. Extensive infiltration of inflammatory
cells was observed in the valves of the RHD group compared to the control group;
magnification, 400
Following the above treatments, we took approximately 2.5 mL of blood from the retinal vein of rats with isoflurane anaesthesia. All rats were then sacrificed by intraperitoneal injection of sodium pentobarbital (150 mg/kg). The absence of a heartbeat or breathing for more than 5 minutes was considered to indicate death of the animal. If the rats had lost more than 15% of their body weight, and their ability to eat and drink also decreased during the experiment, this was considered the humane endpoint. After collection, heart samples were quickly frozen at –80 °C for follow-up tests. In subsequent experiments, a portion of the heart valve were isolated, then mRNA and protein were extracted from valve tissue for reverse transcription-quantitative Polymerase chain reaction (RT-qPCR) and western blot (WB) analysis [7, 14, 41, 42].
A co-culture method of M1 macrophages grown with ECs was used to simulate the
adhesion of M1 macrophages to the endothelium. It was also used to observe
whether this cell combination could trigger downstream cascade reactions that
increased endothelial permeability. Human monocytic (THP-1) cells (Cat. no.
CL-0233; Procell, Wuhan, China) were cultured in THP-1 medium (Cat. no. CM-0233;
Procell, Wuhan, China). Human umbilical vein ECs (Cat. no. PRI-H-00023; ZQXZBIO,
Shanghai, China) were cultured in EC medium (Cat. no. 1001; ScienCell, San Diego,
CA, USA). All cells were cultured in a moist cell incubator at 37
°C and in a 5% CO
RT-qPCR was used to quantify the mRNAs for: M1 macrophage biomarkers [inducible
nitric oxide synthase (iNOS), CD86, TNF-
| Species | Name | F or R | Sequences of primers |
| Rat | iNOS | F | TCTTGGAGCGAGTTGTGGATTGTTC |
| R | AGTGATGTCCAGGAAGTAGGTGAGG | ||
| CD86 | F | GCTGTCTCTTTCTGCTGGTCGTC | |
| R | CTCACAAGTCTTTCTGCTGGGTCTG | ||
| TNF- |
F | AAAGGACACCATGAGCACGGAAAG | |
| R | CGCCACGAGCAGGAATGAGAAG | ||
| Arg1 | F | AGTGTGGTGCTGGGTGGAGAC | |
| R | GCGGAGTGTTGATGTCAGTGTGAG | ||
| TGF- |
F | GACCGCAACAACGCAATCTATGAC | |
| R | CTGGCACTGCTTCCCGAATGTC | ||
| VLA4 | F | TGGCATCTCCTCTACATACTCACAG | |
| R | ACCAACCGCTACATCAACATATCC | ||
| VCAM-1 | F | GCTGCTGTTGGCTGTGACTCTC | |
| R | GCTCAGCGTCAGTGTGGATGTAG | ||
| RAC1 | F | CACACAGCGAGGACTCAAGACAG | |
| R | AAAGGCTCCAGGGACCAAGACC | ||
| FSP1 | F | TGGGGAGAAGGACAGACGAAGC | |
| R | TGGCAATGCAGGACAGGAAGAC | ||
| COL1A1 | F | TGTTGGTCCTGCTGGCAAGAATG | |
| R | GTCACCTTGTTCGCCTGTCTCAC | ||
| COL3A1 | F | AGTCGGAGGAATGGGTGGCTATC | |
| R | CAGGAGATCCAGGATGTCCAGAGG | ||
| F | GCTGTGCTATGTTGCCCTAGACTTC | ||
| R | GGAACCGCTCATTGCCGATAGTG | ||
| Human | CD86 | F | ACTATGGGACTGAGTAACATTCTCTTTG |
| R | GTTCTGGGTAACCGTGTATAGATGAG | ||
| TNF- |
F | TGGCGTGGAGCTGAGAGATAAC | |
| R | GCTGATGGTGTGGGTGAGGAG | ||
| VLA4 | F | CTGTGATGCCTTACATTAGCAC | |
| R | ATCCAAATTTCCAGATATGCGC | ||
| VCAM-1 | F | ACCACATCTACGCTGACAATGAATCC | |
| R | AACACTTGACTGTGATCGGCTTCC | ||
| RAC1 | F | GTCCCAACACTCCCATCATCCTAG | |
| R | TACAGCACCAATCTCCTTAGCCATG | ||
| F | CTACCTCATGAAGATCCTCACC | ||
| R | AGTTGAAGGTAGTTTCGTGGAT |
F, forward; R, reverse; iNOS, inducible nitric oxide synthase; VLA4, very late atigen 4; VCAM-1, vascular cell adhesion molecule-1; RAC1, RAS-related C3
botulinum substrate 1; FSP1, fibroblast-specific protein 1; COL1A1, collagen type I
Primary antibodies used for the detection of proteins by WB analysis were:
CD86-antibody (1:1000 dilution; cat. no. 13395-1-AP; ProteinTech Group, Inc.,
Wuhan, China), TNF-
Haematoxylin and eosin (H&E) staining and Sirius red staining were employed for histological studies. The test object was a 5 µm paraffin section embedded with valve tissue. The valve tissue was fixed with 4% paraformaldehyde at 4 °C for 24 h, dehydrated in alcohol, and then embedded in molten paraffin wax (58–60 °C). H&E staining was performed on paraffin sections at room temperature, with haematoxylin staining for 4–10 min, and eosin staining for 0.5–2 min. Paraffin sections were stained with Sirius red solution for 1 h. A BX43 light microscope (Olympus Corporation, Tokyo, Japan) was used to record images.
After co-culturing M1 macrophages and ECs on a 6-well plate for 24 h, the cells
were washed three times with PBS and then fixed with 4% paraformaldehyde for 10
min at 4 °C. They were then washed three times with PBS, permeabilized
with 0.5% Triton X-100 for 20 min, and washed again three times with PBS. The
cells were then treated for 30 min with blocking solution (Cat no. P0260;
Beyotime, Shanghai, China) and washed. Next, they were incubated overnight at 4
°C with the following primary antibodies: VLA4-antibody (1:100; cat. No.
19676-1-AP; ProteinTech Group, Inc., Wuhan, China), VCAM1-antibody (1:200; cat. No. DF6082;
Affinity Biosciences, Jiangsu, China), RAC1-antibody (1:200; cat. no. 24072-1-AP;
ProteinTech Group, Inc., Wuhan, China), p-PYK2-antibody (1:200; cat. no. 11216; Signalway
Antibody LLC, Greenbelt, MD, USA), and p-VE-cad-antibody (1:200; cat. No. AF3265;
Affinity Biosciences, Jiangsu, China). The next day, the samples were washed three times with
PBST before incubation with fluorescent secondary antibody (Abcam, Cambridge Science Park, UK) for 1 hour at
room temperature in the dark. After washing three times with PBS, 4
The primary antibodies used for immunohistochemistry were as follows: CD86
(1:80; cat. no. 13395-1-AP; ProteinTech Group, Inc., Wuhan, China),
TNF-
Permeability experiments were used to measure the effect of M1 macrophages on
endothelial permeability [45]. ECs were first randomly divided into two groups
(M0+EC group and M1+EC group) and the cell concentration adjusted to 1
The serum levels of IL-6 and IL-17 in rats were measured using ELISA kits (cat. no. E04640r; E07451r; Cusabio Technology LLC, Wuhan, China) according to the manufacturer’s protocols. The absorbance at 450 nm in each well was measured using a microplate reader (Multiskan; Thermo Inc., Waltham, MA, USA).
Statistical analysis of data was performed with SPSS software 17.0 (SPSS, Inc.,
Chicago, IL, USA), and the results presented as the mean
The H&E staining results indicated a greater degree of inflammatory cell
infiltration in the heart valves of RHD rats compared to the control group (Fig. 1B). Sirius red staining also indicated a significantly higher level of fibrosis
in the heart valves of RHD rats (Fig. 1C). According to the results of Sirius red
staining, loosely arranged green fibres represent type 3 collagen (COL3) fibres,
closely packed yellow and red fibres represent type 1 collagen (COL1) fibres, and
the ratio of COL3 to COL1 increases with the degree of tissue fibrosis. The
COL3/COL1 (COL3/1) ratio can therefore be used to represent the degree of
valvular fibrosis [46]. The Sirius red staining also showed the COL3/1 ratio was
significantly greater in the RHD group than in the control group (p
Immunohistochemistry and ELISA also revealed significantly higher levels of IL-6
and IL-17 in the heart valves and serum of RHD rats compared to the control group
(p
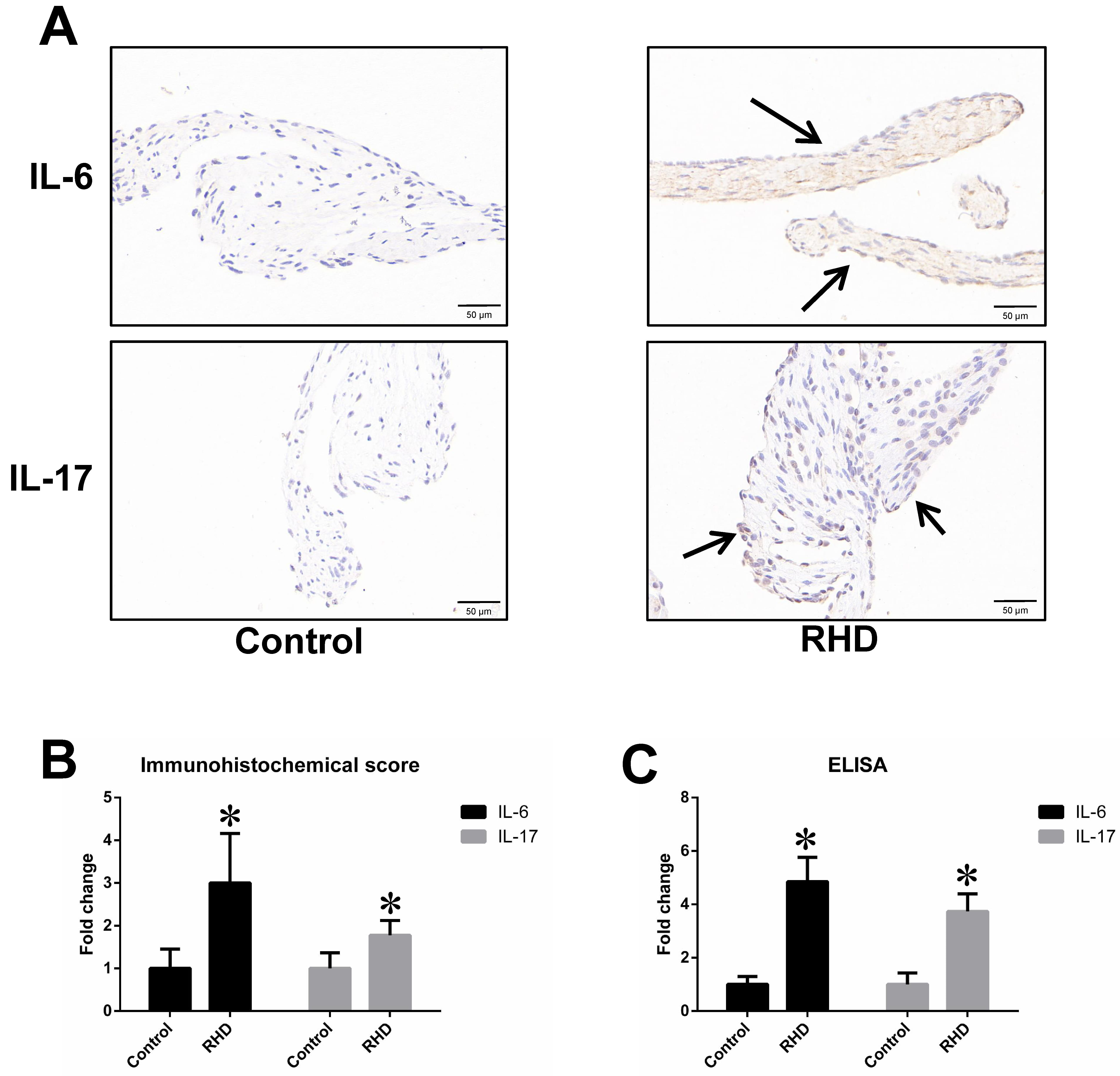 Fig. 2.
Fig. 2.IL-6 and IL-17 expression in the heart valves and serum. (A,B)
Immunohistochemistry results for IL-6 and IL-17. Scale bar: 50 μm. (C) Enzyme-Linked Immunosorbent Assay (ELISA) results for IL-6 and
IL-17. (A), (B) and (C) showed that IL-6 and IL-17 expression in the heart valves
and serum of RHD rats was significantly greater than in the control group. The
data are presented as the mean
The results of RT-qPCR, WB and immunohistochemistry showed that the expression
of M1 macrophage biomarkers (iNOS, CD86, and TNF-
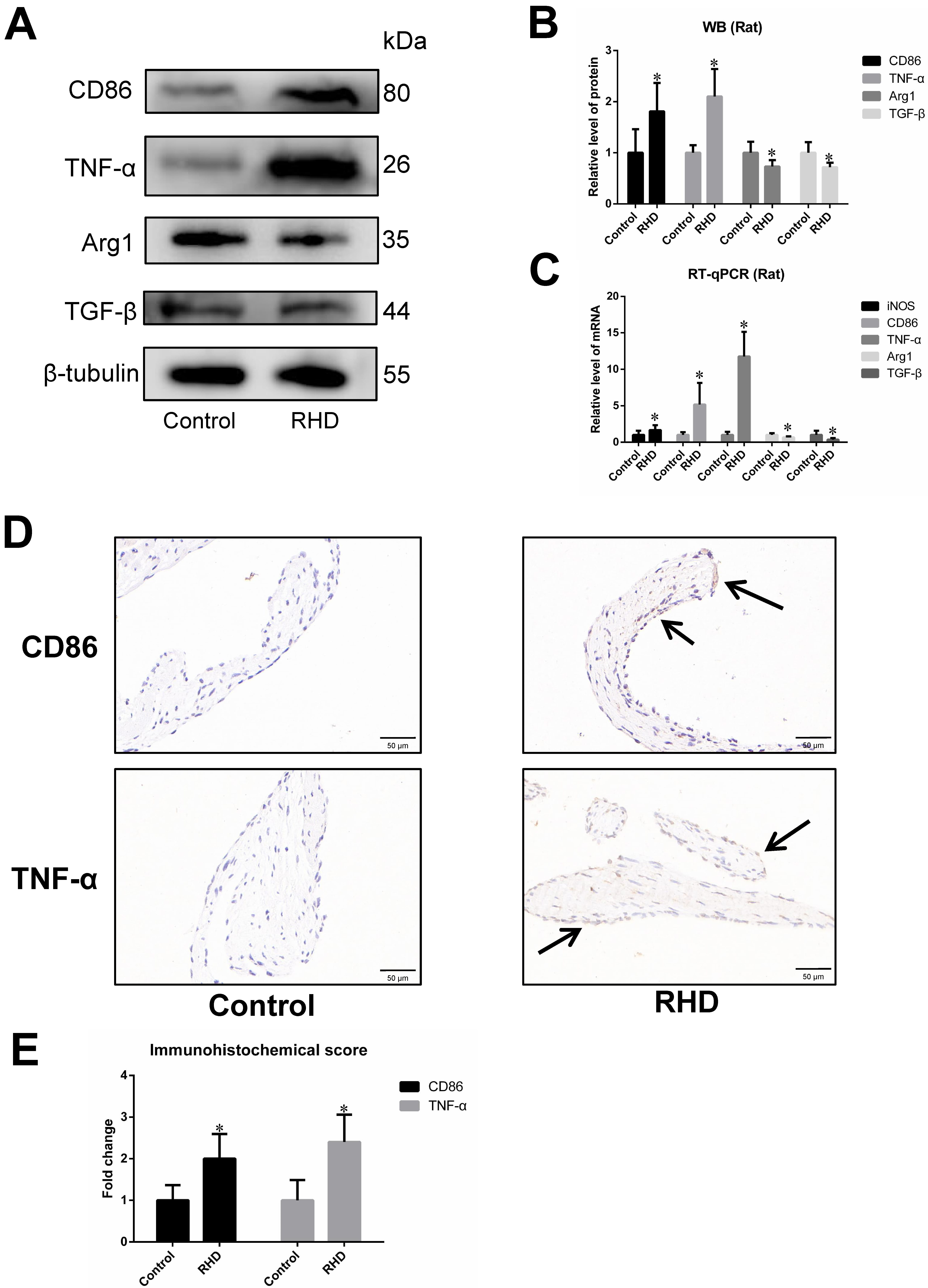 Fig. 3.
Fig. 3.Differences in the expression of M1/M2 macrophage
biomarkers between control group and RHD group. (A,B) WB analysis of CD86,
TNF-
The results obtained with RT-qPCR, WB and immunohistochemistry showed that the
RHD group had significantly higher expression levels of VLA4, VCAM-1, RAC1,
p-PYK2, and p-VE-cad than the control group. The increased expression of p-VE-cad
suggested a possible increase in endothelial permeability, which may be related
to the migration of inflammatory cells across the endothelium (p
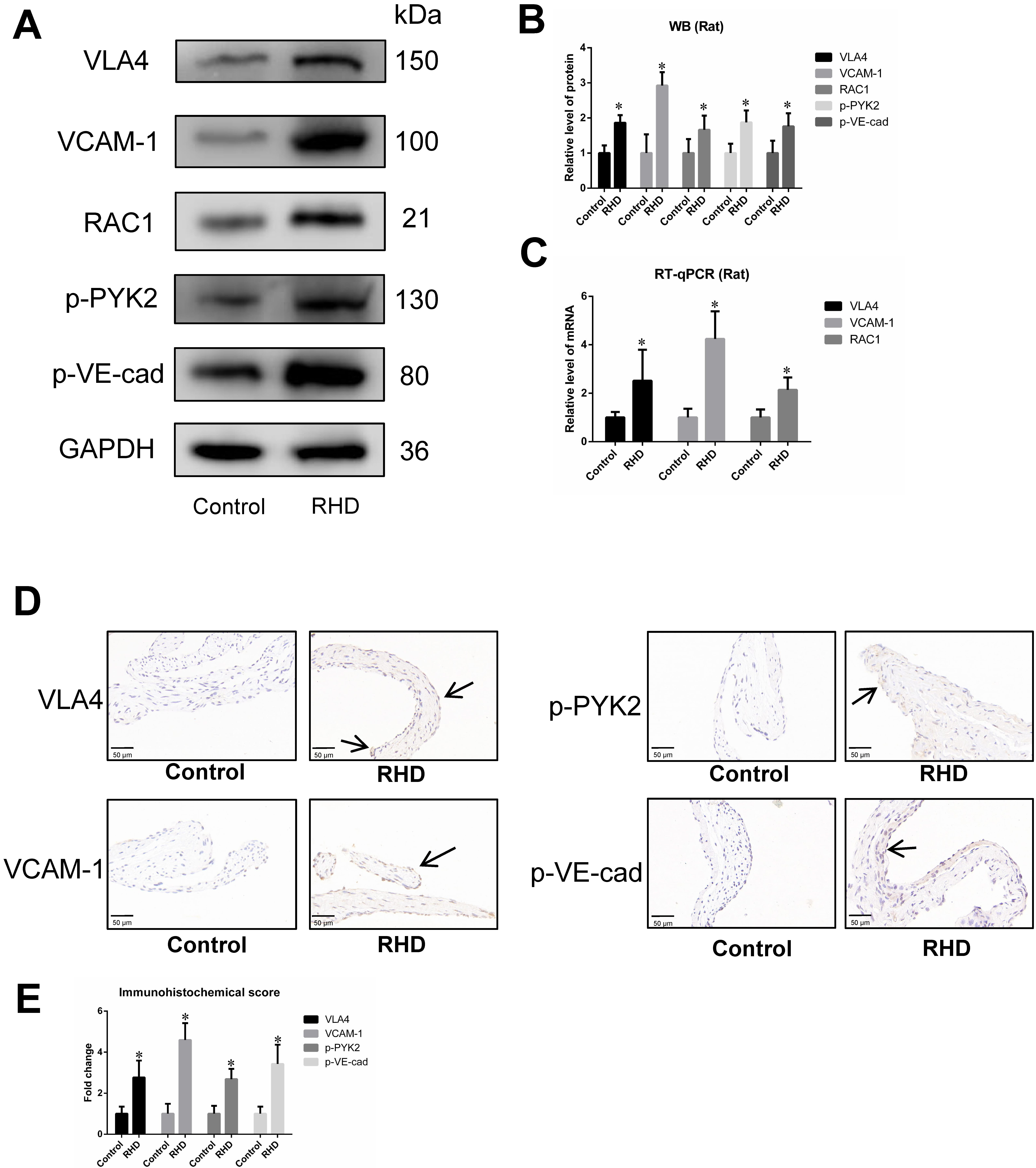 Fig. 4.
Fig. 4.Differences in the expression of
VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad between control group and RHD group. (A,B) WB
analysis of VLA4, VCAM-1, RAC1, p-PYK2 and p-VE-cad. The protein expression
levels of VLA4, VCAM-1, RAC1, p-PYK2 and p-VE-cad were significantly higher in
the RHD group than in the control group. (C) RT‒qPCR results for VLA4, VCAM-1 and
RAC1. The mRNA expression levels of VLA4, VCAM-1 and RAC1 were significantly
higher in the RHD group than in the control group. (D,E) Immunohistochemical
results for VLA4, VCAM-1, p-PYK2 and p-VE-cad in heart valves. Magnification,
400
Results obtained with RT-qPCR, WB and immunofluorescence revealed that the
expression of M1 macrophage markers (CD86, TNF-
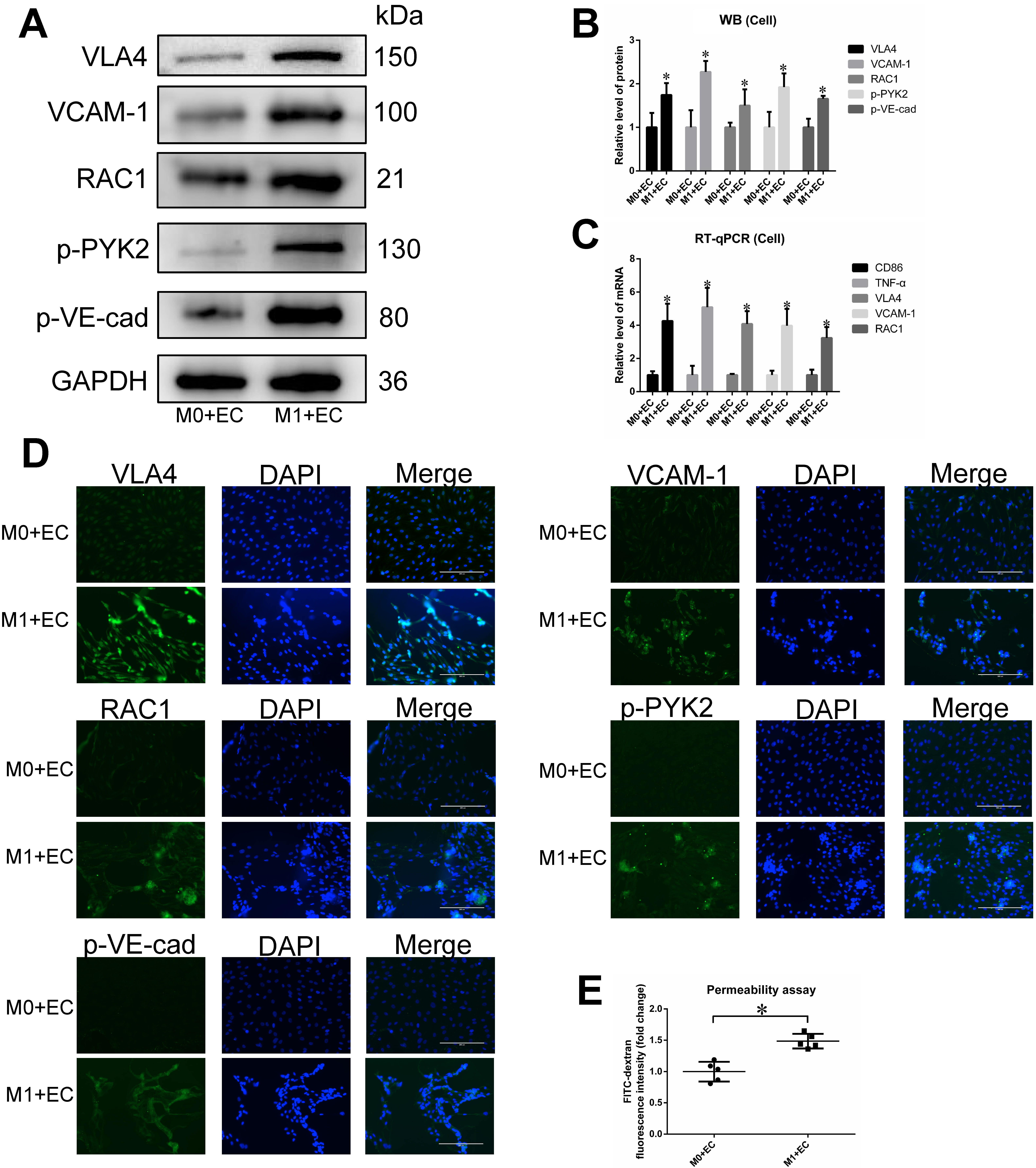 Fig. 5.
Fig. 5.Differences in the expression of
VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad and endothelial permeability between M0+EC group
and M1+EC group. (A,B) WB results for VLA4, VCAM-1, RAC1, p-PYK2 and p-VE-cad.
The protein expression of VLA4, VCAM-1, RAC1, p-PYK2 and p-VE-cad was
significantly higher in the M1+EC group than in the M0+EC group. (C) RT‒qPCR
results for CD86, TNF-
To further verify whether M1 macrophages enhance endothelial permeability, we
conducted a permeability assay. Endothelial permeability in the M1+EC group was
found to be significantly greater than in the M0+EC group. These findings
suggested that M1 macrophages may increase endothelial permeability, thereby
promoting the migration of inflammatory cells across the endothelium and into
target tissue (p
RHD is the result of valvular damage caused by GAS infection. The pathogenesis
of RHD may involve inflammation and fibrosis of heart valves caused by autoimmune
effects [26], and is therefore closely related to autoimmune inflammatory
reactions in the body. Previous studies have shown the existence of cross
antigens between the M protein of GAS and cardiac proteins such as myosin,
laminin and vimentin. The antigen‒antibody cross-reaction triggers a large number
of inflammatory cells to infiltrate the cardiac valve, leading to inflammatory
damage [9, 10, 11, 12, 13]. Previous studies have also reported infiltration of CD4
In the process of autoimmune development, M1 macrophages often play a role in
promoting the inflammatory response [20]. M1 macrophages are typically polarized
by interferon-
Changes in the permeability of the endothelium are extremely important for immune cell migration across the endothelium to the valve. There is a close relationship between macrophages and endothelial permeability. For example, in ovarian cancer, M1 macrophages increase endothelial permeability and promote the development of ascites [27]. Macrophages can also cause tight connections between cochlear ECs to become unstable, leading to increased vascular permeability and finally to hearing loss [50]. Direct interaction between macrophages and ECs is also crucial during pathological processes such as atherosclerosis and inflammation [27]. According to previous studies, the induction of high endothelial permeability by M1 macrophages mainly relies on the firm adhesion between M1 macrophages and the endothelium via VLA4/VCAM-1 pathway, which then triggers downstream signals, ultimately leading to high endothelial permeability [21, 22, 23, 24, 25].
VLA4 is expressed mainly on macrophages and binds to its ligand VCAM-1 on ECs, thereby mediating leukocyte transport during inflammation [27]. VCAM-1 is a protein typically involved in leukocyte adhesion and migration during inflammation. Previous studies have reported that VCAM-1 interacts with VLA4 on macrophages, promoting the migration of inflammatory cells [51, 52].
The high expression of VCAM-1 promotes the expression of p-VE-cad, which is the main endothelial adhesion molecule that controls EC connectivity [53]. Its phosphorylation leads to a decrease in cell adhesion, dysfunction of the vascular barrier, and enhanced leukocyte transport during inflammation [31]. The phosphorylation of VE-cad is related to RAC1 and PYK2, which are potential regulatory factors for p-VE-cad [32]. Previous studies have reported that the downstream cascade reaction RAC1/p-PYK2/p-VE-cad mediated by VLA4/VCAM-1 is important for EC adhesion and endothelial permeability [27].
The above studies suggest important relationships between M1 macrophages and the VLA4/VCAM-1 pathway, and between M1 macrophages and the degree of inflammatory infiltration. Valvular inflammatory infiltration is the core pathological change in RHD. Based on previous findings with regard to the role of M1 macrophages and the VLA4/VCAM-1 pathway, we speculated that during the pathogenesis of RHD, M1 macrophages may activate downstream cascade reactions (RAC1/p-PYK2/p-VE-cad) via the VLA4/VCAM-1 pathway. This reduces EC adhesion and increases permeability, thus promoting the migration of inflammatory cells across the endothelium to the valve.
The current experimental results lend support to our hypothesis. We observed significant infiltration of inflammatory cells and fibrotic lesions in the heart valves of RHD rats. Moreover, the levels of M1 macrophage-related markers were significantly higher than in the control rats, as well as being significantly higher than the levels of M2 macrophage-related markers. In addition, the VLA4/VCAM-1 and RAC1/p-PYK2/p-VE-cad-related indicators were significantly greater than in the control group. We also employed a cell co-culture method involving M1 macrophages and ECs. This simulates the adhesion of M1 macrophages to the endothelium, thus allowing investigation of whether downstream cascade reactions can be triggered to increase endothelial permeability. These cell experiments also showed that co-culture of M1 macrophages with ECs resulted in significantly increased expression of VLA4/VCAM-1 and RAC1/p-PYK2/p-VE-cad-related indicators, as well as increased endothelial permeability. The experimental results suggested that M1 macrophages, VLA4/VCAM-1 and the downstream cascade reactions RAC1/p-PYK2/p-VE-cad may be involved in RHD.
The present results suggest that endothelial activation regulated by M1 macrophages, VLA4/VCAM-1, and downstream cascade reactions (RAC1/p-PYK2/p-VE-cad) may be involved in the pathogenesis of RHD, and may also be involved in altering endothelial permeability to promote inflammatory cell infiltration into the valve. Previous studies have shown that inhibition of VLA4 can have a therapeutic effect in multiple sclerosis, which is also an autoimmune disease, and related drugs have been developed [54]. This finding represents a new perspective in the treatment of RHD, a disease for which effective treatment methods are urgently required. There are many possible intervention targets for RHD, as well as new prevention and treatment ideas that warrant further research.
Limitations of this study: experiments are needed to investigate the specific mechanism involving M1/VLA4/VCAM-1 in RHD like using siRNAs to intervene in the VLA4/VCAM-1 pathway. Future research can be conducted to evaluate the results of this study in the human body, and the therapeutic effects of M1/VLA4/VCAM-1-related intervention drugs in RHD, which have clinical application prospects. Furthermore, this study focused on pathological changes in the valve and did not consider the symptoms of animal. The intervention of RAC1 and PYK2 can deeply interpret the mechanism of VLA4/VCAM-1/RAC1/p-PYK2/p-VE-cad in RHD, which requires further research in the future. In addition, it should isolate and induce the macrophages to detect the biomarkers in vivo.
Inflammation and fibrosis of the heart valve are the core pathologies of RHD. The mechanism by which inflammatory cells infiltrate the heart valve to initiate false attacks is therefore an important part of RHD pathogenesis. Animal and cell experiments conducted in this study found that M1 macrophages and the VLA4/VCAM-1 pathway, which are related to inflammatory infiltration, are potentially involved in the process of valvular inflammatory infiltration in RHD. This study provides new evidence that updates our understanding of the pathogenesis of RHD, and may also spur the development of novel prevention and treatment methods.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
SX and ZZ conceived and designed the study. SX, YL, LB, ST and ZM performed the experiments. SX, HW and FH analyzed the data. SX drafted the manuscript, SX, YL, LB, ST and ZM revised it critically for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
Protocols involving animals were approved by the Animal Care & Welfare Committee of Guangxi Medical University (Approval No. 202209003).
Not applicable.
This work was supported by the National Natural Science Foundation of China (grant no. 81960082), Guangxi Key Laboratory of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention (No. 22-035-18) & Guangxi Clinical Research Center for Cardio-cerebrovascular Diseases (No. AD17129014), and Innovation Project of Guangxi Graduate Education (No. YCBZ2024126).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.