
 , Ekaterina V. Chernyshova 1, Ekaterina P. Krutskikh 1, Irina S. Sadovnikova 1, Elena E. Tekutskaya 3, Anna A. Dorohova 3
, Ekaterina V. Chernyshova 1, Ekaterina P. Krutskikh 1, Irina S. Sadovnikova 1, Elena E. Tekutskaya 3, Anna A. Dorohova 31 Department of Genetics, Cytology and Bioengineering, Voronezh State University, 394018 Voronezh, Russia
2 Laboratory of Metagenomics and Food Biotechnology, Voronezh State University of Engineering Technology, 394036 Voronezh, Russia
3 Laboratory of Problems of Stable Isotope Spreading in Living Systems, Southern Scientific Center of the Russian Academy of Sciences, 344006 Rostov-on-Don, Russia
Abstract
Mitochondrial DNA (mtDNA) is located in the mitochondrial matrix, in close proximity to major sources of reactive oxygen species (ROS) in the cell. This makes mtDNA one of the most susceptible components to damage in the cell. The nuclear factor E2-related factor 2/antioxidant response element (Nrf2/ARE) signaling pathway is an important cytoprotective mechanism. It is well-studied and described that Nrf2 can regulate the expression of mitochondrial-targeted antioxidant systems in the cell, indirectly protecting mtDNA from damage. However, the Nrf2/ARE pathway can also directly impact on the mtDNA repair processes. In this review, we summarize the existing data on the impact of Nrf2 on mtDNA repair, primarily base excision repair (BER), as it is considered the main repair pathway for the mitochondrial genome. We explore the crosstalk between Nrf2/ARE, BRCA1, and p53 signaling pathways in their involvement in maintaining mtDNA integrity. The role of other repair mechanisms in correcting mismatched bases and double-strand breaks is discussed. Additionally, the review addresses the role of Nrf2 in the repair of noncanonical bases, which contribute to an increased number of mutations in mtDNA and can contaminate the nucleotide pool.
Keywords
- Nrf2/ARE pathway
- mtDNA
- base excision repair
- BRCA1
- p53
Mitochondrial dysfunction is responsible for the development of over 200
diseases of various characteristics and is also associated with aging.
Mitochondria possess their unique genetic material known as mitochondrial DNA
(mtDNA), which is responsible for encoding 13 protein subunits involved in the
oxidative phosphorylation system, as well as a complete set of transfer and
ribosomal RNAs. While over 99% of the proteins in mitochondria are encoded by
nuclear DNA, maintaining the integrity of mtDNA is crucial
for mitochondrial functions. This importance is exemplified by the occurrence of
mitochondrial diseases resulting from mtDNA mutations and depletions, as well as
the significance of fragmented mtDNA molecules in various cell signaling pathways
[1]. The greatest contribution to mtDNA damage is attributed to oxidative stress,
which is due to the localization of mtDNA in the mitochondrial matrix, in close
proximity to the major sites of reactive oxygen species (ROS) production [2]. It
is challenging to estimate the exact amount of ROS generated in mitochondria, as
these values can vary depending on cell type and its metabolic status, but it is
often reported that 90% of all ROS is produced in mitochondria [3, 4, 5].
Furthermore, up to 85% of mitochondrial ROS production is attributed to complex
I of electron-transport chain. This is not surprising given that complex I is a
large super complex consisting of 44 subunits, at least 10 of which are
redox-active [6]. ROS-generating enzymes in the inner mitochondrial membrane and
mitochondrial matrix include complex III, aconitase, which contains iron-sulfur
clusters responsible for O
In recent years, new properties of ROS have been discovered that are not associated with toxic effects. It has been shown that ROS can act as important messengers that transmit cellular signals [11]. There is a murburn concept, according that redox proteins mediate catalysis outside their active site, via diffusible reactive oxygen species [12]. However, despite its multifunctionality, there is no doubt that ROS are a major driver of genome instability. The first line of defense for key mitochondrial components, including mtDNA, consists of various ROS scavengers and antioxidants. Superoxide dismutases, catalase, thioredoxin, and glutathione peroxidase systems have their own mitochondrial isoforms [13, 14]. The nuclear factor E2-related factor 2/antioxidant response element (Nrf2/ARE) signaling pathway is a crucial regulator of the expression of key ROS generators and ROS scavengers [15]. Dysregulation of Nrf2 has been shown to lead to various mitochondrial dysfunctions, including impaired mtDNA function [16, 17, 18, 19].
Existing reviews often overlook the fact that Nrf2 also participates in the second line of defense for mtDNA. This defense includes enzymes responsible for repairing damaged bases and nucleotides, as well as the repair of single-strand and double-strand breaks. Although there are some similarities to mechanisms involved in nuclear DNA repair, mtDNA possesses unique error correction mechanisms. In this review, we focus on how the Nrf2/ARE signaling pathway is directly involved in mtDNA repair and how it interacts with p53 and BRCA1, which play a critical role in the regulation of the cell cycle, apoptosis, and DNA repair.
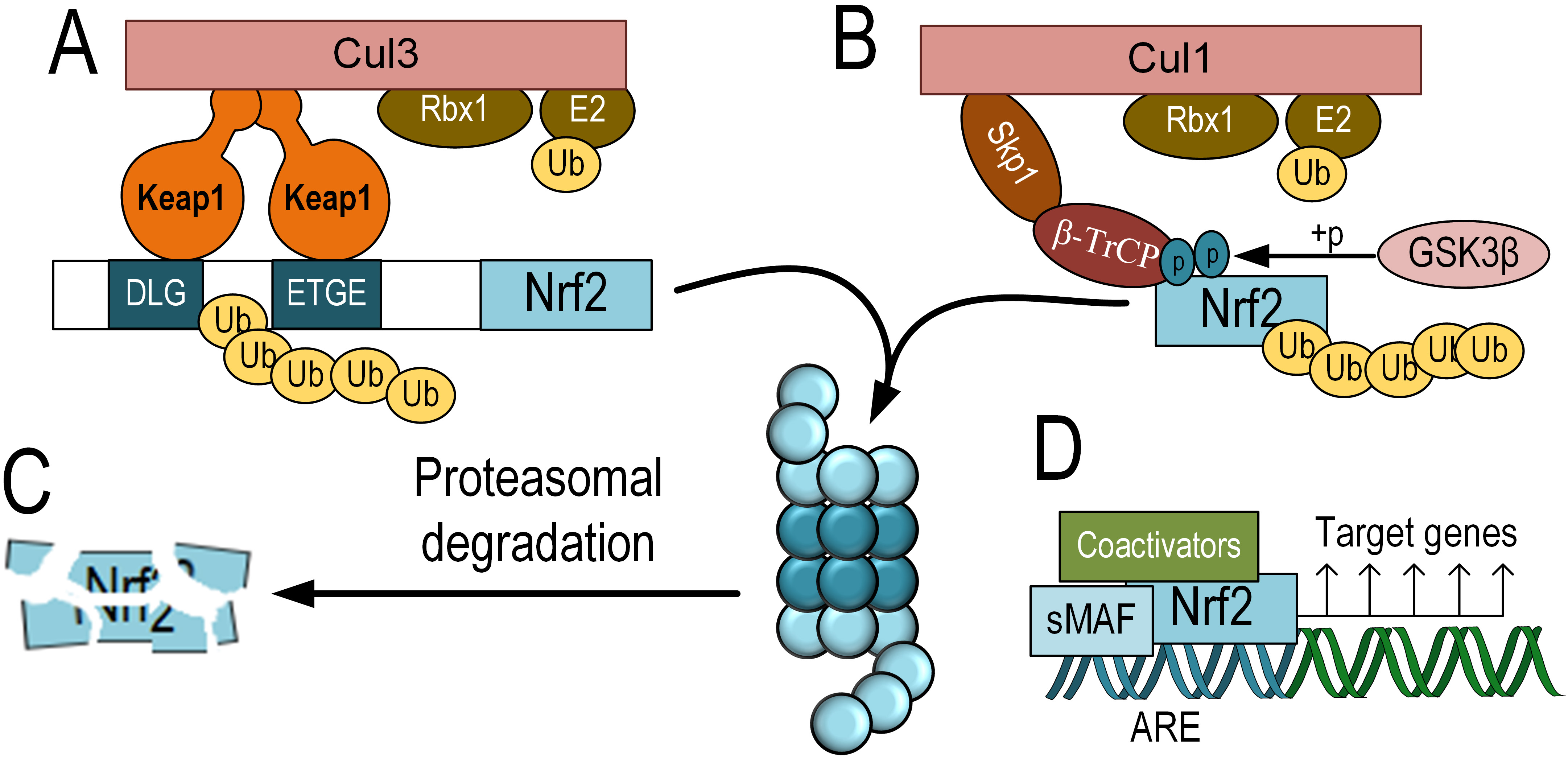
Nrf2 belongs to the cap-’n’-collar (CNC) subfamily of transcription factors,
which are part of the basic leucine zipper (bZIP) family. Nrf2 is a short-lived
protein with an average half-life of about 15 minutes, undergoing ubiquitination
and subsequent proteasomal degradation in the absence of activating factors [20].
The main negative regulator of Nrf2, responsible for its degradation, is
Kelch-like ECH-associated protein 1 (Keap1). Keap1 functions as an adaptor
protein that mediates the interaction of Nrf2 with the E3 ubiquitin ligase
complex Cul3 (Cullin 3) and RING-box protein 1 (Rbx1) for subsequent proteasomal
degradation [21, 22] (Fig. 1A,C). Post-translational modifications,
particularly phosphorylation, play a crucial role in regulating the activity of
Nrf2. Glycogen synthase kinase-3 beta (GSK-3
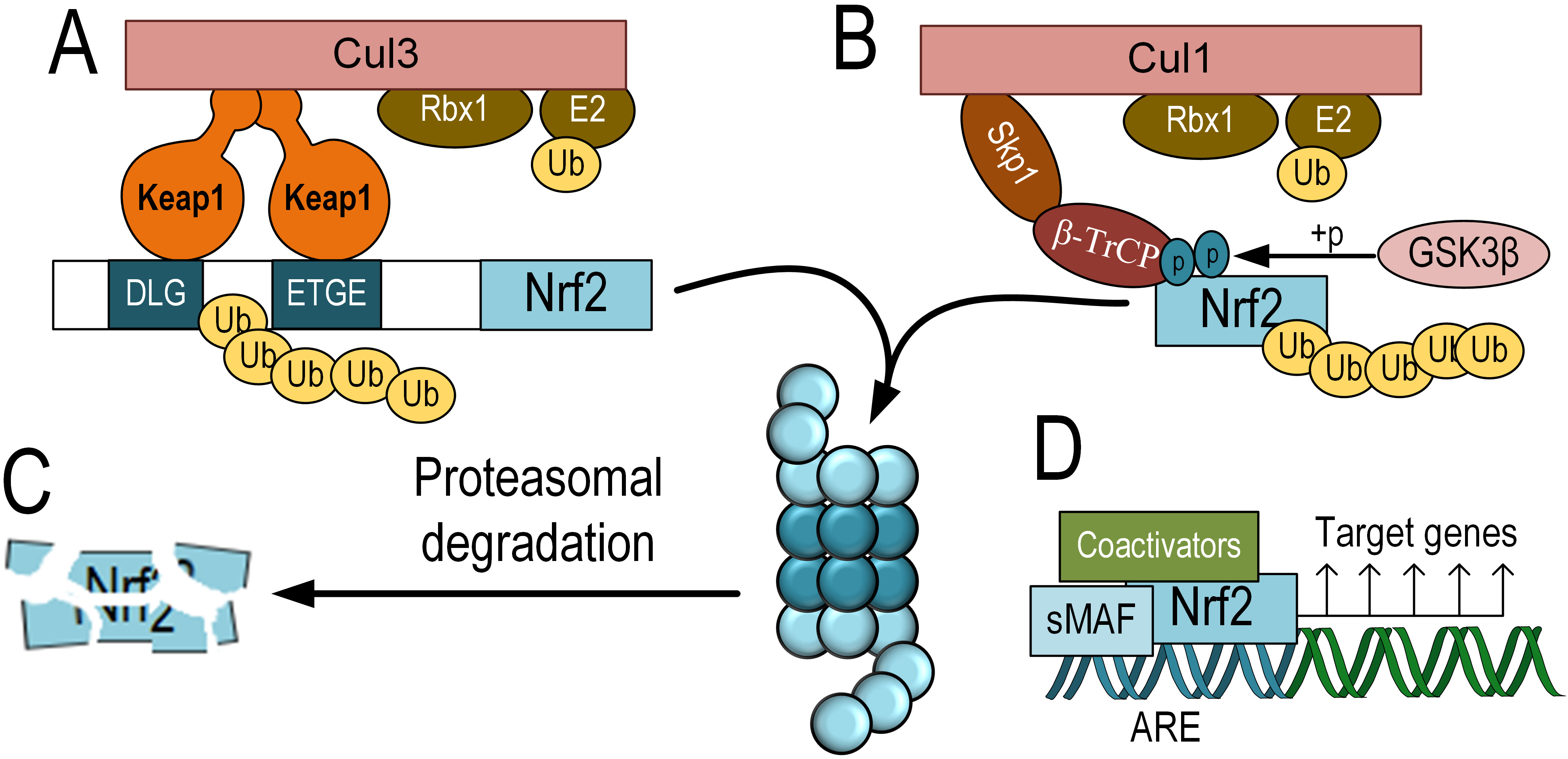 Fig. 1.
Fig. 1.The scheme of KEAP1-dependent and KEAP1-independent degradation
of Nrf2 and its activation. KEAP1-dependent degradation of Nrf2, which involves
binding to Keap1, functioning as an adapter protein for the E3 ubiquitin ligase
complex Cul3 and Rbx1 (A). KEAP1-independent degradation of Nrf2 is mediated by
GSK-3
However, in the presence of elevated levels of oxidative or electrophilic
compounds, Keap1 or GSK-3
Activated Nrf2 translocates into the nucleus. Similar to other bZIP
domain-containing transcription factors, Nrf2 functions as a dimer. However, Nrf2
cannot directly bind to DNA as a homodimer and requires an obligatory coactivator
from the sMaf family. These sMaf proteins are bZIP domain-containing
transcription factors but lack a transactivation domain. There are three members
of the sMaf family that can associate with Nrf2 to initiate transcription: MafF,
MafG, and MafK [25]. Nrf2 binds to ARE – promoter elements that were discovered
during the study of the antioxidant properties of polyphenolic compounds. The
regulatory cis-activating element, ARE, within the promoter regions of genes, is
characterized by the nucleotide sequence 5
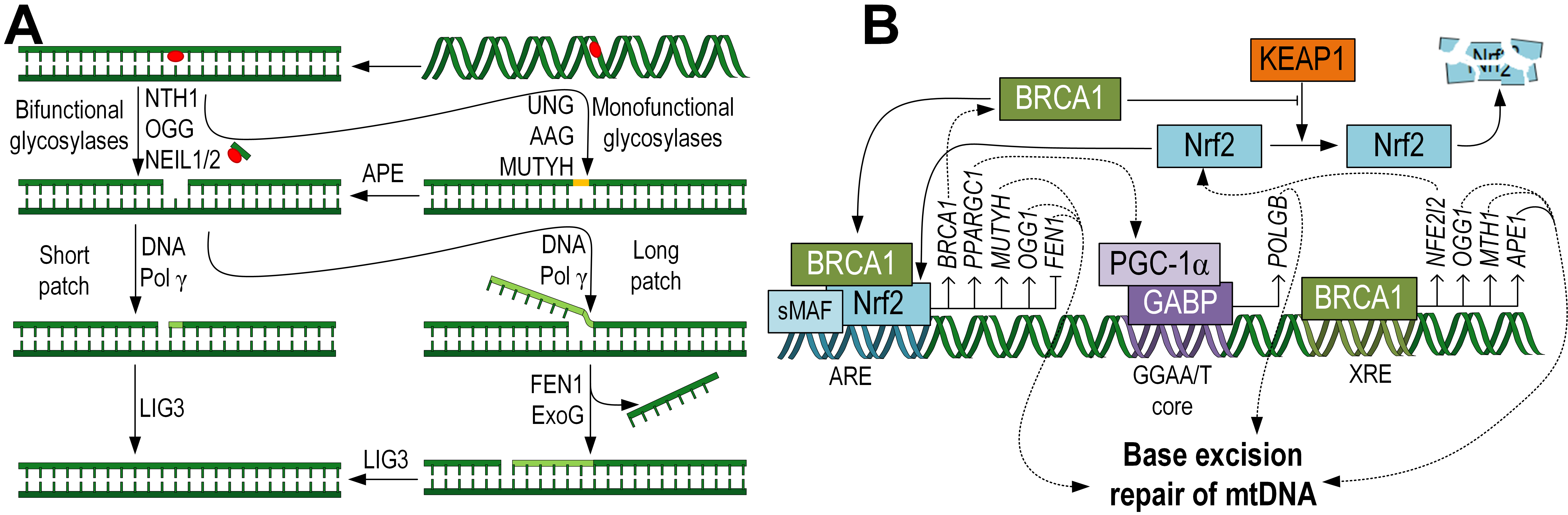
The base excision repair (BER) is a typical mechanism for mtDNA repairing [27]. The initial step of the repair process is carried out by DNA glycosylases. There are two main functional classes of glycosylases: monofunctional and bifunctional. Monofunctional glycosylases only possess glycosylase activity, while bifunctional glycosylases also have apurinic/apyrimidinic (AP) site lyase activity, which allows them to cleave the phosphodiester bond of DNA, creating a single-strand break without the need for an AP endonuclease [28] (Fig. 2A).
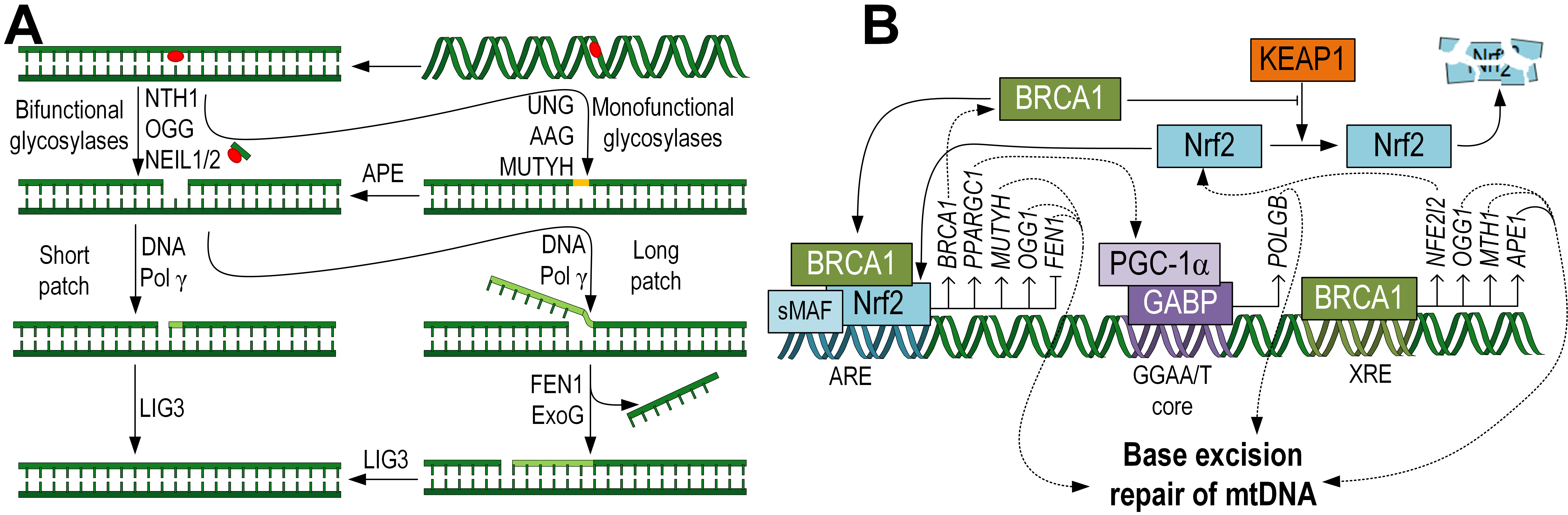 Fig. 2.
Fig. 2.Schematic representation of the BER pathway, specific to the
repair of mtDNA, taking into account the mitochondrial localization of the
enzymes. First, glycosylases excise damaged bases, then DNA Pol
Monofunctional glycosylases are predominantly dual-localized, both in mitochondria and the nucleus. For example, the UNG gene, through alternative splicing, encodes two proteins, UNG1 (mitochondrial form) and UNG2 (nuclear form). This glycosylase removes uracil, which arises due the guanine deamination [29]. Additionally, the alkyladenine DNA glycosylase (AAG) or N-methylpurine DNA glycosylase (MPG), which removes various alkylated bases, is localized in mitochondria [30]. MutY DNA glycosylase (MUTYH) removes adenine, which is often incorporated erroneously opposite 8-oxoG. This enzyme is also dual-localized, both in the nucleus and mitochondria [31]. With western blot analysis showing predominant mitochondrial MUTYH localization [32]. Correlation analysis has demonstrated the influence of Nrf2 on MUTYH expression and the presence of ARE sites in the gene promoter [33] (Fig. 2B). It is worth noting that not all nuclear glycosylases have been detected in mitochondria. For example, thymine DNA glycosylase (TDG), single-strand selective monofunctional uracil DNA glycosylase (SMUG1), and methyl-CpG-binding domain protein 4 (MBD4), which removes thymine from TG pairs at methyl-CpG sites, have not been found in mitochondria [34].
Since monofunctional glycosylases can only excise the damaged base and lack lyase activity, the subsequent hydrolysis of the phosphate backbone is carried out by apurinic/apyrimidinic endodeoxyribonuclease 1 (APE1) [35]. APE1 is found in mitochondria, and the transport of this protein across mitochondrial membranes has been extensively studied [36]. Interestingly, APE1 is a multifunctional enzyme that also interacts with various transcription factors, including Nrf2. It has been shown that APE1 negatively regulates the activity of Nrf2, while APE1 knockdown increases the expression of Nrf2 downstream targets. APE1 and Nrf2 coexist in protein complexes, particularly at defined promoter sites where Nrf2 exerts its transcriptional activity [37].
Bifunctional glycosylases are capable of completely removing both the damaged base and the phosphate backbone from the DNA. Among them, OGG1 is noteworthy as it removes 8-oxoG, the most common type of mtDNA damage arising from oxidative stress or exposure to drugs and xenobiotics [38, 39]. OGG1 contains an N-terminal mitochondrial targeting signal (MTS) that facilitates its transport into mitochondria [40]. Studies have shown that mice overexpressing the Ogg1 exhibited reduced levels of mtDNA damage in lung fibrosis models [41]. Conversely, Ogg1 knockout in neural stem cells resulted in an increased rate of mtDNA damage accumulation [42]. The OGG1 gene contains an ARE, and a positive correlation between Nrf2 and OGG1 expression has been demonstrated [33] (Fig. 2B). Among the bifunctional glycosylases, NTH-1, NEIL1, and NEIL2 are also localized in mitochondria, unlike NEIL3 [40] (Fig. 2A).
The next step is the insertion of the required nucleotide. In nuclear DNA, this
process is carried out by DNA Pol
DNA pol
In the nuclear long-patch BER sub-pathway, DNA pol
BRCA1 plays a role in various cellular pathways involved in maintaining genomic
stability. These pathways include activation of cell cycle checkpoints in
response to DNA damage, repair of DNA damage, ubiquitination of proteins,
remodeling of chromatin structure, transcriptional regulation, and apoptosis
[54]. Coene and colleagues [55] demonstrated that phosphorylated BRCA1 is
predominantly located in the nucleus and mitochondria, indicating its importance
in maintaining mitochondrial genome stability. BRCA1 deficiency leads to
dysfunctional mitochondrial dynamics [56], which further highlights the
importance of BRCA1 in maintaining mitochondrial quality control. It is
well-established that BRCA1 is involved in the regulation of BER. BRCA1 promotes
the expression of glycosylases OGG1, NTH-1, and APE1 [57]. Additionally, BRCA1 is involved in the transcriptional regulation of the
XRCC1 gene [58] (Fig. 2B), which is crucial for the final step of DNA
nick ligation. Similarly, it has been shown that BRCA1 can regulate the
expression of the gene encoding DNA pol
There is a bidirectional regulatory system between Nrf2 and BRCA1. It has been shown that over-expression of Nrf2 stimulates BRCA1 expression. Nrf2 also interacts with CBP and p300 to form an active transcription complex, which can bind to the ARE on the BRCA1 gene promoter and activate its transcription by inducing histone acetylation [60]. In turn, BRCA1 can mechanically interact with Nrf2 through its BRCA1 C-terminal (BRCT) domain and promote its nuclear translocation [61]. It has been shown that BRCA1 affects Keap1-mediated Nrf2 ubiquitination activity, thereby controlling Nrf2 stability and activation [62]. BRCA1 can also regulate Nrf2 activity at the transcriptional level. It has been shown that BRCA1 binds to the xenobiotic response element (XRE) in the Nfe2l2 gene promoter and promotes its expression [61] (Fig. 2B). Therefore, BRCA1 and Nrf2 form a positive feedback loop. This suggests that Nrf2 may mediate BER for mtDNA through a BRCA1-dependent mechanism.
p53 is widely recognized as the guardian of the genome and one of the most well-known proteins that prevents oncogenesis. One way in which p53 suppresses tumorigenesis is by participating in the repair of nuclear DNA. However, evidence is emerging that suggests p53 also plays a role in maintaining the mitochondrial genome [63]. This is achieved through its translocation into mitochondria and interactions with mtDNA repair proteins. Since mtDNA is situated on the mitochondrial matrix, the regulation of mtDNA repair by p53 might occur only once it has translocated to the matrix.
The question of the mechanisms of p53 transport into mitochondria is debated. There is evidence showing that N-terminal cleavage by a specific endoprotease exposes a cryptic mitochondrial localization signal (MTS), which is necessary for interaction with translocase of the outer membrane 20 (TOM20) for further transport into mitochondria [64]. Similarly, it has been demonstrated that RecQ helicase-like protein 4 (RECQL4) is involved in p53 transport into mitochondria. RECQL4 is a nuclear DNA helicase with an N-terminal MTS that binds to TOM20. It has been proposed that the formation of the RECQL4/p53 complex masks the nuclear localization signals (NLS) motif of p53, enabling its specific translocation to mitochondria instead of the nucleus [65]. The third pathway of translocation is redox-dependent. The typical substrate for this system possesses cysteine-rich motifs capable of forming two intramolecular disulfide bonds which have previously been reported to be present in p53 (Cys-135/141 and Cys-275/277). The import receptor (Mia40 in yeasts and CHCHD4 proteins in mammals) covalently binds to its substrate through intermolecular disulfide bonds upon its translocation across the TOM complex. Mia40/CHCHD4 is maintained in an oxidized state by coupling with Erv1/GFER, which transfers electrons from the imported substrate to molecular oxygen via cytochrome c [63].
It is well known that p53 is involved in the BER regulation. p53 regulates the
expression of glycosylases, such as MUTYH [66], and OGG1 [67].
There are studies that show a crucial role of p53 in mtDNA repair through a DNA
pol
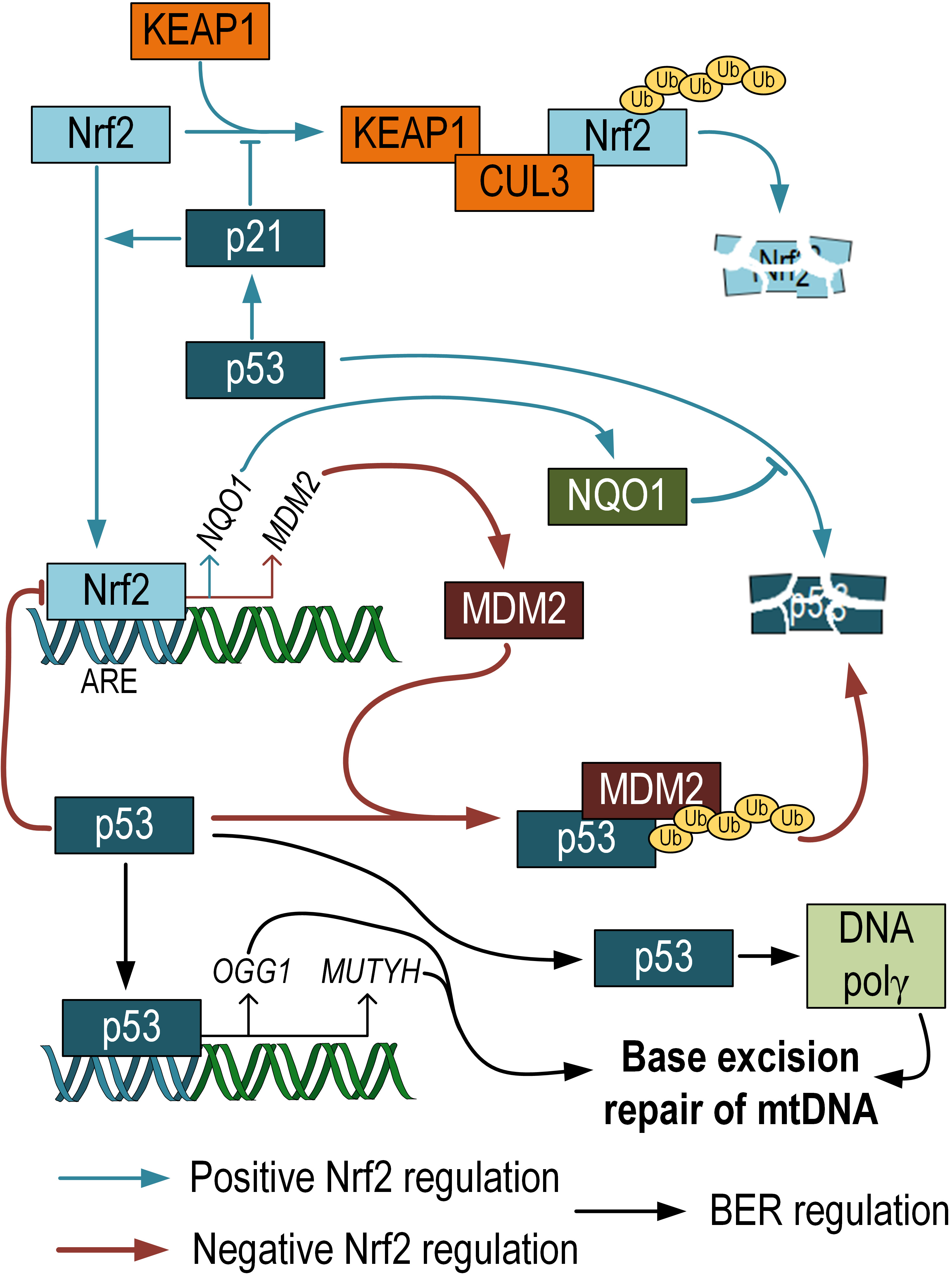
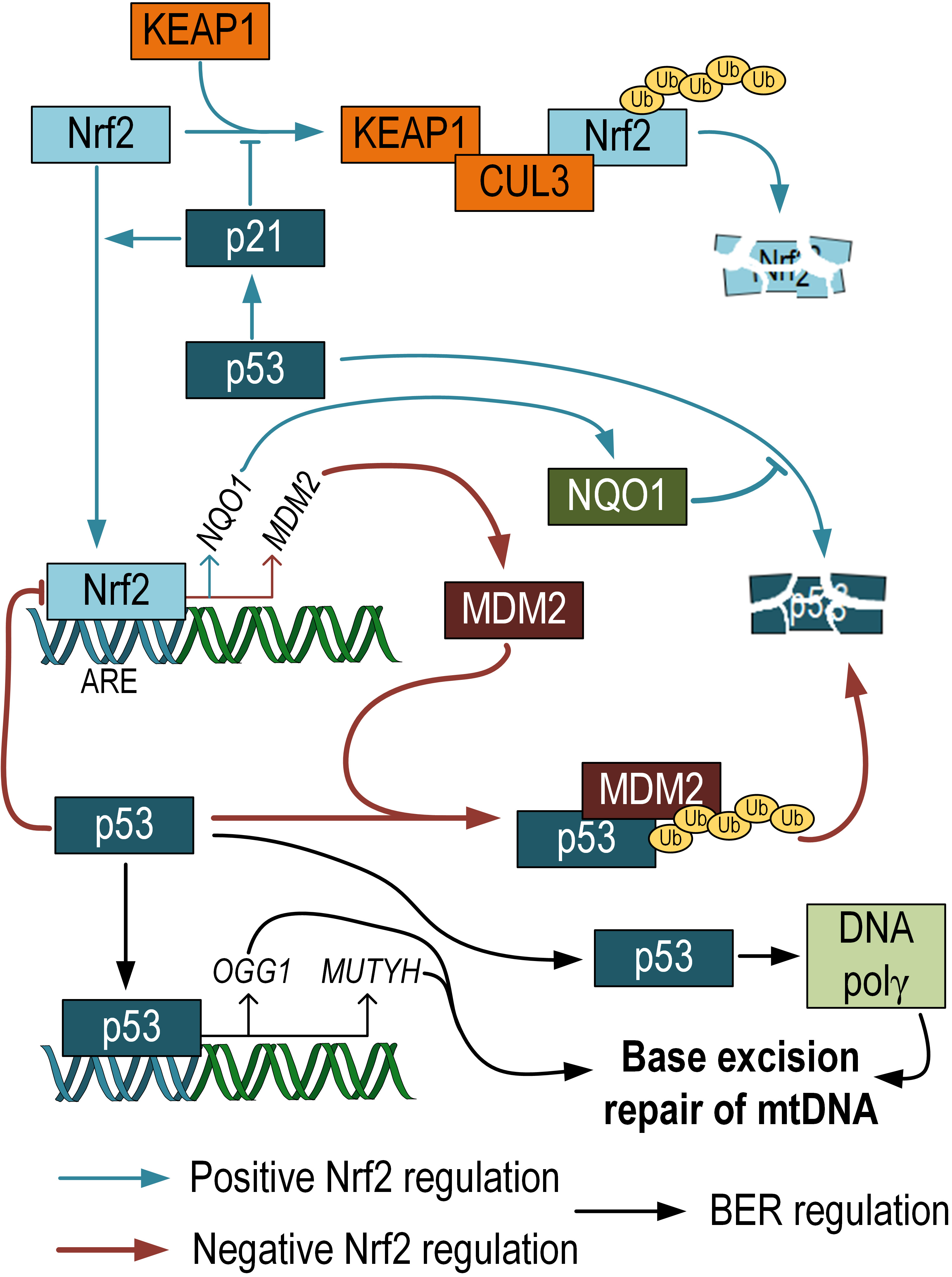 Fig. 3.
Fig. 3.Negative and positive regulation of p53 activity by Nrf2. p53 can prevent proteasomal degradation of Nrf2. Nrf2 positively regulates the expression of NQO1, which prevents p53 degradation. At the same time, Nrf2 also positively regulates the expression of MDM, which, on the contrary, promotes p53 degradation. Additionally, it has been shown that p53 participates in the regulation of mtDNA repair via the BER pathway. Nrf2 and p53 crosstalk scheme adopted from [69]. NQO1, NADH quinone oxidoreductase 1; MDM, mouse double minute.
The crosstalk between Nrf2 and p53 is a highly debated topic. Both transcription factors are known to have similar functions in protecting cells from oxidative stress. It has been shown that p21, a target of p53, prevents the ubiquitination and degradation of Nrf2. The KRR motif in p21 directly interacts with the DLG and ETGE motifs in Nrf2, competing with Keap1 for Nrf2 binding [70]. NADH quinone oxidoreductase 1 (NQO1), an Nrf2 target gene, interacts with p53 and prevents its degradation [71]. These findings suggest the presence of a positive feedback loop between Nrf2 and p53. On the other hand, there are also studies showing that the mouse double minute 2 homolog (MDM2), a repressor of p53, has an ARE and is positively regulated by Nrf2. Inhibition of Nrf2 can suppress Mdm2 expression, which may result in p53 signaling modulation [72]. Additionally, it has been demonstrated that p53 binds to ARE and acts as a transcriptional repressor of Nrf2 target genes [73]. These differences are likely influenced by the different phases in cell life. Under normal conditions, Nrf2 promotes cell survival and protects against oxidative stress, while in cancer cells, Nrf2 provides resistance to apoptosis and therapy [74]. Chen et al. (2012) [75] propose two phases of interaction between Nrf2 and p53. The first phase is induction: when p53 expression is relatively low, it enhances Nrf2 activity and its target genes to promote cell survival. The second phase is repression: when p53 expression is high, the Nrf2-mediated survival response is inhibited by p53. This fine-tuning in the regulation between p53 and Nrf2 allows for strict coordination of cell survival and death pathways based on the needs of the organism (Fig. 3). However, it is currently unknown to what extent Nrf2-dependent alterations in p53 expression affect the level of mtDNA damage. This question requires further research.
The other pathways of DNA repair, such as nucleotide excision repair (NER),
mismatch repair (MMR), non-homologous end joining (NHEJ), and homologous
recombination (HR), which are characteristic of nuclear DNA, are much less
prominent in mtDNA repair. In nuclear DNA, NER is a mechanism for repairing
single-strand breaks and is the only mechanism for removing thymine dimers [76].
Among all the NER proteins, only сockayne syndrome A and B proteins
(CSA and CSB) have been found in mitochondria. MtDNA mutations are highly
increased in cells of aged Csb
MMR is a pathway that detects and corrects errors in the insertion, deletion, and mis-incorporation of bases that can occur during DNA replication and recombination. MutS homolog 2 (MSH2), the main protein involved in MMR, has not been found in mitochondria, although the removal of mismatched bases from mtDNA has been demonstrated [78]. Additionally, MSH1, MSH3, and MSH6 have not been detected in mitochondria [27]. The only protein from the MMR pathway that has been found in mitochondria is the Y Box binding protein 1 (YB-1). YB-1 participates in the recognition of damaged DNA with multiple mechanisms and acts as a scaffold in forming a repair complex at the damaged site. YB-1 depletion in cells increases mitochondrial DNA mutagenesis [79]. Additionally, a relationship between YB-1 and Nrf2 has been shown [80]. Interestingly, YB-1 was found to enhance the NFE2L2 mRNA translation, leading to an increase in the levels of Nrf2 protein [81]. Binding to DNA, YB-1 carries out its functions as a transcription factor controlling the expression of genes involved in cell cycle progression and stress response [80].
Double-strand breaks (DSBs) are the most severe DNA structural damage. Nuclear DNA has two major repair mechanisms: homologous recombination (HR), which is the most accurate repair mechanism involving the readout of a new DNA sequence from a homologous chromosome, and non-homologous end joining (NHEJ), which is a less accurate process. NHEJ is a rapid process, taking approximately 30 minutes to complete, while HR is much slower and can take 7 hours or longer [82]. Due to the haploid nature of mtDNA, there has been no evidence to suggest the possibility of HR occurring in mitochondria. No proteins of the MRN complex, which are essential for initiating HR, have been found in mitochondria [27]. However, there is evidence that Rad51 is present in mitochondria, and its inhibition can lead to increased levels of ROS and abnormal mitochondrial distribution [83]. It has been previously shown that Nrf2 regulates the expression of Rad51 [84]. However, if Rad51 is involved in any way in mtDNA repair, it is likely a secondary role, and it is more likely involved in mtDNA replication and maintenance under stress conditions [85]. Classical NHEJ process is practically absent in mtDNA; however, it appears that the main mechanism for repairing DSBs is microhomology-mediated end joining (MMEJ). Proteins such as CtIP (RBBP8), FEN1, MRE11, and PARP1 have been implicated in mtDNA repair, and DNA ligase III, but not ligase IV or ligase I, is primarily responsible for the final sealing of DSBs [86]. Although it has been previously shown that Nrf2 negatively regulates the expression of FEN1 [50], there is information showing that Nrf2 positively regulates the expression of the ATM kinase [87]. This kinase has mitochondrial localization [88] but does not directly participate in mtDNA repair. However, it phosphorylates MRE11, suggesting its involvement in the repair of mtDNA DSBs [89].
Mutations can arise not only from damage to the existing bases incorporated
into DNA but also from contamination of the nucleotide precursor pool with
non-canonical nucleotides. Such contaminants in the precursor pool primarily
include deoxy- and ribonucleoside triphosphates of inosine (ITP/dITP), xanthine
(XTP/dXTP), and 8-oxoguanine [90] (Fig. 4). Inositol monophosphate (IMP) is used
for the synthesis of adenosine monophosphate (AMP). This synthesis occurs through
two reactions in which aspartate serves as the donor of the NH
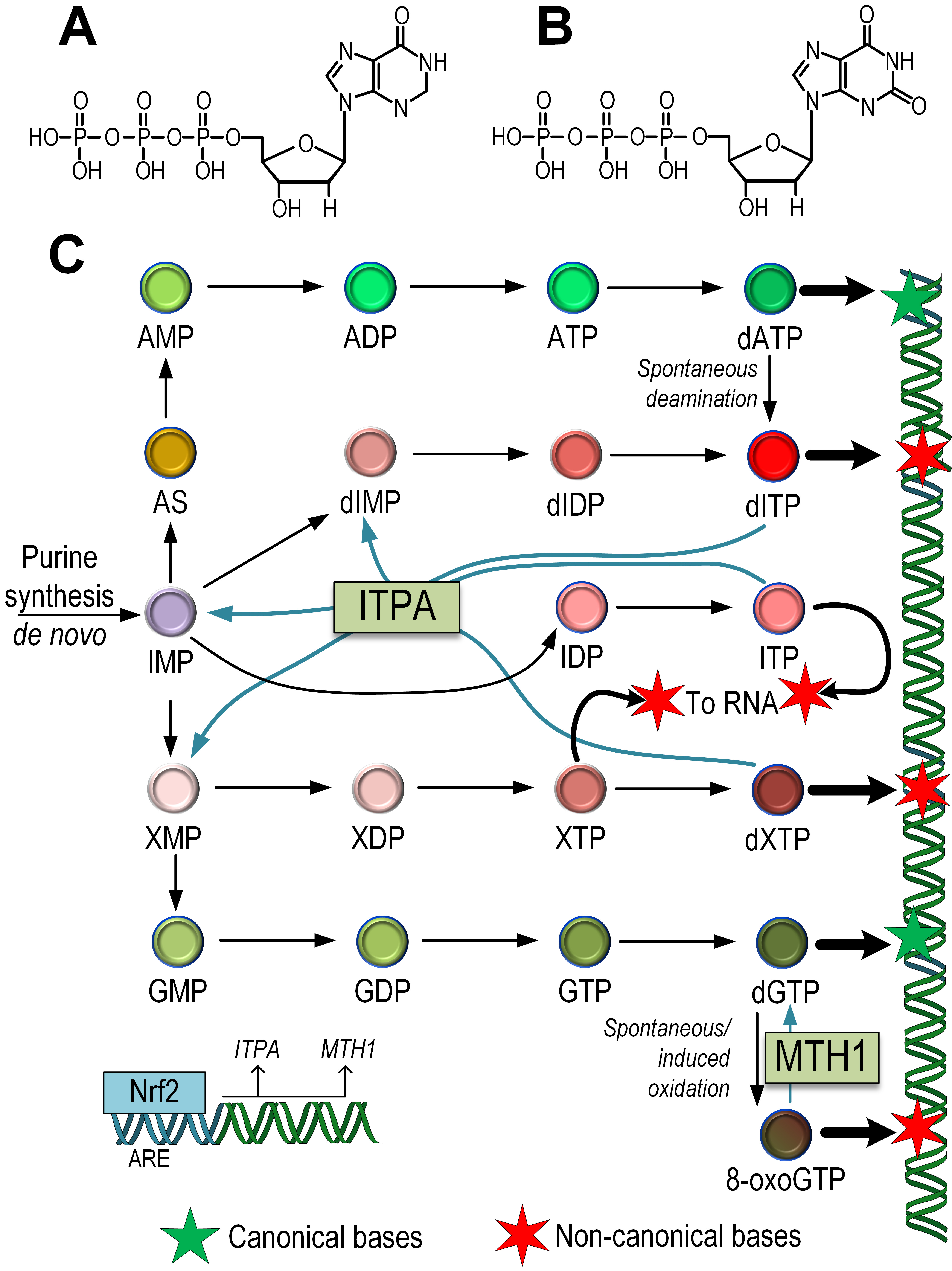
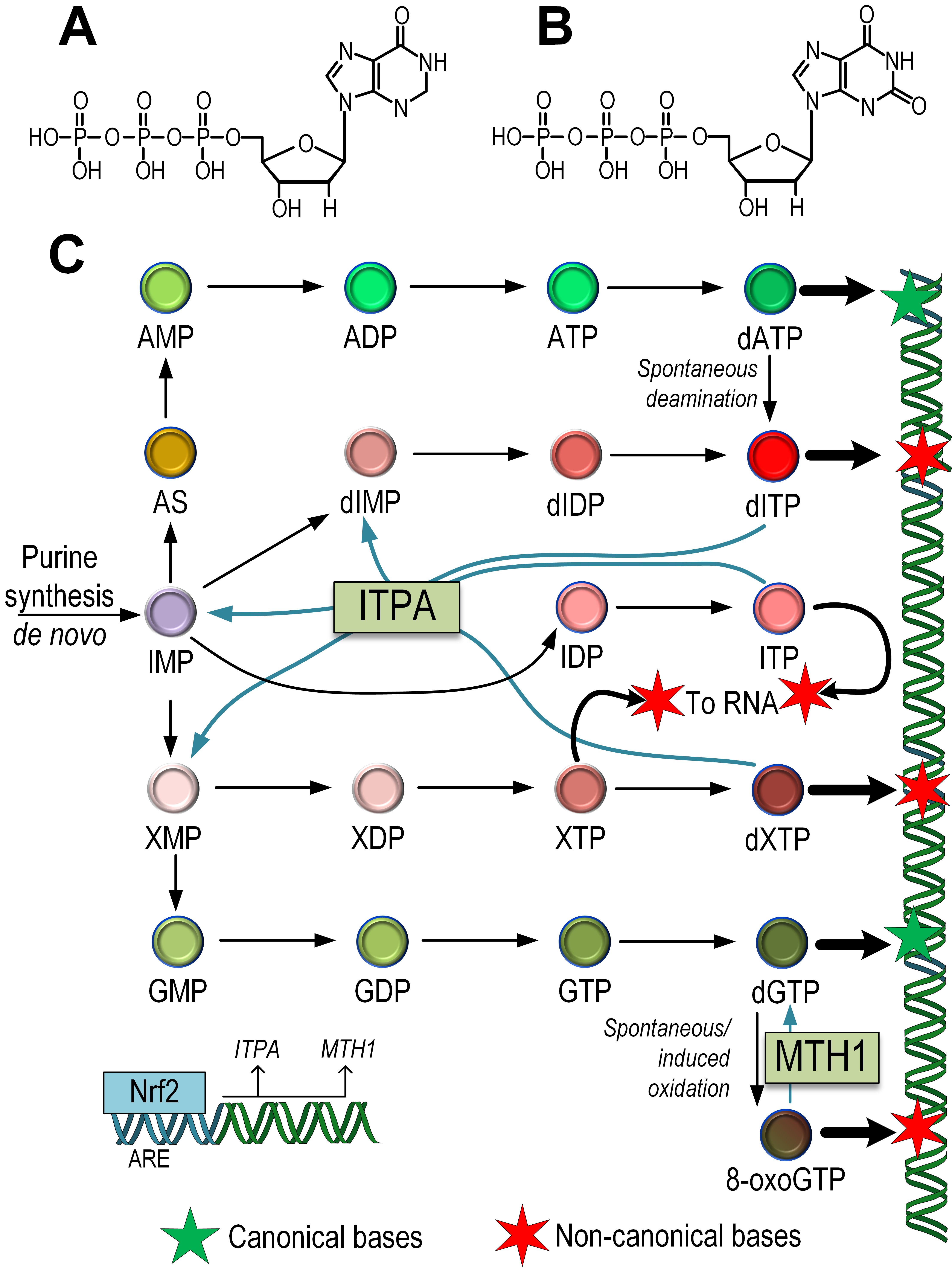 Fig. 4.
Fig. 4.The influence of the Nrf2/ARE pathway on the repair of non-canonical nucleotides that can potentially be integrated into mtDNA. Structural formulas of noncanonical dITP (A), dXTP (B). Scheme of synthesis of dITP, dXTP, and 8-oxoguanine and its incorporation into mtDNA or mRNA. The role of ITPA in the repair of non-canonical nucleotides dITP, dXTP, and ITP, as well as the role of MTH1 in converting 8-oxoGTP into dGTP in the nucleotide pool, is indicated. The role of Nrf2 in the regulation of the expression of the ITPA and MTH1 genes has been demonstrated (C). AMP/ADP/ATP, Adenosine mono-, di-, triphosphate; IMP/IDP/ITP, inositol mono-,di-, triphosphate; dIMP/dIDP/dITP, deoxyinositol mono-, di-, triphosphate; XMP/XDP/XTP, xanthosine mono-, di-, triphosphate; GMP/GDP/GTP, guanidine mono-, di-, triphosphate; dATP, dXTP, dGTP, deoxy-adenosine, -xanthosine, -guanidine triphosphate; AS, adenylosuccinate; 8-oxoGTP, 8-oxoguanine triphosphate.
The synthesis of guanosine monophosphate (GMP) also occurs in a two-step process. First, IMP is oxidized to xanthosine monophosphate (XMP). Then, guanosine monophosphate synthetase (GMPS) uses glutamine as the cellular donor of the NH2 group. The synthesis of GTP occurs in two stages by transferring high-energy phosphate groups from ATP. However, the intermediate product xanthosine monophosphate (XMP) can also undergo the addition of phosphate groups, forming xanthosine triphosphate (XTP) and deoxyxanthosine triphosphate (dXTP), which are aberrant nucleotides capable of being incorporated into DNA [92] (Fig. 4).
ITPase acts by degrading endogenous dITP and dXTP [93]. Mutations in the ITPA gene have been associated with a significant increase in total heteroplasmic/homoplasmic mtDNA mutations compared to wild-type ITPA [94]. This suggests that the “contamination” of the nucleotide pool in the mitochondrial matrix with noncanonical nucleotides contributes to an increased number of mtDNA mutations. Data also indicate that the expression level of Nrf2 correlates with ITPA expression [33] (Fig. 4).
Guanines can be oxidized not only in the DNA chain but also in the dNTP pool [95]. MTH1 (NUDT1) acts to remove 8-oxodGTP from the nucleotide pool, thereby preventing its incorporation into DNA [96]. It has been shown that the expression of Nrf2 correlates with MTH1 gene expression [33]. Alternative splicing generates two forms of the MTH1 protein, with MTH1 localized to mitochondria and MTH2 localized to the nucleus [31] (Fig. 4). Thus, Nrf2 plays a key role in maintaining not only the integrity of mtDNA but also the dNTP pool, which is important for maintaining the stability of mtDNA structure.
Thus, in this review, we have demonstrated that the role of the Nrf2/ARE signaling pathway extends beyond the regulation of antioxidant defense. Nrf2 is involved in the regulation of gene expression involved in BER, the main pathway for mtDNA repair. However, it is important to note that there are other mechanisms of repair in mtDNA for more severe damage, such as double-strand breaks, which are less explored. As mitochondrial dysfunctions, particularly damage and mutations in mtDNA, play a crucial role in the development of various metabolic disorders, a deeper understanding of possible mtDNA repair mechanisms will enable the development of new therapeutic concepts for the treatment of mitochondrial-related diseases. The Nrf2/ARE pathway represents a promising target for both natural and synthetic drugs. Therefore, in further research, it may be valuable to consider not only the antioxidant activity of Nrf2 activators but also their ability to activate repair processes.
APG, EVC, EPK, ISS, EET and AAD jointly conceptualized, searched available literature, wrote the original draft and edited the final draft. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Not applicable.
Not applicable.
The study was supported by the Russian science foundation (grant #22-74-00115 to A.P.G.)
The authors declare no conflict of interest. Given the role of the guest editor, Anna A. Dorohova did not participate in the review of this article and does not have access to information related to its review. Full responsibility for the editorial process for this article was delegated to Gianluca Paventi and Graham Pawelec.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.