
 , Shanshan Luo 1,*, Yu Hu 1,*
, Shanshan Luo 1,*, Yu Hu 1,*1 Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430022 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
The treatment options for multiple myeloma (MM) have undergone significant transformation with the advent of immunotherapy. Novel therapies that focus on tumor antigens now drive advances in MM research. Bispecific antibodies (bsAbs) leverage revolutionary advances in bioengineering techniques and embody the second generation of antibody-based tumor therapy. Recent studies on bsAbs in relapsed/refractory MM cases have revealed remarkable efficacy and acceptable safety profiles. The approval of elranatamab and teclistamab represents the next step in the development of bsAbs for the treatment of MM. This review article addresses the antigen targeting, efficacy, safety, and strategies in the application of bsAbs against treatment-resistant MM, with a focus on clinical trials and real-world data.
Keywords
- bispecific antibodies
- multiple myeloma
- immunotherapy
- tumor antigens
- resistance mechanism
Multiple myeloma (MM) is the second most common hematologic malignancy in the United States, with an estimated 35,730 new cases and an estimated 12,590 deaths in 2023 [1]. The introduction of proteasome inhibitors, immunomodulatory drugs, and autologous stem cell transplant (ASCT) over the past decade has markedly improved the prognosis of MM, resulting in a median survival of 7 to 10 years [2, 3, 4, 5]. Nevertheless, the majority of patients who initially respond eventually progress to a resistant state.
MM employs various strategies to reduce the host immune responses during disease progression, thereby enabling immune escape and uncontrolled cell proliferation. Consequently, immunotherapies, such as monoclonal antibodies (mAbs) and chimeric antigen receptor T-cell (CAR-T) therapies, have emerged as critical new pivotal approaches by reactivating the host immune system to eliminate tumor cells [6, 7, 8]. The dynamic landscape of antibody engineering has sparked interest in the development of bispecific antibodies (bsAbs). BsAbs concurrently target tumor-associated antigens (TAAs) and also engage effector cells through surface-associated molecules, thus triggering potent killing effects and sparking a surge in interest. In contrast to CAR-T therapy, bsAbs are readily scalable “off-the-shelf” products. In 2022, teclistamab received rapid approval from the U.S. Food and Drug Administration, becoming the first bsAb targeting CD3 and BCMA in MM, which has invigorated the field. This review article provides an in-depth examination of the advancements in bsAbs within the context of MM, with an emphasis on clinical studies and on potential strategies for enhancing their antitumor efficacy.
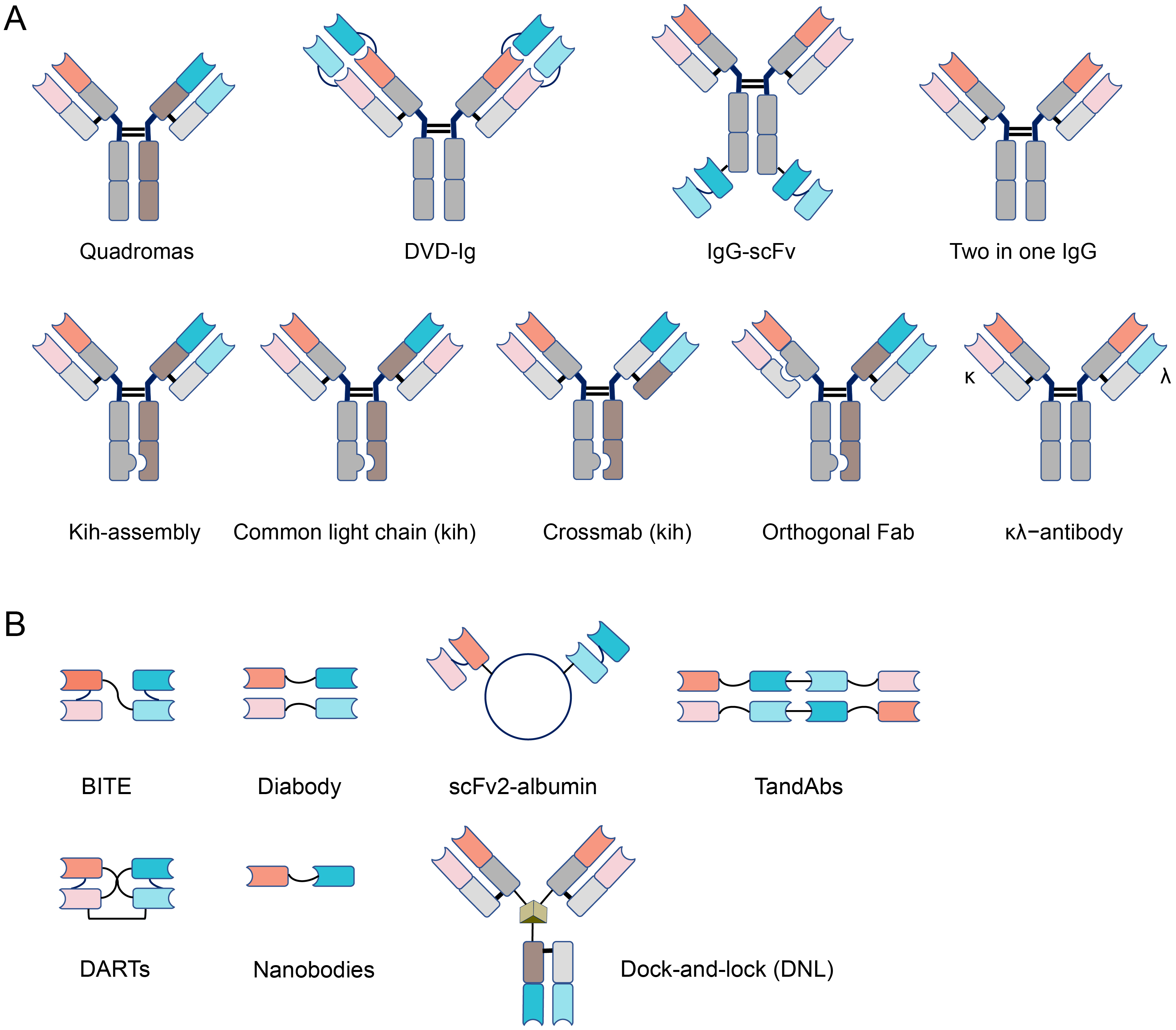
BsAbs are broadly categorized into two main classes, characterized by the presence (IgG-like) or absence (non-IgG-like) of Fc domains [9, 10]. IgG-like bsAbs are larger in size and therefore have a longer serum half-life [11]. Non-IgG-like bsAbs can readily penetrate tissues but must be continuously infused due to their shorter cycle kinetics [9, 12]. Brinkman and Konterman [13, 14] have comprehensively reviewed the ‘Zoo of BsAbs’, which comprises more than 100 distinct bsAb formats. Here we highlight the backbones of IgG-like and non-IgG-like bsAbs in Fig. 1 Ref. [9]. Apart from inhibiting multiple signaling pathways and mediating the formation of protein complexes, the major function of bsAbs is to recruit immunocompetent cells to the tumor environment and induce tumor lysis [15, 16, 17, 18, 19]. T cells play a crucial role in the anti-tumor action of immunotherapy [18]. In MM, the majority of bsAbs focus on T cell engineering through the CD3 receptor. There is also growing interest in the use of alternative constructs to engage with natural killer (NK) cells [20, 21].
 Fig. 1.
Fig. 1.Different formats of bispecific antibodies. (A) IgG-like
structure. IgG-like bispecific antibodies (bsAbs) mainly include Quadromas,
DVD-Ig, IgG-scFv, Two in one IgG, Knobs-into-holes (Kih-assembly, Common light
chain, Crossmab), Orthogonal Fab,
BsAbs generally have two binding sites, with one arm binding to the tumor-associated antigen (TAA) and the
other arm simultaneously binding to CD3 molecules expressed on host T cells [22].
BsAbs mediate cell-cell adhesion forces, thereby promoting the formation of more
stable conjugates between target cells and effector cells, leading to a 3-fold
increase in contact time to fully activate the T cell response [23]. The binding
of bsAbs to target cells and effector cells can induce the formation of an immune
synapse. This process is accompanied by the redistribution of signaling molecules
and secretion granules within the cells, ultimately leading to the release of
perforin, granzyme and cytokines including interleukin (IL)-2, IL-6 and
interferon (IFN)-
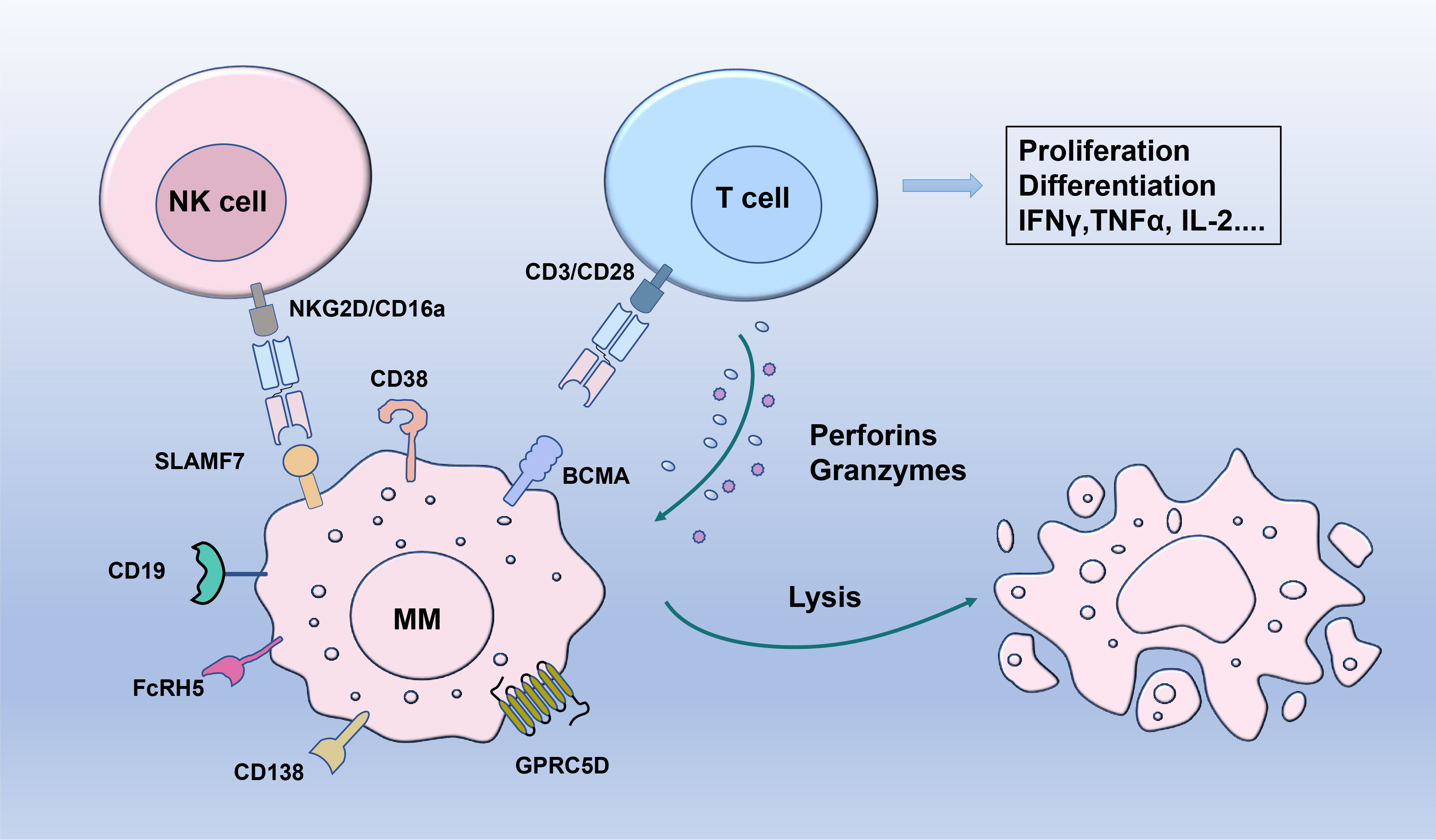
Achieving optimal effectiveness and high specificity hinges on the selection of the appropriate TAA when designing bsAbs. Ideally, the chosen TAA should meet these criteria: (a) Predominant expression on malignant cells, intricately linked to the malignant phenotype; (b) Essential role in tumor biology and/or pathophysiology; (c) Minimal presence in normal healthy cells [28]. This review highlights promising TAA candidates on the surface of MM cells (Fig. 2) that have attracted widespread attention.
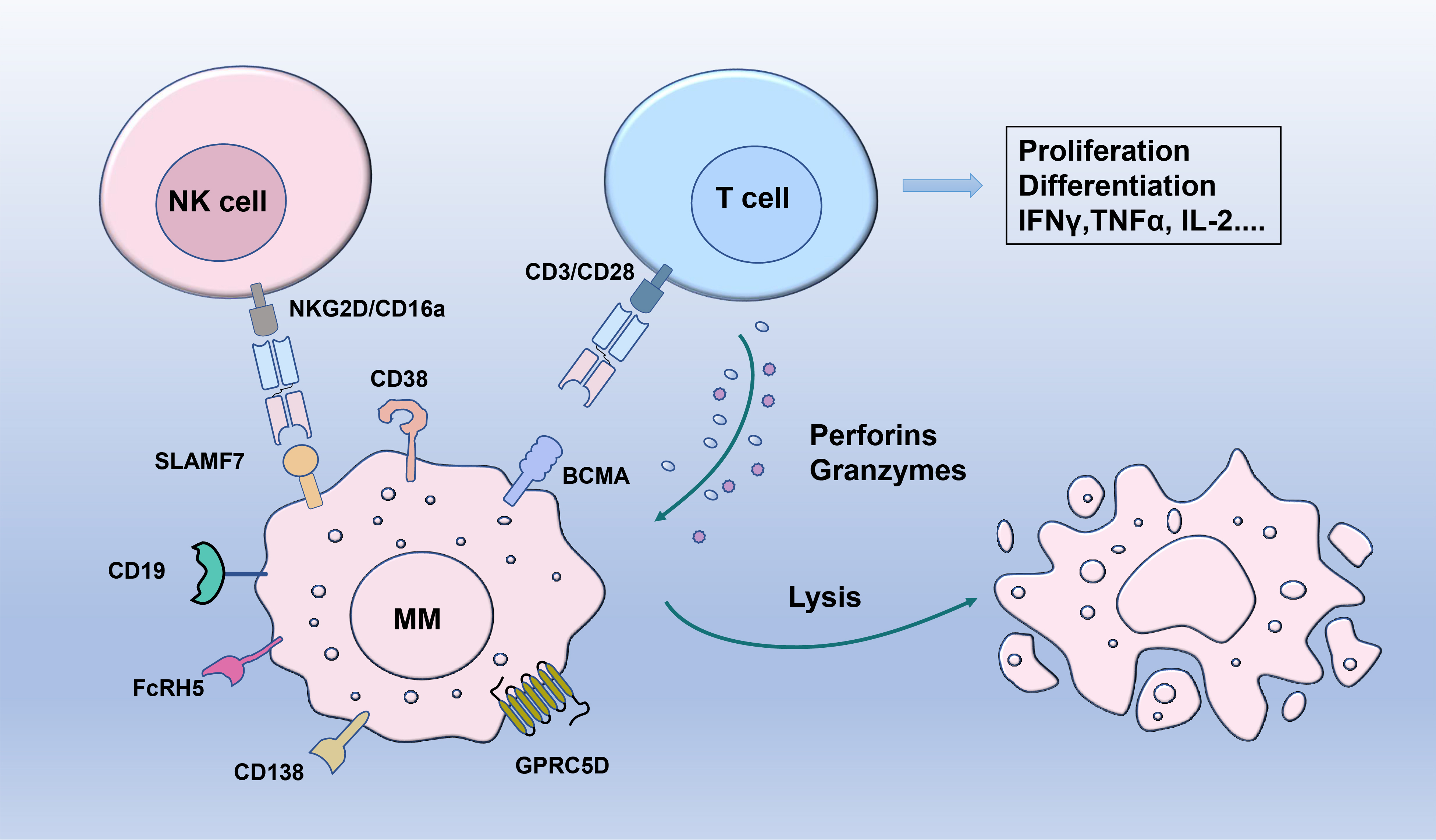 Fig. 2.
Fig. 2.Targeting mechanisms of bispecific antibodies and promising
markers in multiple myeloma. Bispecific antibodies simultaneously engage myeloma
cells through specific tumor antigen (BCMA, GPRC5D, FcRH5, SLAMF7, CD38, CD138,
CD19) and T cells or natural killer (NK) cells via CD3, CD16a or NKG2D and
further promote direct cell-mediated cytotoxicity. IFN
BCMA is a transmembrane glycoprotein belonging to the tumor necrosis factor
receptor superfamily 17. It is selectively expressed on the surfaces of plasma
blasts and differentiated plasma cells. BCMA interacts with two agonist ligands,
a proliferation-inducing ligand and B cell activating factor. This interaction
activates p38/NF-
Elranatamab is a humanized IgG bsAb that
binds to BCMA
Teclistamab is a humanized IgG bsAb that targets BCMA and CD3 [40]. The
MajesTEC-1 trial (NCT04557098) found that teclistamab exhibits promising clinical
activity, with an ORR of 63% and a CR or better achieved in 39.4% of patients.
The most common AEs were CRS (72%), neutropenia (71%), and anemia (55%) [41, 42]. Teclistamab has been approved for patients with RRMM who have received
Alnuctamab is an asymmetric, two-arm, humanized IgG trivalent bsAb that binds
bivalently to BCMA and monovalently to CD3 [49, 50, 51].
A Phase 1 trial (NCT03486067) found that RRMM patients who
received intravenous (IV) alnuctamab had a median response duration of 33.6
months. However, due to the occurrence of CRS in 76% of patients, the study
shifted to SC treatment. As of April 3, 2023, 73 patients undergoing SC
alnuctamab treatment exhibited an ORR of 54% across all doses, with deepening
responses over time. The ORR reached 63% at the target dose of
ABBV-383 is a fully human IgG4 bsAb generated using knob-in-hole technology. ABBV-383 exhibited promising activity in a Phase 1 study (NCT03933735) [54]. An updated study reported the results of the dose expansion study for IV ABBV-383. ABBV-383 demonstrated deep and enduring responses at doses of 40 mg and 60 mg, with a median PFS of 13.7 months and 11.2 months, respectively, and 12-month duration of response (DOR) rates of 70% and 66%, respectively. At doses of 20 mg, 40 mg, and 60 mg, the incidence rates of CRS and of Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) were 50%/3%, 71%/5%, and 70%/5%, respectively. Further clinical evaluation of ABBV-383 is ongoing [55].
Linvoseltamab is a bsAb displays comparable antitumor efficacy to anti-BCMA
CAR-T cells [56]. Preliminary results from the LINKER-MM1 (NCT03761108) study
indicated that treatment with linvoseltamab elicited deep and enduring responses
in patients, with a low incidence of CRS [57]. The updated data
reported the efficacy in the cohort using 200 mg linvoseltamab, with ORR and CR
rates of 70% and 29% for patients with
RO7297089 is a bsAb targeting BCMA and CD16a [59]. The Phase 1 study’s (NCT04434469) findings showed that administering RO7297089 through IV at weekly doses ranging from 60 to 1850 mg in RRMM patients maintained an acceptable safety profile. However, the observed single-agent efficacy of RO7297089 is less robust than that reported with BCMA bsAbs [60]. This possibly results from RO7297089’s extended half-life, which might gradually dampen NK cell function over time, causing a state of hypo-responsiveness and limiting response to repeated infusions [61]. Optimizing dosage or exploring combination strategies might be necessary to advance its clinical development. Combining RO7297089 with agents that enhance NK cell activity presents an intriguing avenue to potentially enhance responses achieved with RO7297089 alone [62].
WVT078 is a potent anti-BCMA
F182112 is an anti-BCMA
GPRC5D is characterized as a C7-type transmembrane receptor and shows highly specific expression in plasma cells [66, 67]. Its close proximity to the plasma membrane enhances the immune synapse between T cells and target cells, fostering more potent cytotoxicity. Given its 7-pass transmembrane nature, GPRC5D is unlikely to shed into the serum. This contrasts with other surface antigens such as BCMA, where shedding can lead to sedimentation effects and reduced efficacy [68]. The mRNA expression of GPRC5D correlates with plasma cell burden and genetic variations, such as chromosome 13q14 deletion and translocation t(4;14) [66]. The exclusive presence of GPRC5D on MM cells suggests it may be a potential target for bsAb therapy [69, 70].
Talquetamab (JNJ-64407564) is a first-in-class bsAb that combines CD3 and
GPRC5D. The activity of talquetamab in H929 cells is not affected by BCMA loss,
suggesting that patients who are resistant to BCMA therapy or have low/absent
BCMA expression may be treated with talquetamab [71]. The Phase 1/2 MonumenTAL-1
study (NCT03399799/NCT04634552) reported an ORR of over 71% in 288 patients who
had not received prior T cell redirecting therapy (TCR) treatment. Among 143
patients treated with 0.4 mg/kg weekly, the ORR was 73%. The most common AEs at
doses of 0.4 mg/kg or 0.8 mg/kg were CRS (79%/72%), dysgeusia (48%/46%), and
anemia (45%/39%) [72]. Of the 135 patients who discontinued talquetamab in
MonumenTAL-1, various treatment modalities including TCR were also found to be
effective, with 25.0% of patients achieving CR or better after CAR-T therapy
[73]. In 70 patients who were previously treated with TCR therapies including
anti-BCMA-CAR-T, anti-BCMA bsAb, or BCMA-ADC, talquetamab demonstrated robust
efficacy, with an ORR of up to 73%. More than 50% of patients previously
exposed to bsAbs showed a response. These results indicate that talquetamab
provides durable responses for TCR/BCMA-naive and TCR/BCMA-exposed patients with
RRMM [74]. In addition to monotherapy, researchers also investigated the efficacy
of talquetamab combined with IMiDs and the anti-CD38 monoclonal antibody
daratumumab. Talquetamab in combination with pomalidomide (MonumenTAL-2,
NCT05050097) at doses of 0.4 mg/kg weekly or 0.8 mg/kg every two weeks
demonstrated ORRs of 86.7% and 83.3%, respectively. The most common AEs were
dysgeusia (77.1%), CRS (74.3%; mostly grade 1/2, 2.9% grade
Another promising anti-GPRC5D
Fc receptor-homolog 5 (FcRH5) is a type I membrane protein belonging to an immunoglobulin superfamily with six genes [80, 81, 82]. FcRH5 is expressed exclusively on the surface of B cells and plasma cells, with an almost 100% prevalence on MM cells [26]. FcRH5 is expressed consistently throughout MM progression [83]. Gain of chromosome 1q21 is one of the most frequent genetic anomalies in MM and is prognostic for invasive disease [84]. FcRH5 was initially identified in clones from this chromosomal region and was found to be deregulated in cell lines exhibiting 1q21 abnormalities [85]. Primary samples from MM patients with 1q21 gain show elevated FcRH5 gene expression, suggesting that targeting FcRH5 may yield clinical benefit for this subgroup [26].
With regard to bsAbs targeting FcRH5, the large size of the FcRH5 extracellular
region could lead to an increased distance between the antigen epitope and the
target membrane, thus potentially hindering the effective formation of T cell
synapses [86]. Li et al. [26] developed a humanized IgG-based bsAb,
cevostamab, that targets the most membrane-proximal domain of FcRH5 on MM cells
and CD3. In a Phase 1 trial (GO39775, NCT03275103), cevostamab showed meaningful
activity and a favorable safety profile when administered Q3W for up to 17
cycles. Patients achieved durable responses (
CD38 is a transmembrane glycoprotein characterized by high surface density and consistent expression on MM cells. CD38 expression is relatively low in normal myeloid and lymphocytes [89, 90]. The clinical success of monoclonal antibodies targeting CD38 has stimulated the corresponding development of bsAbs. [91]. VP301 is a novel bsAb that targets CD38 and intercellular adhesion molecule 1. In vitro and in vivo experiments have shown promising activity of VP301 against primary MM samples. VP301 also exhibits synergistic anti-tumor growth activity when used in combination with the immunomodulatory drug lenalidomide [92]. ISB 1342 is a bsAb that targets CD3 and CD38. In preclinical studies, ISB 1342 demonstrated greater tumor eradication compared to daratumumab [93, 94]. A Phase 1 study (NCT03309111) evaluated 39 subjects receiving IV ISB 1342 and 7 subjects receiving SC ISB 1342. T cell activation was observed following administration of ISB 1342 at doses of 1.0/4.0 mg/kg and higher for 24–48 hours. Moreover, 89% of subjects experienced TRAEs, mostly grades 1–2, including infusion-related reactions (37%), CRS (34%), anemia (24%), neutropenia (24%), and thrombocytopenia (17%) [95]. ISB 1442 is a fully humanized bsAb that targets CD38 and CD47. Preliminary results of a Phase 1/2 study (NCT05427812) in RRMM patients reported on 10 subjects who received weekly SC ISB 1442. ISB 1442 demonstrated good tolerability at the evaluated dose levels. The observed clinical CRS events were moderate and were potentially associated with macrophage activation following ISB 1442 administration [96].
SLAMF7, or CS1/CD319, is a cell surface glycoprotein expressed exclusively on
plasma cells, NK cells and a subset of activated T cells [97, 98]. SLAMF7 is a
vital regulator of normal immune cell function and activates NK cells, promotes
the growth of normal B cells, and inhibits T cell development [99]. Notably,
SLAMF7 can drive tumor progression by facilitating the adhesion of MM cells to
bone marrow stromal cells (BMSCs) within the microenvironment, thus making it a
promising target for MM therapy [98, 100]. Chan engineered a
non-IgG–like, scFv-based bsAb that targets SLAMF7 and NKG2D and can activate all
NKG2-expressing immune cells, including NK cells, CD8
CD138 is a single-pass, type I membrane protein from the syndecan proteoglycan family. It is widely expressed in epithelial cells and plays a role in the proliferation, adhesion, and migration of diverse types of malignant tumors [102]. The notable expression of CD138 on MM cells and its favorable influence on the disease phenotype make it a compelling target for targeted therapy [103, 104]. von Strandmann et al. [21] developed the bsAb ULBP2-BB4, which fuses an anti-CD138 scFv with ULBP2, a ligand for NKG2D receptors on NK cells. ULBP2-BB4 enhanced the NK-mediated cleavage of MM cells and suppressed tumor growth in nude mouse models [21]. STL001 features nanomolar affinity for recombinant CD138 protein and exhibits robust anti-MM activity, both in vitro and in vivo [105]. However, the widespread expression of CD138 on epithelial cells in addition to MM cells presents a challenge. Phase 1 clinical trials of anti-CD138 antibody conjugates have reported related AEs, including hand-foot syndrome, stomatitis, and blurred vision [106]. Additionally, the shedding of sCD138 and its accumulation in the BM may reduce the efficacy of bsAbs that target tumor cells by neutralizing their effects. At present, there are no ongoing clinical trials involving CD138 bsAbs.
CD19 is expressed on B cells during their development, but diminishes during
plasma cell differentiation [107, 108]. The majority of malignant plasma cells
lack CD19 expression, with only around 10% belonging to the CD19
Overall, bsAbs show promising efficacy results with generally manageable AEs. Moreover, the ORR for a monotherapy used in such heavily pretreated patients is impressive, with most trials having a median of 6 prior lines. Studies on teclistamab and talquetamab showed favorable ORR of 63% [42] and 73% [114], respectively. The DOR is evolving in most studies, as many subjects are still alive and responding. In evaluable patients, MRD negativity has ranged from 46% to 100% [42, 52]. With increased clinical testing, the safety data for bsAbs are becoming more comprehensive. While the overall safety of bsAbs is manageable, some side effects require significant attention. Infection and hematologic-related AEs are relatively common, with the latter sometimes causing infections that represent a significant clinical burden. The efficacy and safety of monotherapy trials for some promising bsAbs are summarized in Table 1. MM patients typically experience secondary immune deficiencies such as hypogammaglobulinemia (HGG), with low serum IgG levels and an increased risk of bacterial infections [115]. Studies have found that treatment with bsAbs can prolong the course of HGG and thus increase the risk of infection [116]. Targeting BCMA often leads to profound B-cell depletion. For example, in patients receiving BCMA-targeted therapy, antibody responses to COVID-19 vaccines are particularly weak [117]. Therefore, serious infections in patients who receive bsAbs targeting BCMA should be considered as potentially treatment-related. In MajesTEC-1, 19 patients (11%) died from infections, with 5 of these attributed to teclistamab [42]. The incidence of treatment-related infections in the ABBV-383 trial was 41%, with 20% being grade 3 infections (pneumonia, COVID-19, sepsis, urinary tract infection). However, 7 deaths from COVID-19 and one death from sepsis were considered to be unrelated to ABBV-383 [118].
| Agents | Patient Characteristics | Response Rates | TRAEs |
Elranatumab MagnetisMM-1 NCT03269136 BCMA×CD3 |
(1) RRMM (n = 55), SC monotherapy at dose levels |
(1) ORR: 63.6%, VGPR or better: 56.4%, CR or better: 38.2%; | (1) Hematologic: Neutropenia (74.5%), Anemia (67.3%), Lymphopenia (52.7%), Thrombocytopenia (50.9%); |
| (2) Median age: 64.0 years old; | (2) mPFS: 11.8 months, mOS: 21.2 months, mDOR: 17.1 months, | (2) Non-hematologic: CRS (87.3%, G1–G2), Injection site reaction (56.4%), Fatigue (41.8%), Diarrhea (40%), Dry skin (36.4%), Hypophosphatemia (36.4%), Decreased appetite (34.5%), Nausea (34.5%) | |
| (3) Median lines of prior regimens: five; | (3) 13 patients with confirmed CR or better, all MRD negative | ||
| (4) 90.9% were triple-class refractory, 29.1% had high cytogenetic risk, 23.6% had received prior BCMA-directed therapy | |||
Elranatumab MagnetisMM-3 NCT04649359 BCMA×CD3 |
(1) RRMM (n = 123), SC elranatamab; | (1) ORR: 61%, VGPR or better: 56.1%, CR or better: 35%; | (1) Hematologic: Neutropenia (48.8%), Anemia (48.8%), Thrombocytopenia (30.9%), Lymphopenia (26.8%), Leukopenia (17.6%); |
| (2) Median age: 64.0 years old; | (2) mPFS and mDOR were not reached, estimate of mPFS and mDOR at 15 months was 50.9% and 56.7%, | (2) Non-hematologic: CRS (57.7%, no G3–G4), Diarrhea (42.3%), Fatigue (36.6%), Decreased appetite (33.3%), Pyrexia (30.1%), Covid-19 (29.3%), Injection site reaction (26.8%), Nausea (26.8%), Hypokalemia (26%), Cough (25.2%), Headache (23.6%) | |
| (3) Median lines of prior regimens: five; | (3) MRD negativity was 89.7% in 29 patients with confirmed CR | ||
| (4) No prior BCMA-directed therapy, 96.7% were triple-class refractory, 25.2% had high cytogenetic risk | |||
Teclistamab MajesTEC-1 NCT03145181 NCT04557098 BCMA×CD3 |
(1) RRMM (n = 165), QW SC teclistamab at a dose of 1.5 mg/kg; | (1) ORR: 63%, VGPR or better: 58.8%, CR or better: 39.4%; | (1) Hematologic: Neutropenia (70.9%), Anemia (52.1%), Thrombocytopenia (40%), Lymphopenia (34.5%), Leukopenia (17.6%); |
| (2) Median age: 64 years old; | (2) mPFS: 11.3 months, mOS: 18.3 months, mDOR: 18.4 months, | (2) Non-hematologic: CRS (72.1%, 0.6% in G3–G4), Neurotoxic event (14.5%), Fatigue (27.9%), Diarrhea (28.5%), Nausea (27.3%) Injection site erythema (26.1%), Pyrexia (27.3%), Headache (23.6%), Arthralgia (21.8%), Constipation (20.6%) Cough (20%), Pneumonia (18.2%), Covid-19 (17.6%), Bone pain (17.6%), Back pain (16.4%) | |
| (3) Median lines of prior regimens: five; | (3) MRD negativity was 46% in 65 patients with confirmed CR | ||
| (4) 77.6% were triple-class refractory, 25.7% had high cytogenetic risk | |||
Talquetamab MonumenTAL-1 NCT03399799 GPRC5D×CD3 |
(1) RRMM (n = 30, at 405 µg QW) or (n = 44, at 800 µg Q2W); | dosage of 405/800 | dosage of 405/800 |
| (2) Median age, 64.0 years old; | (1) ORR: 70%/64%, VGPR or better: 57%/52%; | (1) Hematologic: Anemia (60%/43%), Neutropenia (67%/36%), Lymphopenia (40%/39%), Thrombocytopenia (37%/23%), Leukopenia (40%/18%); | |
| (3) Median lines of prior regimens: six; | (2) mPFS: 7.5/11.9 months, mOS: 76.4%/77.4% at 12 months, mDOR: 9.5 months/NR | (2) Non-hematologic: CRS (77%/80%, only one patient in G3–G4 at 405 dosage), Skin-related event (67%/70%), Dysgeusia (63%/57%), Fatigue (33%/27%), Nail-related event (57%/27%), Pyrexia (33%/18%), Headache (20%/25%), Rash-related event (47%/30%), Diarrhea (30%/16%), Cough (20%/11%), Dry mouth (30%/57%), Nausea (30%/16%), Arthralgia (23%/9%), Decreased weight (30%/32%), Increased alanine aminotransferase (20%/30%), Increased aspartate aminotransferase (10%/34%), Back pain (10%/20%), Hypophosphatemia (27%/18%), Dysphagia (37%/27%), Decreased appetite (20%/20%), Constipation (7%/14%), Increased | |
| (4) 99% were triple-class refractory, 16% had high cytogenetic risk | |||
Cevostamab NCT03275103 FcRH5×CD3 |
(1) RRMM (n = 160); | (1) ORR: 54.5%/36.7% (160 mg/90 mg); | (1) Hematologic: Neutropenia (18.1%), Anemia (31.9%); |
| (2) Median age, 64.0 years old; | (2) mDOR: 15.6 months, | (2) Non-hematologic: CRS (80%, 1.3% in G3–G4), Infections (42.5%), Neurological/Psychiatric (40.6%), Diarrhea (26.3%), Cough (23.1%), Nausea (21.9%), Infusion-related reaction (17.5%), Fatigue (16.3%), Increased aspartate aminotransferase (15.6%), Hypomagnesaemia (15.6%), Pyrexia (15.6%), Increased alanine aminotransferas (15%) | |
| (3) Median lines of prior regimens: six; | |||
| (4) 85% were triple-class refractory |
TRAEs, treatment-related adverse events; RRMM, relapsed or refractory multiple myeloma; SC, subcutaneous; ORR, overall response rate; VGPR, very good partial response; CR, complete remission; mPFS, median progression-free survival; mOS, median overall survival; mDOR, median duration of response; MRD, minimal residual disease; NR, not reached; QW, weekly; Q2W, every 2 weeks; CRS, cytokine releasing syndrome.
Current recommendations regarding monotherapy and combination therapy with bsAbs are based primarily on data from studies of teclistamab and elranatamab. BsAbs that target different antigens may have varying infection rates and risks, depending on the dosage, dosing interval, and patient characteristics [119]. Compared to GPRC5D-bsAb monotherapy, BCMA-bsAb monotherapy or GPRC5D-bsAb combination therapy were found to have a higher cumulative incidence of infections, and were associated with more grade 3 infections [120]. In the small phase I/II TRIMM-2 study, the incidence of infections with teclistamab or talquetamab in combination with daratumumab was similar to that of monotherapy [121, 122]. However, in the MajesTEC-2 trial in which teclistamab was combined with daratumumab and lenalidomide, 90.6% of patients experienced any grade infection, and 37.5% experienced grade 3/4 infections [123]. Expert consensus has suggested similarities in infection monitoring and prevention between bsAb and CAR-T cell therapy [124]. However, it should be noted that bsAbs carry their own individual infection risk [119]. The mechanism of cytopenia is unclear, with one hypothesis postulating a bystander effect of cytokine release and marrow plasma cell destruction [125]. CRS is a key toxicity reported for all bsAbs, with an overall incidence as high as 87% [126]. Almost all grade 3 CRS cases were confined to 3% [42, 114, 127]. Moreover, CRS associated with alnuctamab treatment can result in fatal outcomes [51]. CRS manifests primarily after the initial injection and is self-limiting [57]. It necessitates only supportive care with antipyretics and IV fluids. In some intensive cases, CRS can be controlled by dexamethasone and tocilizumab [42, 52, 128]. The major risk factors for CRS are tumor load and the initial dose of bsAbs [129]. Hypoxia and abnormal liver function were also found to be drivers of grade 3 CRS [130], and the use of an incremental dosing regimen can reduce the risk of severe CRS [131]. Following the use of teclistamab, patients with a history of TCR therapy had a significantly lower incidence of CRS than those with no prior TCR therapy, possibly due to T-cell exhaustion [132]. With elranatamab, the presence of extramedullary disease and prophylactic use of tocilizumab were associated with a reduced incidence of CRS [133]. ICANS is also a common side effect, and has been documented with the use of elranatamab [36], teclistamab [42], linvoseltamab [57], talquetamab [134] and cevostamab [127]. Some cases with ICANS were observed concurrently with CRS, but subsided once the CRS had resolved. In RRMM patients receiving teclistamab, the prophylactic use of tocilizumab before dose escalation reduced the incidence and severity of CRS and decreased the occurrence of ICANS [135, 136]. Serious AEs such as fatigue [36, 42, 57], pyrexia [42], back pain [42, 59], diarrhea [36] and hypokalemia [36] were also reported. Additionally, because some cells in the oral mucosa express GPRC5D, patients treated with talquetamab may experience taste changes, thereby affecting their quality of life and nutritional status [137]. Researchers found that patients who received GPRC5D bsAbs experienced significant taste impairment, leading to reduced appetite, weight loss, and dry mouth [138]. Despite these toxicities, the health-related quality of life of RRMM patients treated with bsAbs was found to improve [139]. As bsAbs become more widely used in the clinic, their efficacy and safety profiles will be better defined in the coming years.
Cancer is a heterogeneous disease that employs diverse evasion strategies over time to adapt to its environment. The potent selective pressure exerted by bsAbs can drive myeloma blasts to adopt specific and extreme escape strategies. Plausible mechanisms for this include antigen loss, an immunosuppressive tumor microenvironment (TME), and the absence of costimulatory domains [22]. The short cycle kinetics of bsAbs could be perceived as a limitation. Researchers are exploring genetic approaches for the in-situ generation of bsAbs that circumvent the need for continuous drug infusion and partly counter the immunosuppressive TME. This section explores the resistance mechanisms to bsAbs, as well as strategies that could enhance their efficacy.
Tumor antigens play a pivotal role as distinctive markers that serve as the primary target for various immunotherapies. The selection of a suitable target antigen is a pivotal step in ensuring the efficacy and precision of immunotherapy. However, to evade immune attack by bsAbs, tumor cells often manipulate their surface properties by reducing antigen expression, or even translocating the tumor antigen to the effector T cell surface [140]. At the gene level, monoallelic deletions, biallelic deletions, or mutations can reduce expression of the target antigen. In addition, quantitative and structural changes at the chromosomal level can reduce TAA expression, or even cause total loss of expression. So far, three cases of BCMA genomic loss have been reported, all of which exhibited a similar mechanism of biallelic loss as the TNFRSF17 gene. This included extensive prior loss of chromosome 16p, followed by a second focal genomic hit on the remaining allele [141, 142, 143]. Monoallelic GPRC5D frameshift and missense mutations can occur in up to 15% of MM patients, and patients with monoallelic GPRC5D deletion may have a specific risk of developing resistance to teclistamab [141, 144]. In the study by Lee et al. [145], 6 of 14 patients (42.8%) experienced disease progression following anti-BCMA bsAb treatment, with several TNFRSF17 mutation events observed. Moreover, GPRC5D biallelic mutations leading to GPRC5D loss were found among the 4 patients who relapsed after anti-GPRC5D bsAb treatment [145].
Another potential mechanism associated with BCMA loss is interference by sBCMA.
A recent study by Chen et al. [146] suggested that high serum levels of
sBCMA may decrease the ability of anti-BCMA antibodies to bind to tumor cells in
patients with RRMM. Most RRMM patients have elevated sBCMA, which can
significantly reduce BCMA on the surface of MM cells and thus downregulate the
functionality of BCMA in these patients. Inhibition of
The intricate interaction between myeloma cells and the BM microenvironment is
pivotal for sustaining the growth of myeloma cells and suppressing apoptosis
[151, 152]. The accumulation of malignant myeloma cells contributes to a high
tumor burden and promotes tumor progression and drug resistance. Longitudinal
analysis of the bone marrow T-cell repertoire and its response to bsAb
therapy-induced perturbations revealed that the state of T cells and tumor
recognition are prerequisites for the expansion of clone T cells and clinical
responses. Most CD4
Checkpoint signaling, particularly the PD-1/PD-L1 pathway, plays a crucial role
in immunosuppression [159]. Increased PD-L1 expression on MM cells can in part be
attributed to the production of IFN-
Continuous stimulation of T cell receptor-CD3 signaling in the absence of costimulatory molecules such as CD28 or CD137 was identified as another key factor in the bsAb-induced nonresponse or apoptosis of effector T cells. A tri-specific antibody against CD3, CD28 and CD38 was found to enhance T cell activation and improve the effectiveness of tumor cell targeting. This tri-specific molecule inhibited myeloma growth in a humanized mouse model, stimulated the proliferation of memory/effector T cells, and reduced Treg levels in non-human primates [170].
The therapeutic potential of exogenous bsAbs is hindered by their limited serum half-life and the risk of off-target tumor toxicity. The concept of in-situ bsAb generation aims to circumvent the immunosuppressive TME and eliminate the need for continuous drug administration. Specific approaches for the in-situ generation of bsAbs within the tumor tissue include engineered oncolytic viruses (OVs), transferred autologous tumor specific T cells, and transfected mesenchymal stem cells [171]. OVs represent a promising platform for the delivery of bsAb to tumor sites via their leverage of virus-mediated T-cell recruitment. Yu et al. [172] developed an OV that targets CD3 and EPHA2. This was shown to eliminate infected tumor cells and to induce bystander killing of non-infected cells, thereby demonstrating the potential of bsAb-armed OVs to enhance oncolytic immunotherapy [172]. The combination of CAR-T cells with bsAbs to create autologous tumor-specific T cells is therefore of great interest. Liu et al. [173] created CD19-bsAb transferred T cells that showed superior anti-tumor responses compared to CD19 CAR RNA-transferred T cells. Moreover, due to their inherent migratory capacity, mesenchymal stromal cells have emerged as a “cell platform” for delivering bsAbs to tumor sites in vivo [174].
The success of elranatamab and teclistamab has inspired the expansion of bsAbs for the treatment of MM. BsAbs are currently undergoing intensive clinical evaluation for the management of advanced MM patients. However, critical factors for an ideal bsAb include the selection of optimal antigen, the ability to stimulate effector cells while mitigating excessive immune responses, and overcoming the challenges of the immunosuppressive microenvironment to ensure durability without compromising patient safety. These factors present formidable challenges for scientific researchers. While the journey from laboratory to patient care is still ongoing, the advent of bsAbs as an “off-the-shelf” therapeutic has reshaped the landscape of MM immunotherapy. Future research should focus on strategic combinations with bsAbs to potentially achieve a functional “cure” following frontline treatments.
ASCT, Autologous stem cell transplant; BMSCs, Bone marrow stromal cells; BsAb, Bispecific antibodies; CAR-T, Chimeric antigen receptor T-cell; CR, Complete response; CRS, Cytokine release syndrome; DOR, Duration of response; EMD, Extramedullary disease; IV, Intravenous; mAb, Monoclonal antibodies; MM, Multiple myeloma; MRD, Minimal residual disease; ORR, Objective response rate; OS, Overall response; OVs, Oncolytic viruses; PFS, Progression-free survival; SC, Subcutaneous; TAA, Tumor-associated antigen; TME, Tumor microenvironment; TRAEs, Treatment-related adverse events; VGPR, Very good partial response.
MW and CW: Conceptualization, Writing-original draft, Writing-editing. CS, SL, and YH: Conceptualization, Supervision, Writing-review & editing. JD and HW: Data curation, Writing-review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.