
 , Guangwei Sun 1,*, Jun Shang 1,6,*
, Guangwei Sun 1,*, Jun Shang 1,6,*1 Department of Orthopedics, Shanxi Medical College Seventh Affiliated Hospital: Linfen People's Hospital, 041000 Linfen, Shanxi, China
2 Department of Orthopedics, Qilu Hospital of Shandong University, Shandong University, 250033 Jinan, Shandong, China
3 Shandong University Centre for Orthopedics, Advanced Medical Research Institute, Shandong University, 250012 Jinan, Shandong, China
4 Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmaceutical Sciences and Research Center of Basic Medical Sciences, Tianjin Medical University, 300070 Tianjin, China
5 Department of Medicine, Nantong University Xinglin College, 226008 Nantong, Jiangsu, China
6 Department of Orthopaedics, The Second Hospital of Shandong University, 250033 Jinan, Shandong, China
†These authors contributed equally.
Abstract
Traumatic spinal cord injury (SCI) is a serious disease of the central nervous system. Aside from the limited intrinsic regenerative capacity of neurons, complex microenvironmental disturbances can also lead to further cellular damage and growth inhibition. Programmed cell death regulated by pyroptosis has an important role in the pathogenesis of SCI. While there has been a wealth of new knowledge regarding cellular pyroptosis, a detailed understanding of its role in SCI and possible therapeutic strategies is still lacking. This review summarizes current advances in the regulatory role of pyroptosis-regulated cell death and inflammasome components in the inhibitory microenvironment following SCI, as well as recent therapeutic advances.
Keywords
- spinal cord injury
- pyroptosis
- gasdermin D
- inflammasome
- NLRP3
Spinal cord injury (SCI) causes permanent and devastating loss of sensory-motor and autonomic function and greatly reduces patient quality of life. SCI increases public health costs and presents a challenge to clinics and public health professionals globally. There are 759,302 patients with traumatic SCI in China, with 66,374 new cases reported each year [1]. Moreover, about 17,000 people are diagnosed with SCI each year in the United States, with the financial cost of a high-grade quadriplegic patient being approximately $1 million in the first year [2]. Following a strike to the spinal cord, deleterious pathophysiological changes are induced by a variety of traumatic injuries such as shear stress, contusion, torsion, and compression. This results in transient or permanent impairment of normal sensory, motor, and autonomic functions of the spinal cord. In addition, the death of neurons in damaged tissue and the activation of inflammatory cells in damaged areas may cause secondary damage that induces axonal degeneration, accumulation of pro-inflammatory cytokines and chemokines, demyelination, and cellular damage. Due to the irreversible and unavoidable nature of primary SCI, therapeutic modalities have focused on ameliorating disturbances of the spinal cord microenvironment caused by secondary injuries, as well as promoting the intrinsic regeneration of host nerves in order to improve the clinical outcome and prognosis of patients with SCI. Several pathophysiological changes occur after SCI, including oxidative stress, inflammation, pyroptosis, and apoptosis. Moreover, there is growing evidence that modulation of cellular pyroptosis following secondary injury may prove beneficial in the treatment of SCI.
Previous studies have identified cellular pyroptosis as an important event
leading to increased cell death and neuroinflammatory responses after SCI.
Pyroptosis is a novel, pro-inflammatory type of cell death regulated mainly by
Caspase-1 and Gasdermin D (GSDMD). It is also accompanied by the production of
different inflammatory mediators, such as interleukin (IL)-1
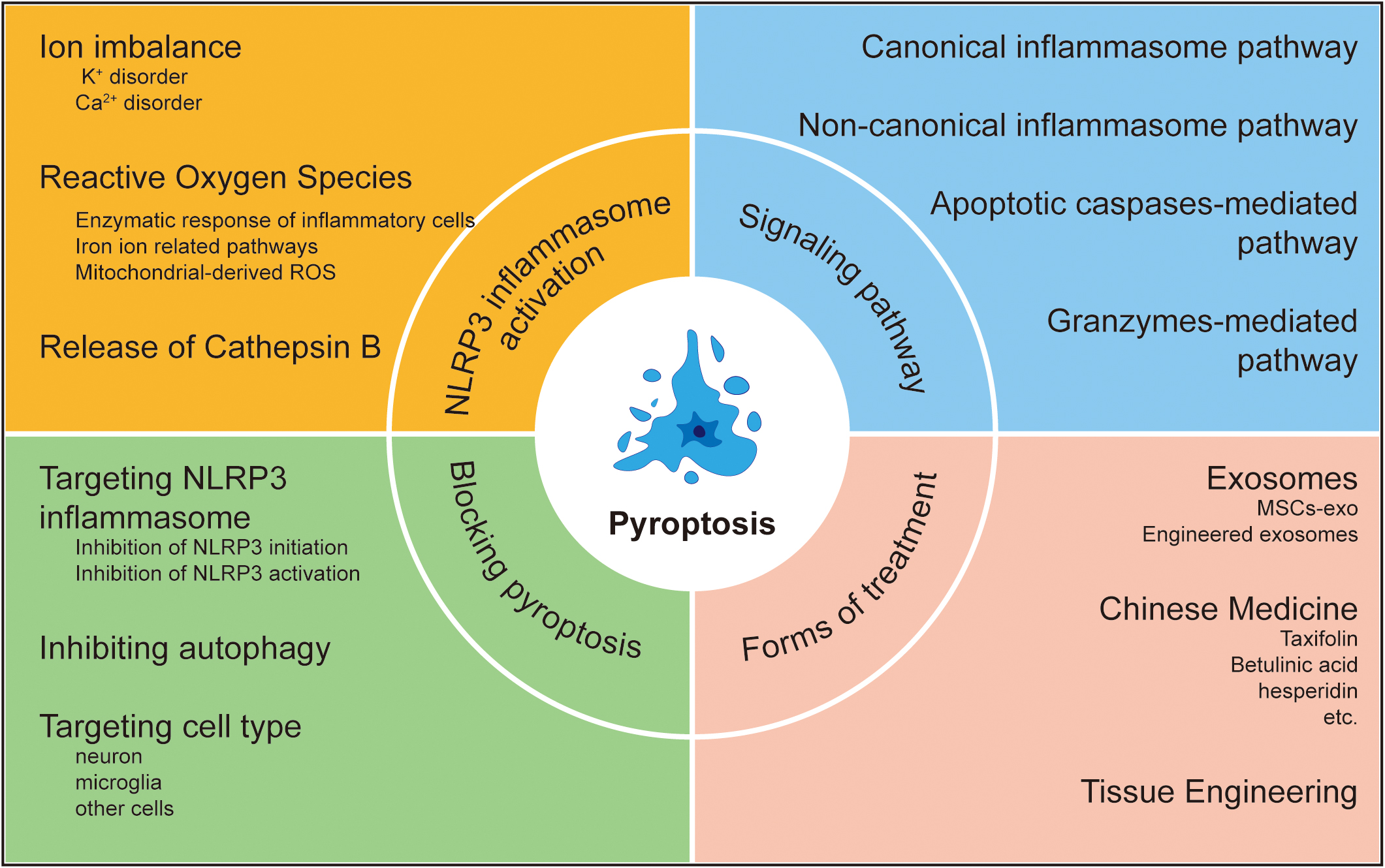
Despite the wealth of newly discovered information on pyroptosis, its regulatory role in SCI is still not fully understood. Moreover, it remains unclear how the inhibition of pyroptosis can improve clinical outcomes. Therefore, this review will systematically summarize new findings on the role of pyroptosis in the pathogenesis of SCI. Furthermore, we discuss potential biomarkers and drugs for therapeutic strategies involving pyroptosis in SCI (Fig. 1).
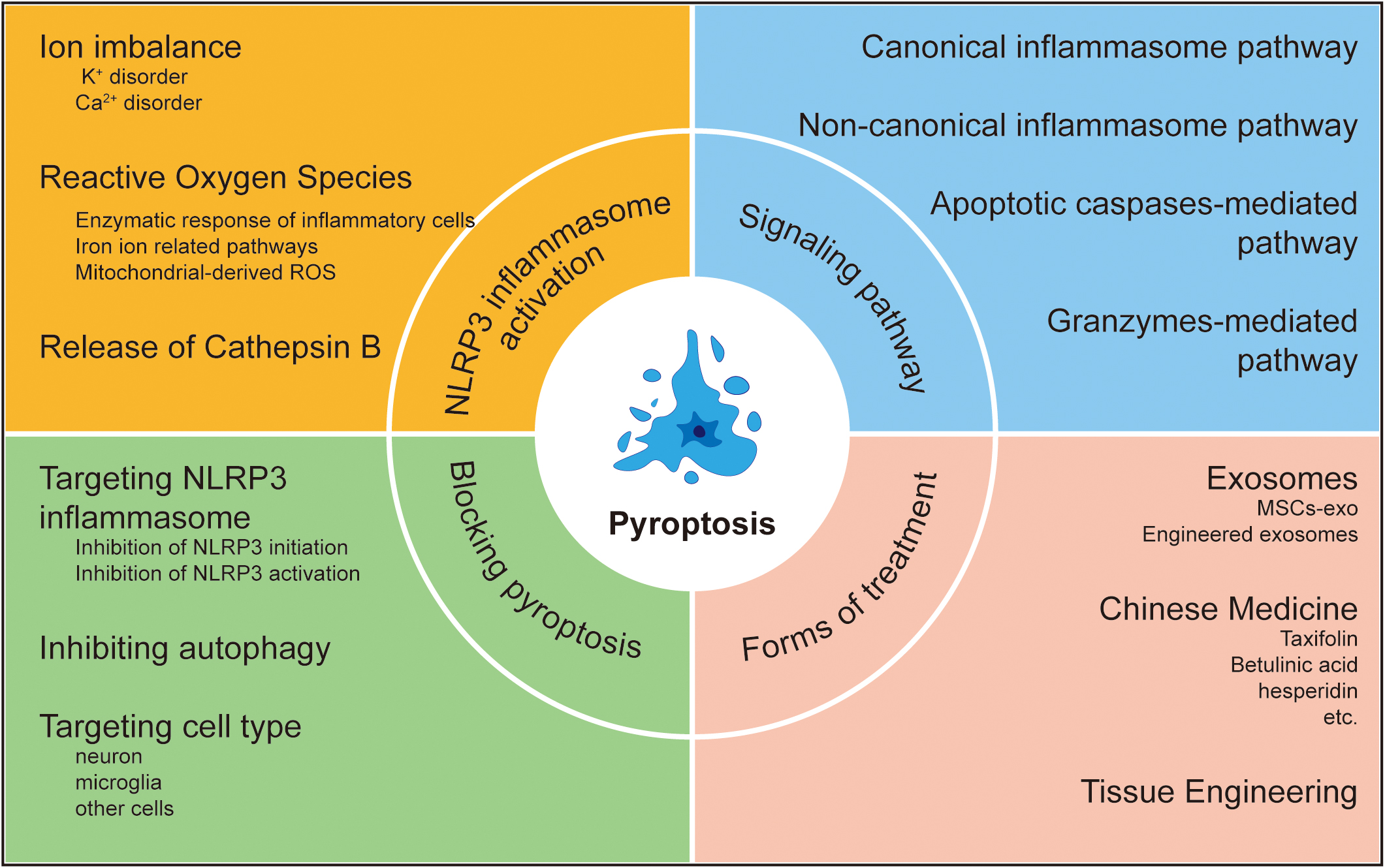 Fig. 1.
Fig. 1.Pyroptosis in spinal cord injury. ROS, Reactive Oxygen Species.
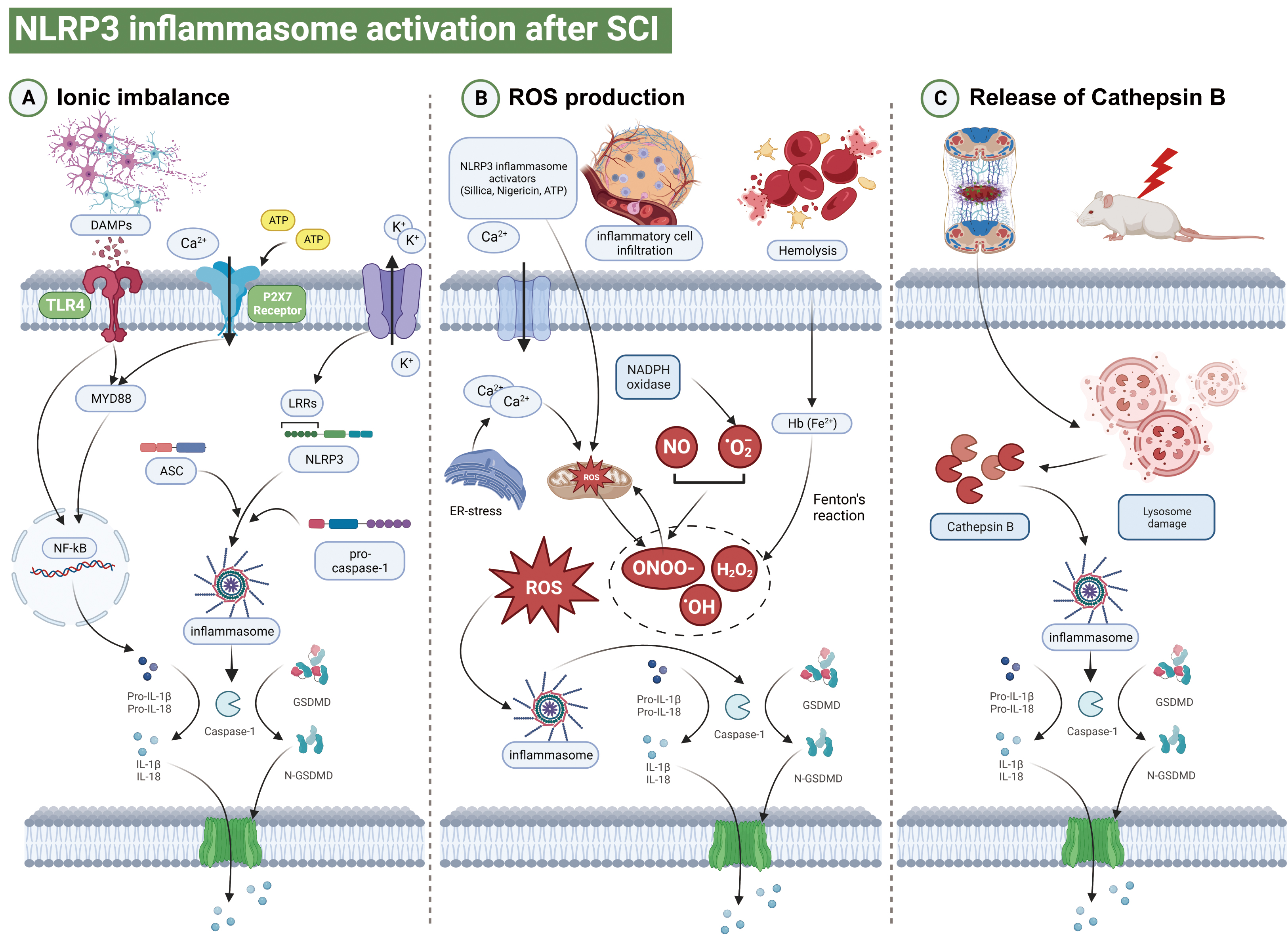
Immediate contusion and compression of the spinal cord tissue as a result of the impact of a direct external force is accompanied by vascular injury and disruption of the blood-brain barrier [3]. The “primary injury” to spinal cord tissue results in the local and systemic release of damage-associated molecular patterns (DAMP), and binding to leucine-rich repeat sequence (LRR) domains. This directly disrupts the spinal cord microenvironment and induces local cellular pyroptosis [4]. The mechanism underlying NOD-like receptor protein 3 (NLRP3) inflammasome formation remains unclear. Previous reports suggest that ionic imbalance, oxidative stress, and cathepsin B (CTSB) leakage are the three main activators of NLRP3 inflammasomes. Moreover, the pathways linking ionic imbalance, oxidative stress and CTSB leakage with cellular death are not mutually exclusive. Each of these signals may interact independently with NLRP3, leading to different conformational changes in the molecules involved in inflammasome assembly [5]. Here, we discuss in detail how ion imbalance, ROS, and CTSB activate NLRP3 inflammasomes after SCI (Fig. 2).
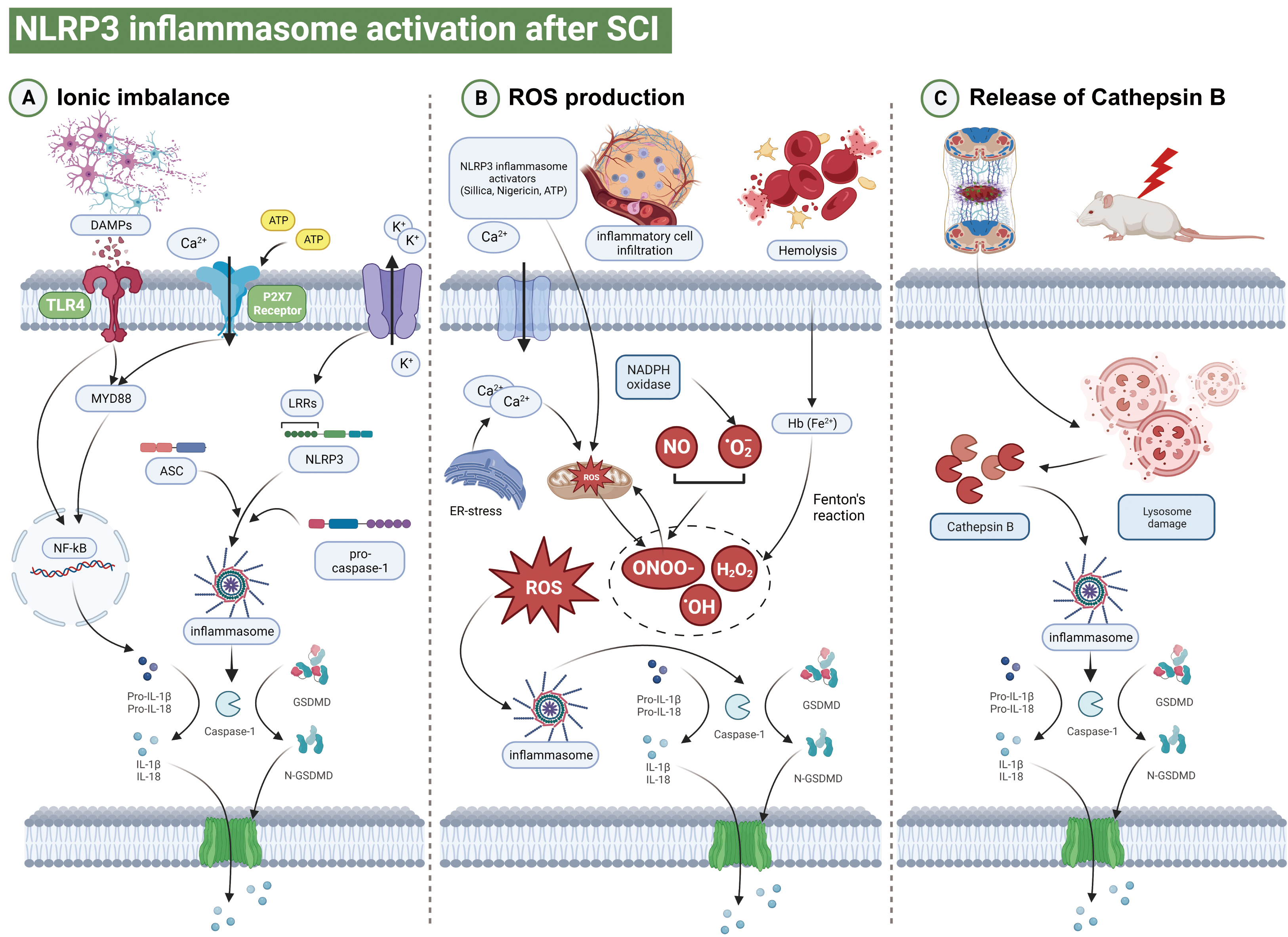 Fig. 2.
Fig. 2.Schematic illustration of different pyroptosis pathways. (A)
Damage associated molecular patterns (DAMPs) such as ATP or Reactive Oxygen Species (ROS) are released from parenchymal cells undergoing death
after primary spinal cord injury (SCI). These promote the transcription of pro-IL-1
Potassium (K
Indeed, the impact of changes in ion channels on cellular pyroptosis following
SCI remains unclear. The P2X7 receptor (P2X7R) is a member of the P2X purinergic
receptor family. P2X7R is preferentially expressed in microglia and specifically
detects ATP signaling in DAMP after SCI. This in turn promotes K
Excessive accumulation of intracellular Ca
Two mechanisms have been proposed for the Ca
ROS are highly reactive, oxygen-containing substances produced mainly by redox
reactions during normal physiological activities. Oxidative damage to proteins,
lipids and nucleic acids is mainly driven by ROS and reactive nitrogen species
(RNS), which can in turn induce pyroptosis, inflammation, autophagy, and
apoptosis [27]. In the minutes or hours after SCI, the reduced activity of
superoxide dismutase (SOD), catalase, and glutathione peroxidase results in
oxidative stress because superoxide anion (i.e., O
2.2.1.1 Enzymatic Response of Inflammatory Cells
Neutrophils are the first inflammatory cells of the immune system to be
activated following SCI and to reach the lesion, with their number peaking 24 h
after injury [27, 29]. Neutrophils and monocytes migrate to the injury site due
to the release of chemokines and cytokines, and to the upregulation of several
adhesion molecules on endothelial cells. The three main functions of these immune
cells include phagocytosis and scavenging of cellular debris, secretion of
myeloperoxidase proteases and elastase, and the release of ROS [30]. Activation
of microglia also occurs immediately after SCI, with the cells exhibiting altered
protein expression profiles and morphology, while also migrating towards the
lesion where they localize and proliferate. In addition, circulating monocytes
infiltrate the injury site and differentiate into macrophages. These cells are
very similar to activated microglia in terms of their protein expression profile,
morphology, and function [31, 32]. The rapid increase in the number of
macrophages generates substantial quantities of O
2.2.1.2 Ferritin and Transferrin
Iron homeostasis plays a crucial role in the metabolic activities of the CNS,
including oxidative phosphorylation, neurotransmitter production, and myelin
synthesis [36]. Under physiological conditions, iron exists as a complex within
ferritin and transferrin. However, due to the significant decrease in tissue pH
in the injured area after SCI, ferric ions tend to be released from ferritin and
transferrin. In the presence of iron, the Fenton reaction generates
O
2.2.1.3 Mitochondria and the Endoplasmic Reticulum
The mitochondrial respiratory chain is a prominent intracellular generator of
ROS. As a significant site of ROS synthesis, mitochondria can trigger and
regulate both pyroptosis and autophagy following SCI [40, 41]. Under normal
physiological conditions, the energy-dependent intra-axonal Ca
Previous studies have demonstrated that ROS, particularly those produced by
mitochondria, contribute to the activation of NLRP3 inflammasomes [51, 52, 53, 54]. Many NLRP3 inflammasome activators have been reported to trigger
mitochondrial ROS production in a variety of cell types, and NADPH oxidase was
originally thought to be the source of ROS production. ROS is therefore thought
to be a common signal for NLRP3 inflammasome activation [55]. Palmitate, a
saturated fatty acid, has been shown to activate NLRP3 inflammasomes, leading to
the release of active IL-1
Several studies have shown that macrophages induce the activation of NLRP3 inflammasomes following the phagocytosis of particulate matter such as amyloid beta, silica, MSU, calcium crystals, and cholesterol crystals [55, 57, 58, 59]. The phagocytosis of particulate matter damages lysosomes and causes leakage of lysosomal contents into the cytoplasm. Release of CTSB through lysosomal disruption appears to be a crucial step in the particulate-induced activation of NLRP3 inflammasomes, as this is inhibited in macrophages treated with CA-074-Me, a chemical inhibitor of CTSB [58, 60]. Ellis et al. [61] have shown that CTSB expression is upregulated following SCI, suggesting that it may be involved in the secondary injury cascade that lasts for up to a week. However, the source of CTSB after SCI has not yet been clearly identified. Further elucidation of the source of CTSB after SCI and its relationship with cellular pyroptosis should therefore help to understand its mechanism of action.
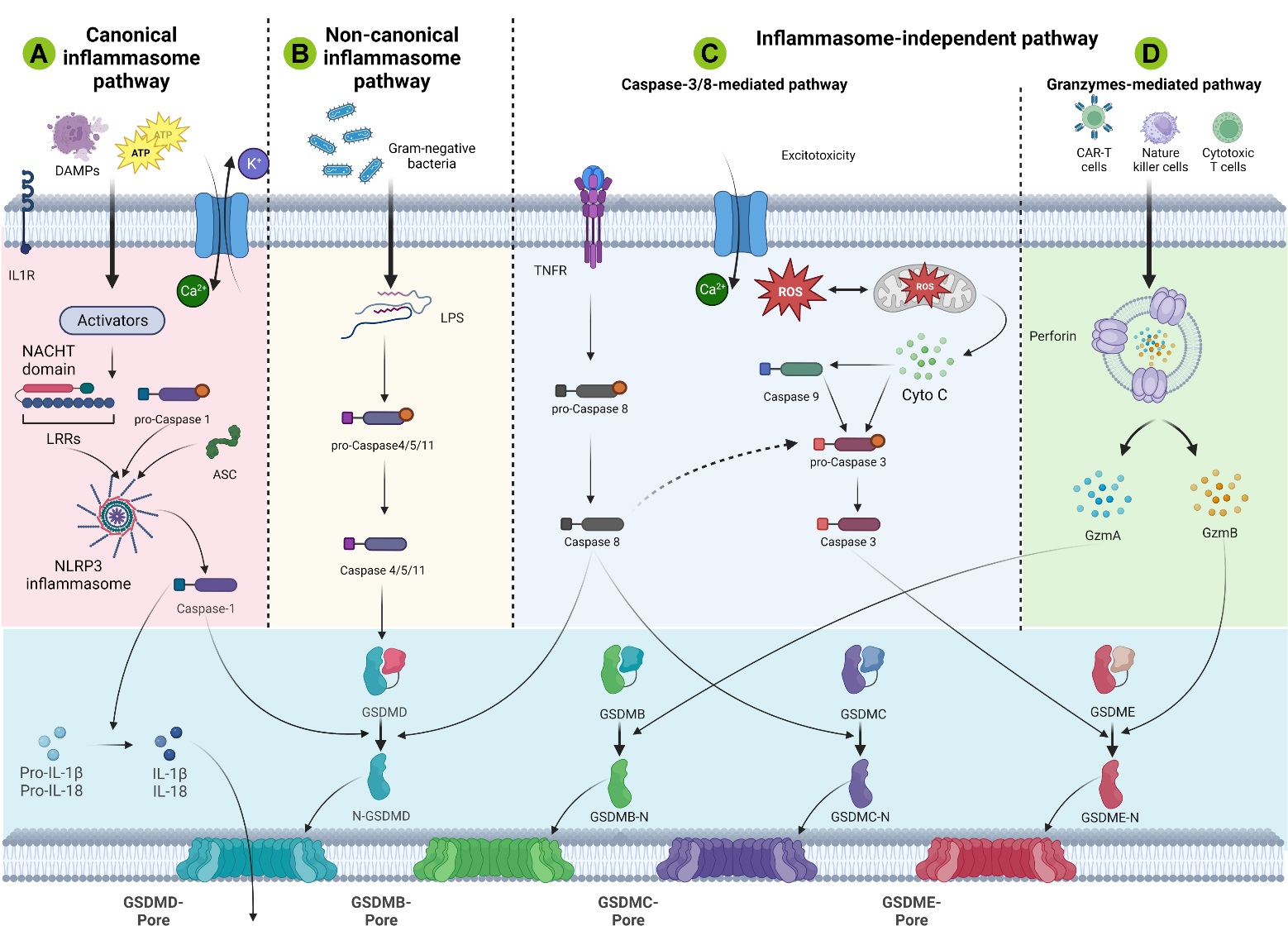
Pyroptosis plays a crucial role in the pathophysiological mechanisms of multicellular organisms by maintaining a dynamic balance between cell proliferation and cell death. Currently, four distinct cellular pathways of pyroptosis have been identified: canonical inflammasomal pathways, noncanonical inflammasomal pathways, apoptotic caspase-mediated pathways, and granzyme-based pathways (Fig. 3). In all four pathways, GSDM protein acts as the final target and is cut by upstream caspase or granzyme. In general, inflammasome-dependent pyroptosis includes caspase-1-dependent pathways (canonical) and caspase-4/5/11-dependent pathways (noncanonical) [62, 63]. Recent studies have identified novel pathways that are not reliant on inflammasomes, including the caspase-3/8-mediated pathway, as well as other GSDM-mediated pathways [64, 65].
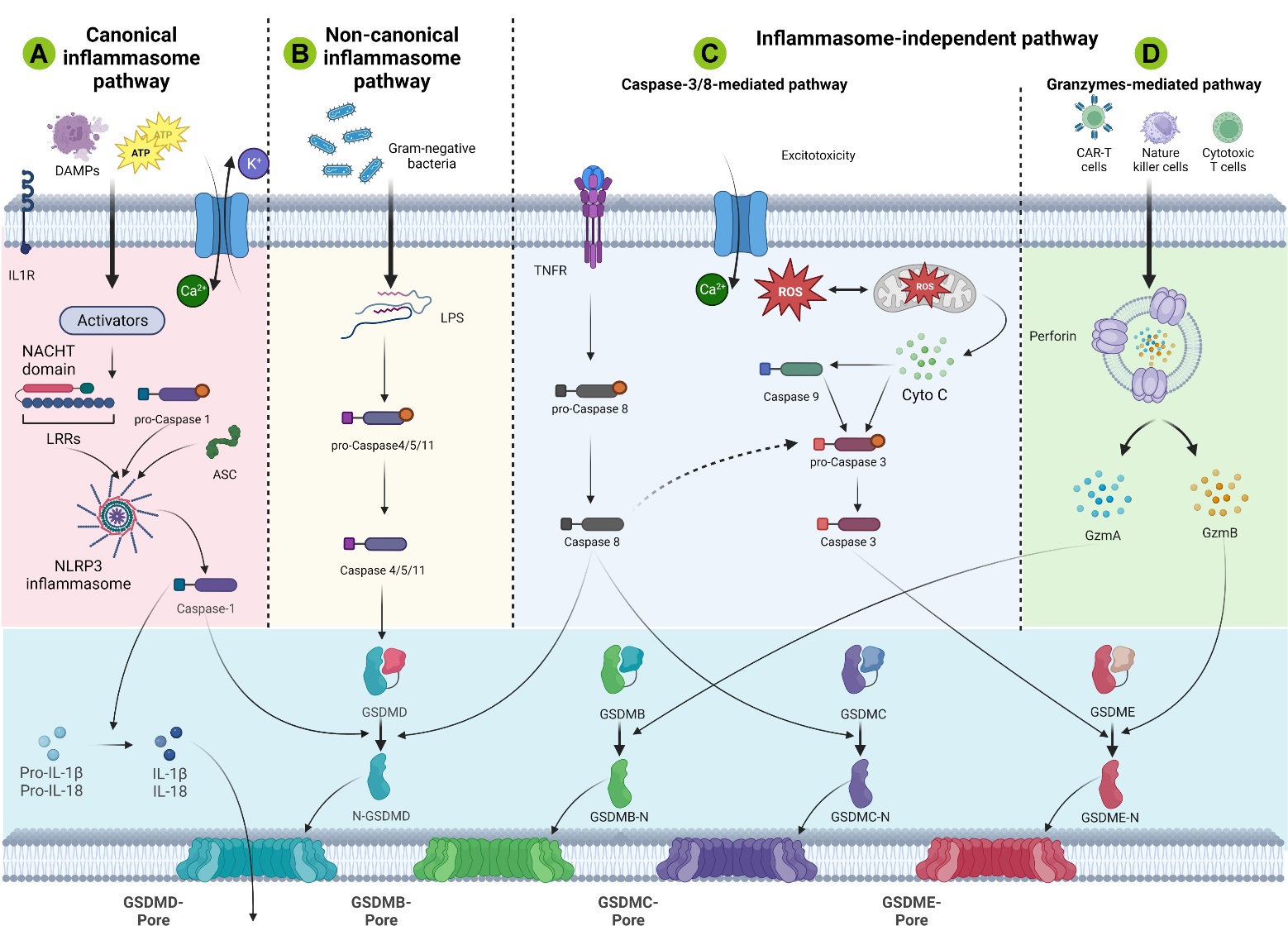 Fig. 3.
Fig. 3.Schematic illustration of the molecular mechanisms of NLRP3 activation after SCI. (A) In the canonical inflammasome pathway of SCI, DAMPs such as ATP or ROS stimulate inflammasomes, which then activate caspase-1 to cleave Gasdermin D (GSDMD) for pore formation. (B) In vitro simulation of post-SCI inflammation. Lipopolysaccharide (LPS) from Gram-negative bacteria is used to directly activate caspase-4/5/11, followed by GSDMD-mediated lysis leading to atypical inflammasome pyroptosis pathways. However, this pathway is rarely reported in mouse SCI models. (C) The caspase-mediated apoptosis pathway following SCI involves caspase-3/Gasdermin E (GSDME), caspase-8/Gasdermin C (GSDMC), GSDMD, and other mechanisms. (D) In the granzyme-mediated pathway, Granzyme A (GZMA) or GZMB derived from cytotoxic lymphocytes, Chimeric Antigen Receptor (CAR) T cells and natural killer cells can cleave GSDMB or GSDME to cause pore formation or pyroptosis, respectively. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; TNFR, tumor necrosis factor receptor. This figure was created with Biorender (https://www.biorender.com/).
The classical inflammasome pathway was the first to be discovered. Inflammasomes
are multiprotein complexes that assemble in response to either
pathogen-associated molecular patterns (PAMPs), or non-pathogen-related
damage-associated molecular patterns (DAMPs). In general, inflammasomes are
composed of intracellular pattern recognition receptor (PRR),
apoptosis-associated speck-like protein containing CARD (ASC), and inflammatory
caspase [66]. In the canonical pyroptosis pathway, PRR forms functional
inflammasomes by sensing DAMPs after SCI. The PRR, which recognizes the
pathogenic stimulus, then binds to pro-caspase 1 with the help of the adaptor
protein ASC, thus eventually forming the inflammasome. After formation of the
inflammasome, caspase-1 is activated and cleaves inactive pro-IL-1
The majority of Gram-negative bacteria activate atypical inflammasomes via the
noncanonical inflammasome pathway, which is separate from the classical
inflammasome complex. Caspase-4/5/11 in host cells is activated upon recognition
of Lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria, which
subsequently initiates GSDMD cleavage and pyroptosis [68, 69]. Although
caspase-11 is able to lyse GSDMD, it is unable to convert the precursors of
IL-1
Caspase-3 has long been regarded as a key marker of cell apoptosis and is generally considered to be a pivotal effector of apoptosis-mediated cell death. However, recent studies have shown that it can also stimulate GSDME to perform pyroptosis. Members of the gasdermin protein family are structurally highly conserved. All gasdermins except DFNB59 contain C-terminal and N-terminal domains, with the latter being an activator of pyroptosis [72]. Notably, activation of caspase-3 results in the cleavage of GSDME and subsequent formation of GSDME-N termini. This leads to the creation of pores in the cell membrane region, ultimately resulting in pyroptosis in a process akin to GSDMD-N [64, 73].
Furthermore, pro-caspase-8 serves as an initiating caspase that can undergo self-processing upon binding to the Fas tumour necrosis factor family of death receptors. Upon binding the Fas ligand (FasL or CD95L), the Fas receptor (a 45 kDa membrane receptor) associates with the adaptor protein FADD (a member of the death domain superfamily) and pro-caspase-8 to form a death-inducing signaling complex (DISC). Subsequently, either directly or through a mitochondria-dependent mechanism, the activation of caspase-8 can trigger cleavage of downstream targets such as caspase-3 [74]. Thus, the regulation of cell pyroptosis by GSDME and GSDMD may provide new insights into the inflammatory cell death process. Further studies are needed to investigate the underlying mechanisms and significance of this process in various diseases, including CNS disorders such as SCI.
Recent work indicates that granzymes derived from cytotoxic T lymphocytes and natural killer cells can be delivered to target cells via perforin to induce cancer cell pyroptosis through the cleavage of specific gasdermin family members [75]. Granzyme A (GZMA) is the most prevalent serine protease in the granzyme family and can promote the release of inflammatory factors and induce pyroptosis by cleaving GSDMB. Furthermore, granzyme B (GZMB) from natural killer cells can trigger GSDME-dependent pyroptosis in tumor cell targets through direct cleavage of GSDME and indirect activation of caspase-3 [76]. Recent studies by Wu et al. [77] have shown that ferric ions can induce the pyroptosis of B cells after SCI by activating the Tom20-Bax-caspase-GSDME pathway. However, these pathways have yet to be explored in other immune cell types following SCI.
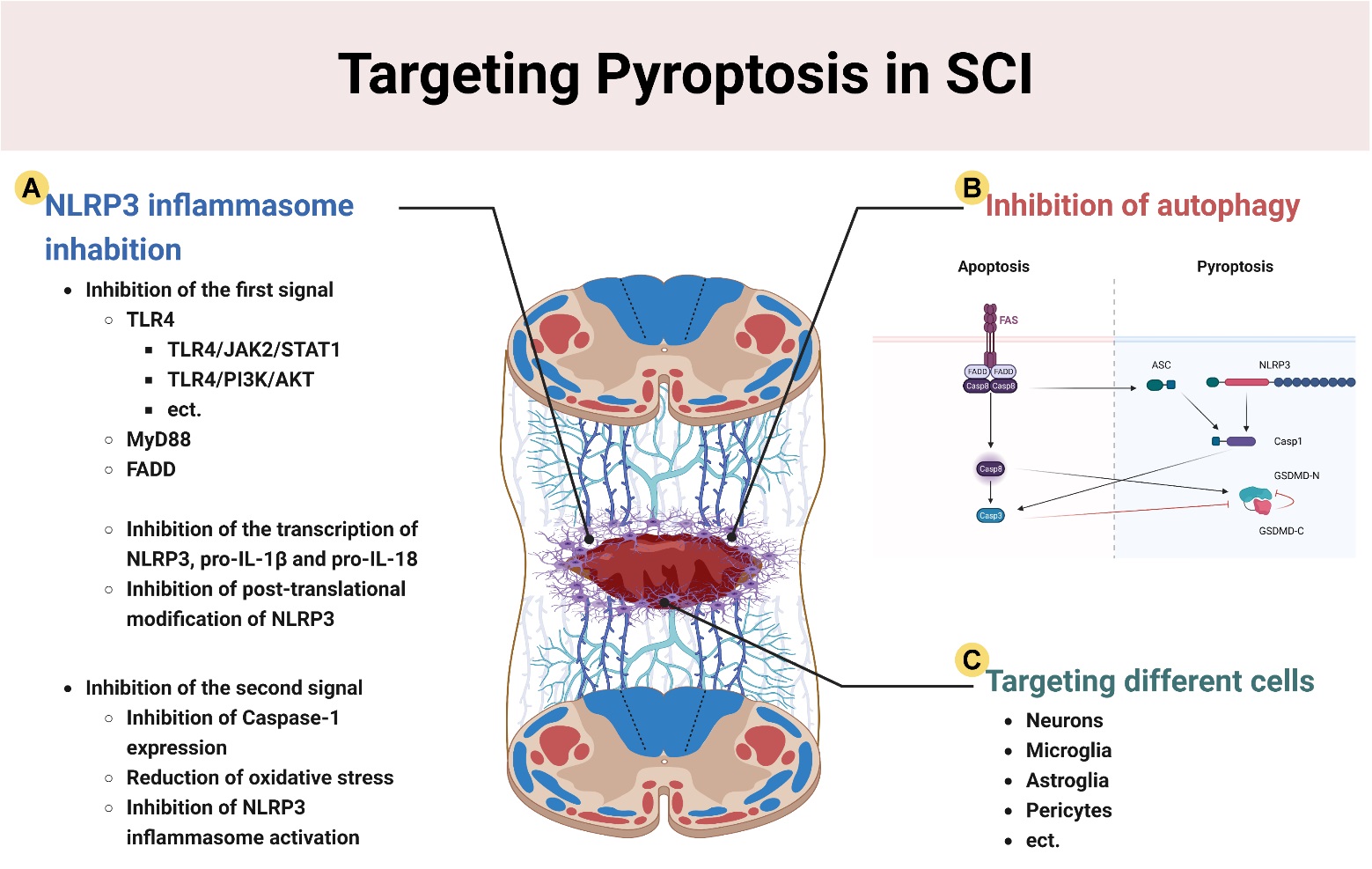
Therapeutic strategies that target cellular pyroptosis after SCI are still relatively limited and focus on the following areas: (1) inhibition of priming and activation of the NLRP3 inflammasome; (2) the promotion of cellular autophagy to inhibit pyroptosis based on autophagy-pyroptosis crosstalk; and (3) targeted inhibition of the pyroptosis of different cell types in the spinal cord (Fig. 4).
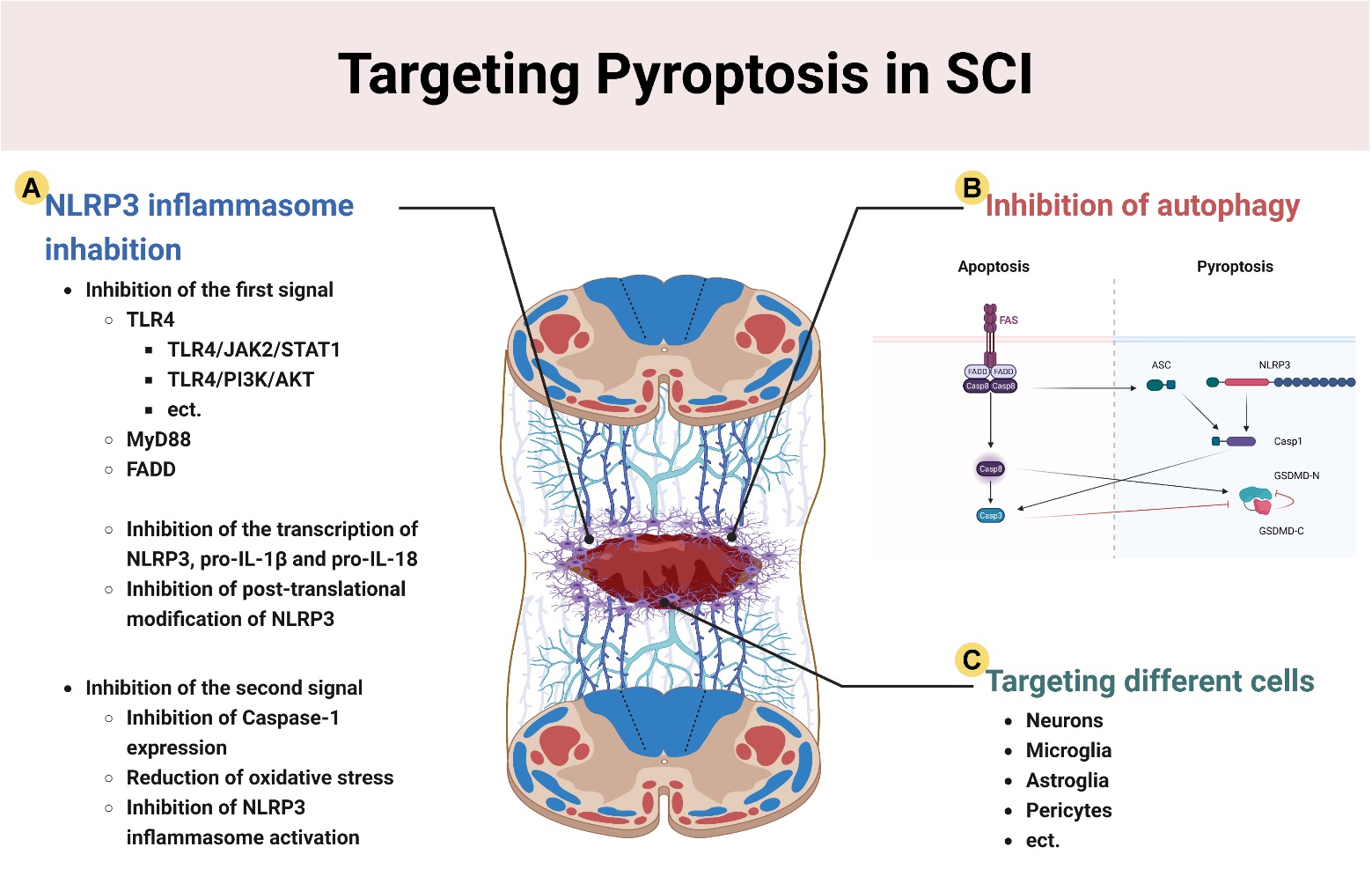 Fig. 4.
Fig. 4.Therapeutic strategies for pyroptosis after SCI. (A) Inhibition of NLRP3 inflammasomes through: (1) Inhibition of NLRP3 transcription and post-transcriptional modification; and (2) Inhibition of the expression of caspase-1, and activation of NLRP3. (B) The promotion of autophagy after SCI inhibits pyroptosis through the caspase-3/8 pathway. (C) The targeting of neurons, microglia, astrocytes, pericytes and other cell types to attenuate parenchymal cell pyroptosis after SCI. This figure was created with Biorender (https://www.biorender.com/).
NLRP3 is the most well-studied inflammasome sensor and is comprised of the
nucleotide-binding domain (NACHT), the amino terminal PYRIN domain (PYD), and the
carboxy-terminal LRR domain [78]. NLRP3 is a crucial integrator of cellular
stress due to its capacity to react to multiple stimuli in cases of SCI. Stimuli
such as extracellular ATP, lysosomal damage, and K
It is also worth noting that Nek7 plays a crucial role in the formation of mouse NLRP3 inflammasomes by binding to the NAHT of both NLRP3 and LRR [79]. Nek7 is essential for activation of NLRP3 inflammasomes, but not for activation of NLRC4 and AIM2 inflammasomes. Nek7 also regulates the formation of ASC specks, the oligomerization of NLRP3, and the activation of caspase-1 [80]. However, NLRP3 activation cannot be induced by the mere presence of Nek7. Both priming and activation are obligatory steps to achieve complete activation of NLRP3 [79]. The priming step enables cells to increase NLRP3 expression by stimulating its transcription and post-transcriptional modification. Subsequently, the secondary step results in complete formation and activation of the inflammasome [80].
Two main signals are involved in the priming stage of NLRP3 [5]. Firstly, NLRP3
is upregulated by promoting transcription [78, 79]. Once PAMP or DAMP molecules
are recognized by PRR, the nuclear factor-
Several mechanisms of NLRP3 activation are now widely recognized. These can act
together or independently, and include pore formation and metabolic disorders,
mitochondrial dysfunction, ion redistribution, lysosomal disorders and
noncanonical models [78, 84]. NLRP3 begins to activate and assemble when primed
cells are subjected to activation stimuli under appropriate conditions [85].
Multiple factors can act as second signals for the activation and assembly of
NLRP3 after SCI, including cholesterol and ATP levels, cytoarchitectural
instability such as lysosomal rupture and mitochondrial dysfunction, and ionic or
molecular perturbations such as K
Autophagy is a well-recognized lysosomal degradation process that breaks down protein aggregates and damages organelles and cytoplasmic proteins [90]. Autophagy-induced neuroprotection is initiated in both neurons and microglia during SCI and other CNS disorders [91, 92]. This is a crucial process that helps to refresh cells and maintain homeostasis, which is especially vital for the health of terminally differentiated cells like neurons [93]. Many studies have demonstrated the vital role of autophagy in neurons [94, 95]. Increased autophagy has been observed after SCI, and there is accumulating evidence that autophagy has pro-survival, neuroprotective effects by regulating the death of nerve cells [96]. In support of this, the stimulation of autophagy hinders both pyroptosis and necrotic apoptosis [97].
Necrotic apoptosis is similar to cellular pyroptosis in that both are inflammatory cell death pathways [98]. Mechanistically, RIPK1 and RIPK3 assemble to form a multi-protein complex known as the RIPK1/RIPK3 necrosome following stress-induced activation of the TNF receptor 1 (TNFR1) [99]. Subsequently, phosphorylation of the downstream molecule MLKL is facilitated by the necrosomes. p-MLKL then oligomerizes and moves to the plasma membrane, where it increases permeability to induce cell death [100]. Caspase-8 is also thought to be an important inhibitor of necrotic apoptosis [101].
A recent study based on Gene Expression Omnibus (GEO) analysis and RNA sequencing revealed the molecular mechanism by which cyclic helix B peptide improves functional recovery after SCI. It does this by inhibiting pyroptosis and reducing necroptosis through the enhancement of autophagy. Support for this mechanism was obtained using the autophagy inhibitor 3-methyladenine (3-MA), with regulation of autophagy shown to involve the transcription factor EB (TFEB) [102]. Two other studies also reported that promotion of autophagy by facilitating dephosphorylation and nuclear translocation of TFEB was effective in ameliorating pyroptosis and necrotic apoptosis in cells after SCI [103, 104]. In addition, Wang et al. [105] showed that Bexarotene enhances the nuclear translocation of transcription factor E3, activates autophagy and mitochondrial autophagy, inhibits ROS production, and suppresses pyroptosis through stimulation of the AMPK-mTOR pathway in the cytoplasm and the AMPK-SKP2-CARM1 pathway in the nucleus. In conclusion, an effective therapeutic strategy after SCI may be to improve downstream pyroptosis by inhibiting autophagic flow through multiple pathways (Fig. 3).
One of the main current therapeutic strategies is to maximize the preservation of residual neurons in secondary spinal cord injuries with microenvironmental imbalances. Pyroptosis is closely related to neuronal loss and is a key pathway for neuronal death following SCI [106, 107]. Therefore, it is important to find a feasible treatment for neuronal pyroptosis after SCI. Jiang et al. [108] demonstrated that inhibiting the activation of NLRP3 inflammasomes and reducing GSDMD expression in neurons after SCI could inhibit the onset of pyroptosis and decrease the levels of pro-inflammatory cytokines. Similarly, Zheng et al. [107] showed that carbon monoxide inhibits signaling from inflammasomes and cell pyroptosis by reducing the activation of inositol-requiring enzyme 1 (IRE1), thereby reducing neuronal death and improving motor function recovery after SCI. Furthermore, substantial evidence indicates that inhibition of neuronal pyroptosis at the lesion site can improve the prognosis of rats with SCI [104, 109, 110].
Pyroptosis is a stimulus-associated, programmed cell death pathway that can significantly influence neuroinflammation. It is a key factor driving selective injury after SCI and is activated by inflammasomes. Microglia are intrinsic immune cells in the CNS. They are significantly activated after SCI and are major participants in the subsequent neuroinflammatory process. There is substantial evidence showing increased microglial pyroptosis in CNS disorders such as SCI. An important step in the neuroinflammation observed following CNS injury is the activation of cytoplasmic inflammasome complexes leading to cell pyroptosis [111, 112, 113]. Xiong et al. [114] showed that application of Treg cell-derived exosomes after SCI significantly improved local neuroinflammation and facilitated functional recovery by inhibiting microglial pyroptosis in vivo.
Pyroptosis occurs widely after SCI, even in cell types that are less numerous than neurons and microglia. Therefore, the targeting of pyroptosis in other cell types may improve histological and functional outcomes after SCI. Zhou et al. [115] reported that exosomes derived from bone marrow mesenchymal stem cells effectively reduced pericellular pyroptosis, improved the integrity of the blood-spinal barrier, and promoted functional recovery after SCI. It was also reported that astrocytes play a role in neuroinflammation. Neuroinflammation induced by pyroptosis of astrocytes has been shown in several CNS diseases. A bioinformatics analysis by Shan et al. [116] demonstrated that erythropoietin reduces astrocyte pyroptosis both in vivo and in vitro by targeting the miR-325-3p/Gsdmd axis, thereby promoting functional recovery in a rat model of SCI.
Extensive neuroprotection studies have demonstrated the involvement of exosomes sourced from cells of the CNS, PNS, mesenchymal, and other tissues involved in neural regeneration (Table 1, Ref. [104, 114, 115, 117, 118, 119, 120, 121, 122]). Compared to cell transplantation, exosomes are easier to obtain and store, and are subject to fewer ethical restrictions [123]. In addition, the outer body size is much smaller than the cell secretion, and therefore will not be captured in lung and liver tissue. Moreover, exosomes can penetrate the blood-spinal cord barrier [124]. Recent attention has therefore focused on the use of exosomes to treat pyroptosis in SCI. Sheng et al. [117] showed that loading miRNA-22 on MSCs-EV could inhibit the occurrence of microglial pyroptosis, reduce the expression of GSDMD, and reduce the opening of cell membrane pores and subsequent release of inflammatory factors. Similarly, another study found that netrin-1 enrichment with engineered exosomes alleviated LPS-induced inflammation and pyroptosis, while promoting recovery in SCI rats [118].
| Treatments | Examples | Animals | Experimental Models | Methods of administration | Mechanisms of action | Therapeutic effects | Reference |
| Exosomes | Treg cell-derived exosomes | Foxp3DTR mice | Contusion SCI model | tail vein injection | miR-709/NKAP/NF-κB | Inhibition of microglia pyroptosis | [114] |
| BMSC-EXO | Sprague-Dawley rats | Contusion SCI model | tail vein injection | miR-21/PTEN/PDCD4 | Improvement of neuronal survival; inhibition of pericyte pyroptosis | [115] | |
| MSCs-EV | Sprague-Dawley rats | Crush SCI model | tail vein injection | Inhibition of GSDMD | Inhibition of GSDMD; inhibition of microglia activation and pyroptosis | [117] | |
| TCM | Hesperetin | Sprague-Dawley rats | C5 Contusion of SCI model | gastric gavage | increased Nrf2 signaling | Reduction of oxidative stress, inhibition of NLRP3 inflammasomes activation and pyroptosis | [119] |
| Taxifolin | Sprague-Dawley rats | Crush SCI model | gastric gavage | PI3K/AKT | Inhibition of pyroptosis-related gene and inflammatory factor expression; promotion of axonal regeneration | [120] | |
| Betulinic acid | C57/BL6 mice | Contusion SCI model | intraperitoneal injection | AMPK-mTOR-TFEB signaling pathway | Enhancement of autophagy and mitochondrial autophagy; reduction of ROS accumulation; inhibition of pyroptosis | [104] | |
| Tissue engineering | RM-LIP/MC | Mice | / | injected into tail veins | / | Inhibition of activation of inflammasomes; inhibition of pyroptosis | [121] |
| H2S@SF hydrogel | Male ICR mice | Control cortical impact | local coverage | H2S antioxidant effect | Reduction of endothelial cell pyroptosis; amelioration of oxidative stress | [122] | |
| EX-netrin1 | Sprague-Dawley rats | Contusion SCI model | tail vein injection | Unc5b/PI3K/AKT/mTOR pathway | Reduction of inflammatory response and pyroptosis; promotion of axonal growth | [118] |
Abbreviations: BMSC, Bone marrow mesenchymal stem cell; EXO, exosomes; TCM, Traditional Chinese Medicine; RM-LIP/MC, fabricated macrophage membrane-c[amouflaged liposomes; H2S@SF, H2S-releasing silk fibroin; EX-netrin1, engineered EX enriched in netrin-1; SCI, Spinal cord injury; TBI, Traumatic brain injury.
Traditional Chinese medicine (TCM) is a promising supplementary treatment method
that has attracted cosiderable attention in recent years. Studies have
demonstrated that herbal active extracts, metabolites, and traditional botanical
formulas are effective and can play an important role in the prevention and
treatment of SCI, especially in reducing pyroptosis [125]. Hu et al.
[120] showed that Taxifolin significantly reduced oxidative stress mediated by
microglial activation, and inhibited the post-SCI expression of
pyroptosis-associated proteins (NLRP3, Caspase-1, GSDMD, and ASC) and
inflammatory cytokines (IL-1
With the rapid advances in tissue engineering, several biological substances have been extensively studied with regard to the treatment of SCI. These include natural elements such as alginate, hyaluronic acid, chitosan, etc., as well as synthetic materials such as polymers, nitrocellulose membranes, and biodegradable synthetic materials. The purpose of tissue engineering therapy is to reconstruct damaged tissues by embedding living cells or loading drugs, improving the injured microenvironment, and promoting nerve regeneration. Biomaterials employed for SCI typically require that they be biocompatible [129]. Moreover, they should possess adequate softness to avoid compressing the adjacent spinal cord tissue, while being sufficiently robust structurally to maintain local fixation [130]. In addition, they should have an appropriate rate of degradation [131]. Xiao et al. [132] implanted miR-138-modified umbilical cord mesenchymal stem cell-derived exosomes in a thermo-responsive hydrogel system into rats with SCI. This material was found to be biocompatible and able to decrease inflammation in BV-2 cells via the NLRP3-caspase-1 signaling pathway, while reducing neuronal apoptosis through down-regulation of intracellular ROS levels by Nrf2 [132]. Tang et al. [121] employed macrophage membrane-modified, liposome-loaded minocycline to effectively attenuate the pyroptosis process after SCI with high biocompatibility and advanced targeting efficacy. Although extensive data are still lacking, tissue engineering has shown great therapeutic potential for the inhibition of pyroptosis after SCI.
The important role of programmed cell death in SCI, and in particular
pyroptosis, is gradually being uncovered. This article has reviewed the
mechanisms and pathways of pyroptosis following SCI, including key players such
as inflammasomes and gasdermin. In addition, we reviewed current therapeutic
strategies for targeting pyroptosis after SCI. The inflammatory response and
parenchymal cell death are important components of secondary SCI. Pyroptosis is a
pro-inflammatory mode of cell death characterized by rapid rupture of the plasma
membrane and subsequent leakage of cellular contents and cytokines (IL-1
Although newly developed pyroptosis therapeutic strategies have shown great promise for SCI, the research and potential applications of pyroptosis in SCI are still quite limited. For experimental purposes, the use of TCM, exosomes, miRNAs, etc., in combination with tissue-engineered materials, may further improve therapeutic efficacy. Further research should focus on the flexibility of pyroptosis in regulating cell death, and explore potential mechanisms of pyroptosis in other cell death types in SCI, rather than just a single function of pyroptosis such as lethality or mediation of one mode of death. In addition, there are few studies showing cell specific effects in vitro. Specially in neuronal cultures, thus cell specific targets of the drugs an in vitro experiment with neuronal cultures will be essential. Moreover, the discovery of marker molecules and associated metabolic mechanisms of pyroptosis-related death, the exploration of clinically applicable targeted agents, and the clinical application of these markers in combination with pyroptosis regulation may transform disease treatment.
In summary, there is growing evidence that pyroptosis plays a dual role in promoting inflammation and cell death after SCI. Elucidation of the pathogenic mechanism of pyroptosis, and the potential benefit of inhibiting pyroptosis, could provide a rational, mechanism-guided model for treating SCI.
JS, GWS and YNZ were responsible for the study conception and design. GJinG, HQY, and HSZ performed the literature review. GJinG, HQY, HSZ, WJK, and PPZ wrote the original manuscript. PPZ, GJieG, JL, WHS and PCC contributed to preparation of the table and figures. JS, WHS and PCC revised the manuscript. All authors have contributed to the acquisition and analysis of literatures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Some of the figures were created with biorender.com (https://www.biorender.com/). We apologize to authors whose excellent studies were within the scope of this review, but were not mentioned owing to space limitations.
The authors are grateful to the following organizations for partial funding of this research: the Scientific Research Project of Traditional Chinese Medicine in Shanxi Province (No.2023ZYYB2048), key medical research projects of Shanxi Province (2020XM51), and Basic Research Project of Shanxi Province (202203021221295).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.