





 , Ignazio Alberto Barbagallo 1
, Ignazio Alberto Barbagallo 11 Department of Biomedical and Biotechnological Sciences, University of Catania, 95123 Catania, Italy
2 Department of Drug Sciences, University of Catania, 95123 Catania, Italy
3 Unità Operativa Complessa di Ematologia con Trapianto di Midollo Osseo, Azienda Ospedaliero-Universitaria Policlinico “G.Rodolico San Marco", 95123 Catania, Italy
4 Hospital Pharmacy Unit, Ospedale Cannizzaro, 95125 Catania, Italy
5 Laboratorio Arpa Sicilia, Viale Sicilia, 11, 97100 Ragusa (RG), Italy
6 Pharmaceutical Biotechnology Laboratory, Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia
7 Department of General Surgery and Medical-Surgical Specialties, University of Catania, 95123 Catania, Italy
Abstract
Background: Nonalcoholic fatty liver disease (NAFLD) is a prevalent condition characterized by hepatic fat accumulation, often progressing to severe liver injury, for which approved treatments are currently lacking. This study explores the potential therapeutic impact of alpha-lipoic acid (ALA), a natural compound crucial in lipid metabolism, on NAFLD using an in vitro model. Methods: HepG2 cells were treated with a palmitic acid:oleic acid (PA:OA) mixture, representing a cellular model of steatosis. Subsequent treatment with ALA at concentrations of 1 µM and 5 µM aimed to evaluate its effects on lipid content and metabolism. Real-time polymerase chain reaction (PCR), BODIPY staining, cytofluorimetric analysis, and lipidomics were used to assess gene expression, lipid droplet accumulation, and fatty acid profiles. Results: Our results showed that ALA significantly reduced lipid droplets in PA:OA-treated HepG2 cells, with a concentration-dependent effect. Analysis of fatty acid profiles demonstrated a decrease in palmitic acid levels with ALA treatment, while oleic acid reduction was observed only at the higher concentration. Moreover, ALA modulated the expression of genes involved in cholesterol biosynthesis and low-density lipoprotein (LDL) metabolism, indicating a potential role in lipid homeostasis. Further insights into molecular mechanisms revealed that ALA modulated peroxisome proliferator activated receptors (PPARs), specifically PPAR-alpha and PPAR-gamma, involved in fatty acid metabolism and insulin sensitivity. Finally, ALA counteracted the overexpression of thermogenic genes induced by exogenous fatty acids, suggesting a regulatory role in energy dissipation pathways. Conclusion: In conclusion, this study highlights ALA as a therapeutic agent in mitigating lipid accumulation and dysregulation in NAFLD.
Keywords
- lipoic acid
- liver steatosis
- fatty acid
- lipid metabolism
Nonalcoholic fatty liver disease (NAFLD) is a condition characterized by the accumulation of fat in the liver, which can progress to more severe liver injury [1]. The liver is a central organ in lipid metabolism, responsible for processing dietary fats, synthesizing lipoproteins, and regulating lipid storage and release. Despite numerous clinical trials exploring responses to various medications and supplements, no approved treatment has yet been suggested for managing NAFLD.
Lipoic acid, also known as alpha-lipoic acid (ALA), is a natural compound that plays a crucial role in lipid metabolism and various other biological processes [2]. It has been investigated for its potential to improve NAFLD by promoting the breakdown of triglycerides (a type of lipid) in liver cells [3]. Additionally, lipoic acid exhibits antioxidant properties that can reduce oxidative stress in the liver, a factor in the development and progression of NAFLD [4, 5, 6]. The pharmacological effects of ALA are multifaceted, as it influences several aspects of lipid metabolism, including energy production, antioxidant defense, fatty acid oxidation, and insulin sensitivity regulation [7].
Previous research showed that ALA serves as a cofactor for various enzymes
involved in mitochondrial energy production, particularly in the citric acid
cycle (Krebs cycle) and oxidative phosphorylation [2]. These processes are
critical for breaking down macronutrients, including lipids. Furthermore, ALA has
demonstrated potential benefits in improving insulin sensitivity, a factor
associated with abnormal lipid metabolism and elevated circulating lipids
(hyperlipidemia) in conditions such as type 2 diabetes and obesity [7]. In
particular, previous studies showed that ALA exhibited similar effects to
metformin, significantly upregulating the glycolytic enzymes glucokinase (GCK),
hesokinase-1 (HK-1) and pyruvate kinase (PK), and the glycogen synthesis enzyme
GS, and downregulating the gluconeogenic enzymes PEPCK and G6Pase, thus decreased
glucose production, and promoted glycogen synthesis and glucose utilization in
livers. Moreover, ALA markedly increased PKB/Akt and GSK3
HepG2 cells (American Type Culture Collection, Manassas, VA, USA; ATCC HB-8065
™) were a kind gift from Prof. Maurizio Parola of the University
of Turin. Briefly, low-passage cells were grown in DMEM (Sigma-Aldrich, Milan,
Italy) supplemented with 10% FBS (EuroClone, Milan, Italy), 100 U/mL penicillin
(Life Technologies, Milan, Italy), and 100 µg/mL streptomycin (Life
Technologies) at 37 °C in a humidified incubator in an atmosphere of
95% air and 5% CO
The choice of mixture (PA:OA - 250 µM:500 µM) was based on a previous study that showed that the FFA mixture containing a low proportion of palmitic acid (palmitate/oleate 1:2 ratio) is associated with minor toxic and apoptotic effects, thus representing a cellular model of steatosis [15]. The choice of ALA concentration was based on a dose-response curve, which showed that the concentration of 1 and 5 µM is not toxic to cells, and furthermore, it is in a clinically relevant range. Cell culture was tested for possible mycoplasma contamination before all the experimental procedures with a (PlasmoTest™, rep-pt1, InvivoGen, San Diego, CA, USA) according to manufacturer’s protocol. Additional STR profiling is available at the following link: https://www.atcc.org/products/hb-8065.
HepG2 cells were pretreated with PA:OA to induce steatosis and, in the following
6 h, treated with ALA. RNA was extracted using Trizol® reagent
(Invitrogen, Carlsbad, CA, USA). First-strand complementary DNA (cDNA) was then
synthesized with a reverse transcription reagent from Applied Biosystems (Foster
City, CA, USA). Quantitative real-time PCR (qRT-PCR) was performed in a StepOne
Fast Real-Time PCR System (Applied Biosystems) using the SYBR Green PCR MasterMix
(Life Technologies, Monza, Italy). The specific PCR products were detected with
SYBR green fluorescence. The relative messenger RNA (mRNA) expression level was
calculated by the threshold cycle (Ct) value of each PCR product and normalized
with that of actin using a comparative 2
| Gene | Forward 5 |
Reverse 5 |
| COX1 | CGCCAGTGAATCCCTGTTGTT | AAGGTGGCATTGACAAACTCC |
| COX2 | CTGGCGCTCAGCCATACAG | CGCACTTATACTGGTCAAATCCC |
| HMGCR | TTCGGTGGCCTCTAGTGAGA | TGTGAGTTGGAACTGAGGGC |
| DHCR7 | GCTGCAAAATCGCAACCCAA | GCTCGCCAGTGAAAACCAGT |
| CYP51A1 | GAAACGCAGACAGTCTCAAGA | ACGCCCATCCTTGTATGTAGC |
| SREBF2 | CCTGGGAGACATCGACGAGAT | TGAATGACCGTTGCACTGAAG |
| PCSK9 | AGACCCACCTCTCGCAGTC | GGAGTCCTCCTCGATGTAGTC |
| LDLR | GAGAGCTTGTGCCGAGATGTG | CCGCAGTTGTTAGTGCCATCA |
| PPARa | AAGAGCTTGGAGCTCGGC | TGAAAGCGTGTCCGTGATGA |
| PPARd | GGGACAGGCTGATGGGAAC | TGAACACCGTAGTGGAAGCC |
| UCP1 | TGTCCTGGGAACAATCACCG | TCCAGGATCCAAGTCGCAAG |
| UCP2 | GCCTCTACAATGGGCTGGTT | GAGCATGGTAAGGGCACAGT |
| CCTTTGCCGATCCGCCG | AACATGATCTGGGTCATCTTCTCGC |
BODIPY staining and cytofluorimetric analysis were used for lipid droplet
measurement. Briefly, HepG2 cells were seeded in 24-well plates and treated with
PA:OA and ALA. Following 24 h of ALA treatment, cells were washed using PBS to
remove media/serum and incubated with BODIPY staining solution in the dark for 15
min at 37 °C. Cells were washed with PBS to remove staining solution and
then trypsinized to generate a single cell suspension. This was then
centrifugated at 250
Liquid chromatography coupled with mass spectrometry (HPLC-Ms/Ms) (Waters
Alliance 2695/Waters Micromass Quattro Micro API) was used to analyze the fatty
acid content. The separation was performed using water:methanol as mobile phase
with a gradient elution as follows: 0–8 min, 50:50 (v/v); 8–12 min, 2:98 (v/v);
and 12–15 min, 50:50 (v/v). The elution rate was 300 µL/min for 15 min on
a C18 column (Atlantis Waters) 3 µm C18, 150
Statistical analysis was performed using GraphPad Prism software, version 9.0
(GraphPad Software Inc., San Diego, CA, USA, RRID: rid_000081). For comparison
of n
In order to measure the accumulation of fatty acids within hepatocytes treated with PA:PO, we measured lipid droplets through staining with BODIPY. Our data show that PA:PO significantly increased the lipid content in HepG2 cells compared to controls and that a 1 µm dose of ALA increased this fat accumulation. Different results were obtained with the dose of 5 µm of ALA, this concentration was able to reduce the accumulation of fats inside the cells previously treated with PA:OA. No effects were found on cells treated with ALA alone at both concentrations (Fig. 1A,B).
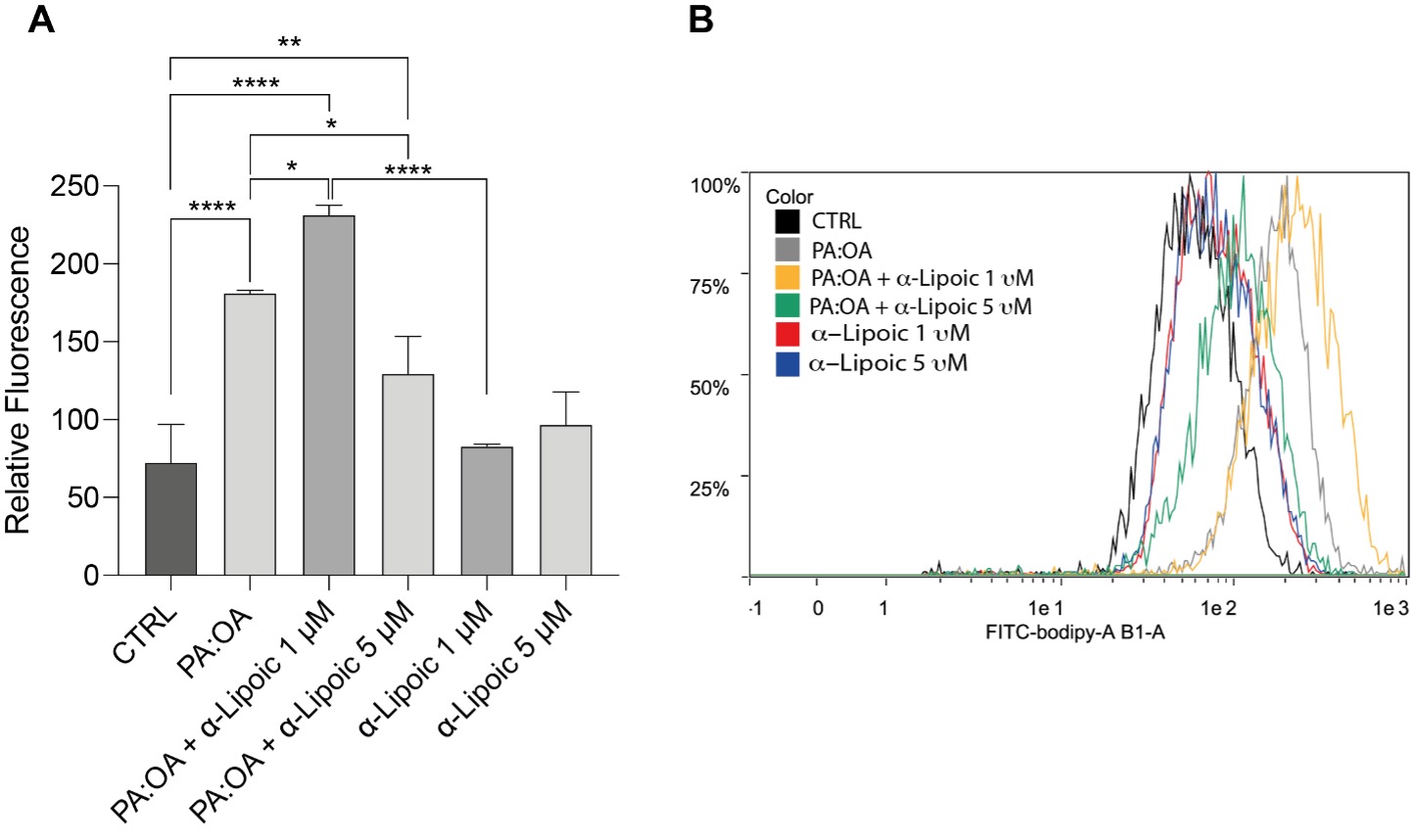 Fig. 1.
Fig. 1.Lipid droplets are reduced by alpha-lipoic acid (ALA). (A)
Relative fluorescence of Fluorinated Boron-Dipyrromethene (BODIPY) staining in
HepG2 cells treated with palmitic acid:oleic acid (PA:OA) and/or ALA. (B) Representative histogram of BODIPY
staining. Data are presented as mean
To analyze fatty acid profiles in cell content, we performed HPLC to measure the concentration of different fatty acids. Our results show that palmitic acid was increased in PA:OA-Treated HepG2 cells, confirming the accumulation of exogen addition while both ALA concentrations were able to decrease PA after PA:OA treatment. Interestingly, Fig. 2 shows that oleic acid induces no significant differences in PA:OA cells with respect to control treatment. Moreover, ALA at both concentrations reduced OA cell content after PA:OA treatment. In our investigation we also analyzed the cell content of linoleic acid and arachidonic acid (Fig. 2C,D). Our results showed that, increasing linoleic acid concentrations were found in PA:OA treated HepG2 cells while only 1 µm of ALA was able to decrease this accumulation with respect to the control treatment. Moreover, both doses of ALA alone were able to increase the content of this fatty acid. Inversely, the concentration of arachidonic acid decreased in all treatments with only 1 µm ALA showing no significant change with respect the control treatment.
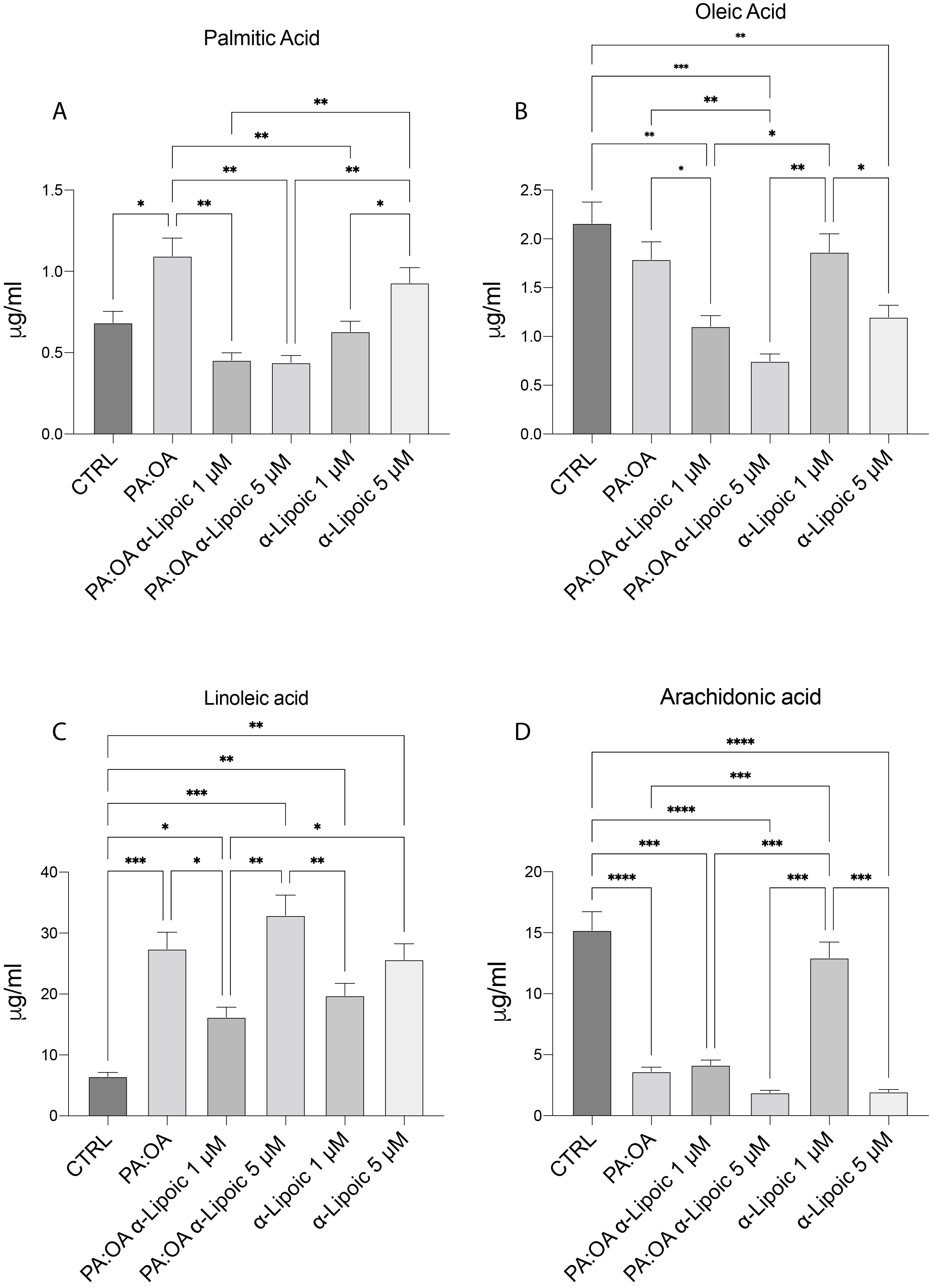 Fig. 2.
Fig. 2.PA:OA treatment in HepG2 cells modulates free fatty acid
profile. HPLC analysis of free fatty acid in HepG2 cells treated with PA:OA
and/or ALA. (A) Palmitic Acid. (B) Oleic acid. (C) Linoleic acid. (D) Arachidonic
acid. Data are presented as mean
In order to investigate the impact of cyclooxygenases on lipid profile, we analyzed the expression of COX1 and COX2 (Fig. 3A,B). Both genes were overexpressed in the treatment with exogenous fatty acids while both concentrations of ALA were able to reduce them to similar values to the PA:OA treatment.
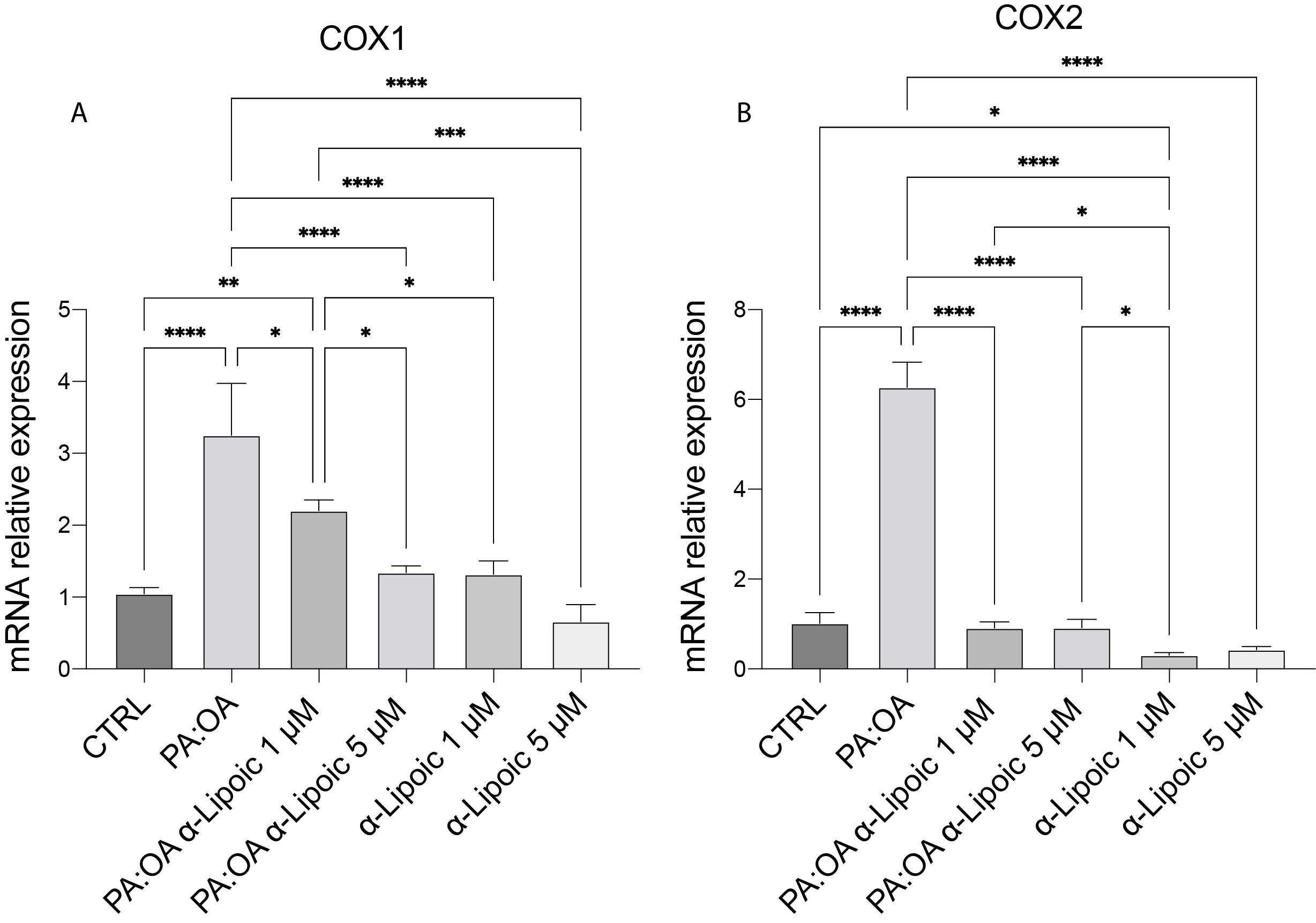 Fig. 3.
Fig. 3.The expression of cyclooxygenase genes is reduced by treatment
with ALA. (A) mRNA expression levels of COX1. (B) mRNA expression levels of
COX2. Data are presented as mean
In order to investigate cholesterol biosynthesis, we analyzed the gene expression of its enzyme pathway. Fig. 4 shows gene expression of SREBF2, HMGCR, DHCR7 and CYP51A1. Our data show that all these genes were significantly increased after PA:OA treatment and that both doses of ALA were able to decrease this gene pathway. In accordance with a previous study, we also investigated the expression of PCSK9 and low-density lipoprotein (LDL) receptor genes (Fig. 4E,F). We found PCSK9 and LDLR both increased after PA:OA treatment while both doses of ALA decreased their expression. ALA treatments alone decreased PCSK9 gene expression, confirming its role as a systemic hypolipidemic agent.
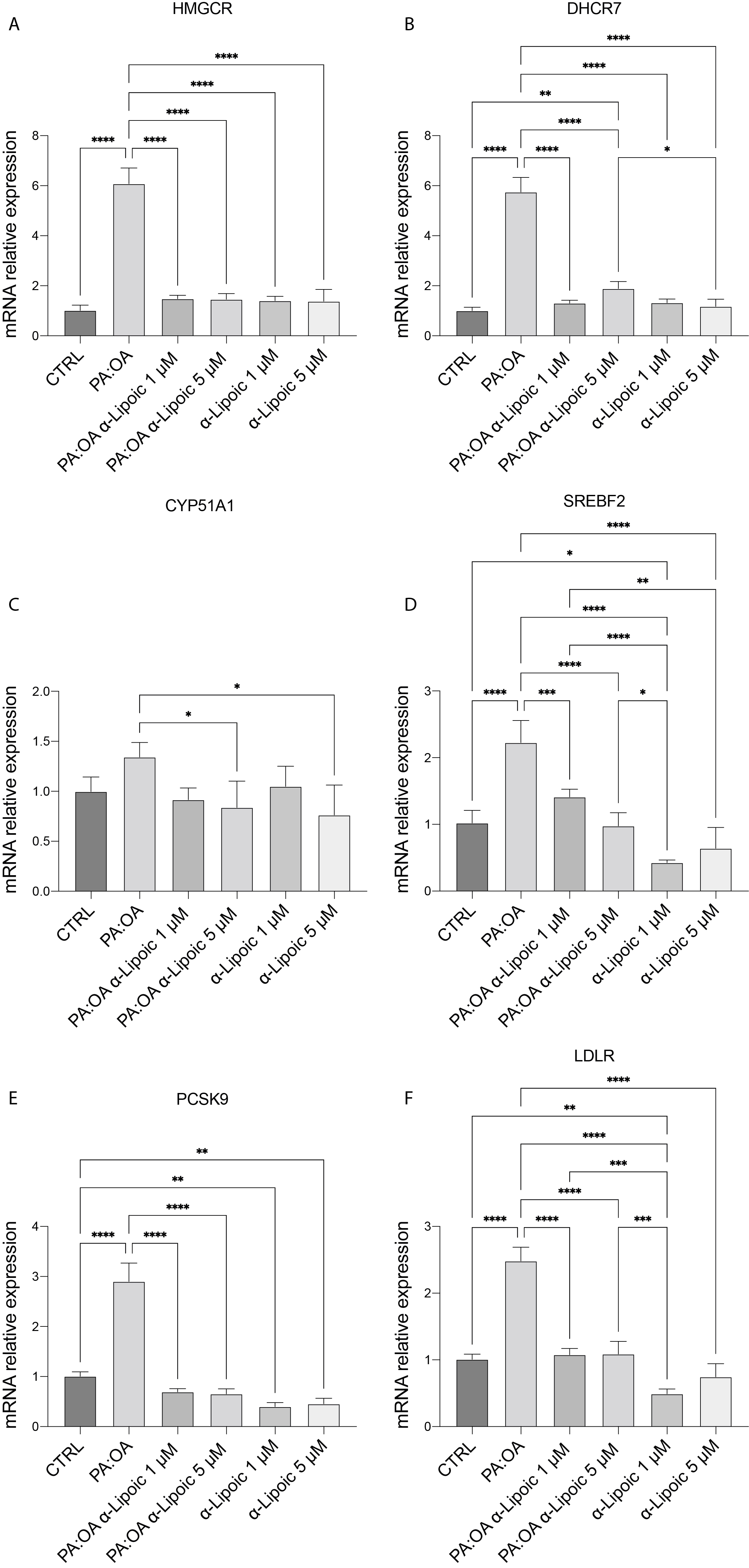 Fig. 4.
Fig. 4.The cholesterol biosynthesis and LDL intake were reduced by ALA
treatment. (A) mRNA expression levels of HMGCR. (B) mRNA expression levels of
DHCR7. (C) mRNA expression levels of CYP51A1. (D) mRNA expression levels of
SREBF2. (E) mRNA expression levels of PCSK9. (F) mRNA expression levels of LDLR.
Data are presented as mean
As shown in Fig. 5, PPARa and PPARd gene expression were increased in PA:OA-treated HepG2 cells while both doses of ALA were able to restore this gene’s overexpression to control values (Fig. 5A,B).
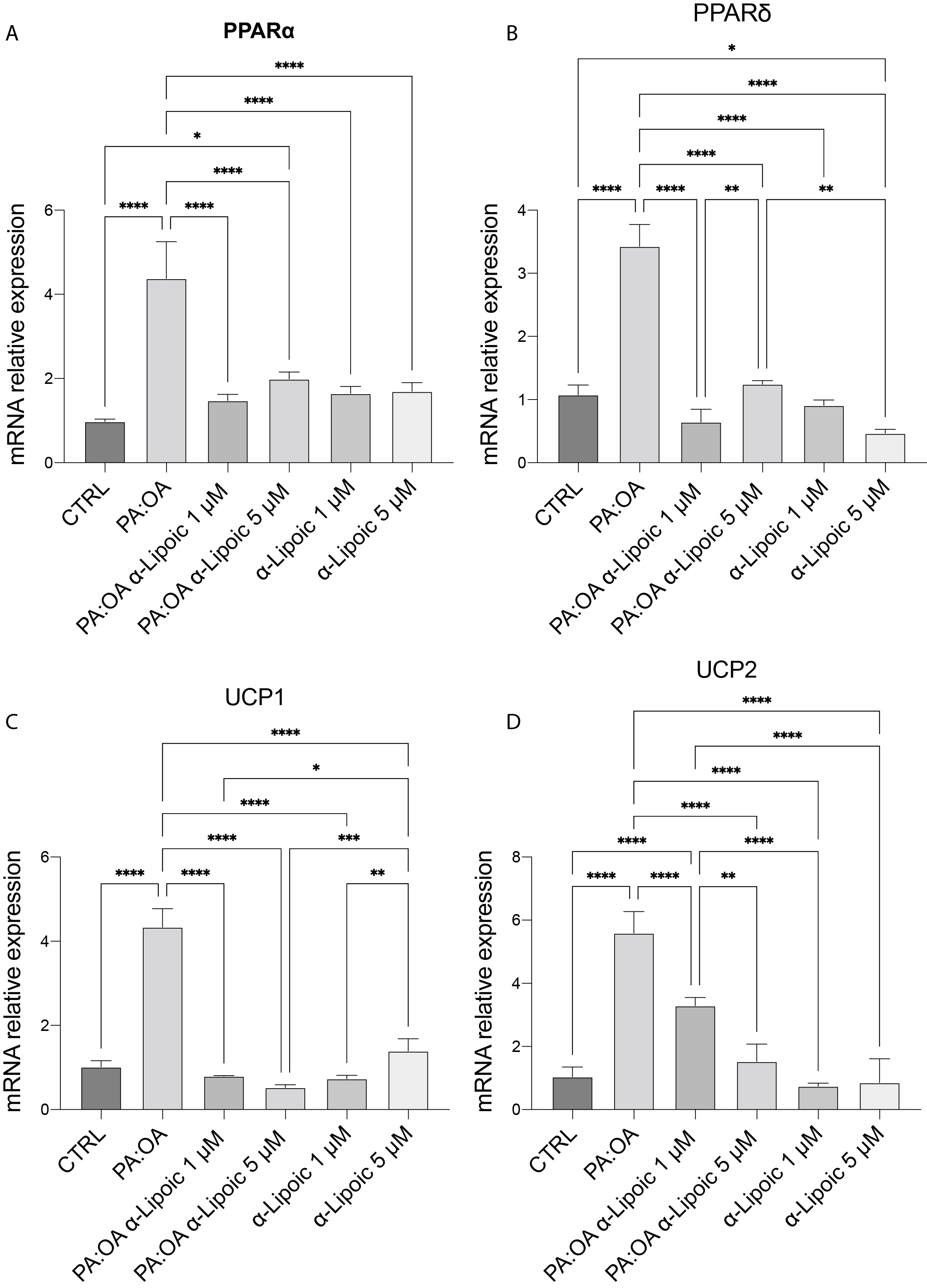 Fig. 5.
Fig. 5.PPARs and thermogenesis genes were induced by PA:OA treatment.
(A) mRNA expression levels of PPAR
Furthermore, in order to investigate the thermogenic pathway that dissipates excess energy, we analyzed the expression of uncoupling proteins (UCPs) 1 and 2. Both UCP genes were overexpressed in the treatment with exogenous fatty acids while both concentrations of ALA were able to reduce it to similar values to the control treatment (Fig. 5C,D).
NAFLD is a persistent disorder associated with liver damage and closely linked
to obesity, insulin resistance, and metabolic syndrome. We recently showed a
significant association between steatosis and alterations in mitochondrial
dynamics, impacting the metabolic function of liver cells and contributing to
intracellular lipid accumulation [4]. However, the direct effects of ALA on
hepatic liver metabolism have not been investigated. This study investigates the
impact of ALA on gene expression within the primary lipid regulatory pathways
using a model of hepatic steatosis induced by exogenous fatty acids [15]. In our
experimental model, we observed an accumulation of lipid droplets in cells
exposed to the PA:OA mixture. There was an increase in free palmitic acid levels,
while oleic acid concentrations decreased. This is consistent with previous
studies indicating that oleic acid induces more effective steatogenic effects and
reduces cytoplasmic oleic acid concentrations [17]. The accumulation of
cytoplasmic palmitic acid has been linked to increased apoptosis through
PPAR
Furthermore, previous studies have indicated that PPAR
To this regard, we found that ALA mitigates the overexpression of UCPs induced by PA:OA treatment. While UCPs activate thermogenesis pathways to catabolize excess fatty acids, overexpression, particularly of UCP2, may contribute to compromised mitochondrial physiology and liver diseases [25, 26]. Finally, in order to analyze the genetic pathway of cholesterol biosynthesis, our results suggest that the PA:OA model was able to upregulate the gene expression of the main target enzymes of the biosynthetic pathway such as SREBF2, HMGCR, DHCR7 and CYP51A1 and that ALA restores this upregulation confirming a regression in cholesterol biosynthesis.
Furthermore, the expression of the LDL receptor and PCSK9 genes were analyzed. LDLR mediate LDL clearance and increased LDLR expression improves LDL hepatic absorption and decreases plasma LDL. Conversely, PCSK9 functions as a chaperone, guiding LDLR to internal degradation and preventing its recycling to the cell surface [27]. Our results showed an overexpression of both genes in PA:OA treated cells and a decrease mediated by ALA treatment. Elevated HMGCR activity and PCSK9 are associated with diminished LDLR expression and LDL uptake in the liver. Although the expected expression of PCSK9 and LDLR should be inversely proportional, the increase in LDLR could be the effect mediated by PPARs which allows the influx of fatty acids into the hepatocyte or a secondary mechanism affected by the consequence of feedback.
Our data showed other free fatty acid profiles present in the cytoplasm such as linoleic acid and arachidonic acid. Both fatty acids are responsible for activating the prostaglandin pathway through the action of cyclooxygenases. PA:OA treatment highlighted an accumulation of linoleic acid while arachidonic acid concentration was significantly downregulated compared to control cells. Interestingly, 1 µm ALA showed a reduction in free linoleic acid concentration. Therefore, in order to investigate cyclooxygenases, we measured the gene expression of COX1 and COX2. Both genes were overexpressed following PA:PO treatment and ALA administration significantly downregulated their expression. To this regard, COX-1 and COX-2 are heme proteins, and such a prosthetic group is crucial for the expression of catalytic activity [28]. Recent studies demonstrated that ALA reduces lipotoxicity and induces the heme oxygenase system, the enzymes responsible for regulating intracellular heme content, in acute lung injury and an in vitro model of steatosis [29, 30]. Consistently with this hypothesis, previous reports showed that prostaglandin synthesis reduces in response to treatments that upregulate the expression of heme oxygenase (HO)-1 [31, 32]. Our results reveal that ALA acts by inhibiting cyclooxygenases, playing a role in the inflammation exhibited in hepatic steatosis.
In conclusion, our results demonstrated a direct effect of ALA on hepatic liver metabolism and lipid profile that combined with the other systemic effects of improved insulin resistance, oxidative stress and systemic inflammation may represent a strong rationale for the use of ALA in metabolic syndrome treatment or NAFLD. Since ALA is commercially available for several clinical purposes, its use could be easily and highly recommended for patients with NAFLD.
The datasets used and/or analysed in this study are reported within the manuscript and are available from the corresponding authors.
LL, DT, TZ, GLV and IAB designed the research study. LL, ELS, TZ, GZ and SR performed the research. AN, AMA, AS, ET and SR contributed to formal analysis, data interpretation and in editing the final version of the manuscript. LL, FMS, MA, WC and FG analyzed the data. GLV and IAB wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank all the present and past members of the Giovanni Li Volti’s lab for their technical support.
This research was funded by the researchers supporting project number (RSP2024R261, to AMA) King Saud University, Riyadh, Saudi Arabia. This work was supported by Piano di Incentivi per la Ricerca di Ateneo 2020-2022, Linea di Intervento 2, “IMYTRA” to GLV.
Given their role as Guest Editor, GLV had no involvement in the peer-review of this article and have no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Margaret O. James. The authors declare no other conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.