

1 School of Systems Biomedical Science and Integrative Institute of Basic Science, Soongsil University, 06978 Seoul, Republic of Korea
Abstract
Accurate gene expression is fundamental for sustaining life, enabling adaptive
responses to routine tasks and management of urgent cellular environments. RNA
polymerases (RNAP I, RNAP II, and RNAP III) and ribosomal proteins (RPs) play
pivotal roles in the precise synthesis of proteins from DNA sequences. In this
review, we briefly examined the structure and function of their constituent
proteins and explored to characterize these proteins and the genes encoding them,
particularly in terms of their expression quantitative trait loci (eQTL)
associated with complex human traits. We gathered a comprehensive set of 4007
genome-wide association study (GWAS) signal–eQTL pairs, aligning GWAS Catalog
signals with eQTLs across various tissues for the genes involved. These pairs
spanned 16 experimental factor ontology (EFO) parent terms defined in European
Bioinformatics Institute (EBI). A substantial majority (83.4%) of the pairs were
attributed to the genes encoding RPs, especially RPS26 (32.9%). This large
proportion was consistent across all tissues (15.5~81.9%),
underscoring its extensive impact on complex human traits. Notably, these
proportions of EFO terms differed significantly (p
Keywords
- expression quantitative trait loci
- GWAS signal
- ribosomal protein
- RNA polymerase
Gene expression is the most fundamental process in creating human traits from genetic information. Transcription involves copying DNA into RNA molecules by RNA polymerases (RNAPs), and translation decodes mRNA molecules into polypeptides in ribosomes. In transcription process, RNAPs can synthesize a variety of RNA molecules in the nucleus, including mRNA for protein synthesis as well as non-coding RNA (ncRNA) that function as RNA molecules itself. Eukaryotic RNAPs undertake specialized roles in transcribing nonoverlapping gene groups. RNAP I synthesizes 5.8S, 18S, and 28S rRNAs, RNAP II synthesizes mRNAs, long noncoding RNAs (lncRNAs), and small nuclear RNAs (snRNAs), and RNAP III synthesizes 5S rRNAs and tRNAs. Given their critical roles, the defects in the biogenesis of RNAPs can disrupt the essential cellular processes, resulting in impaired growth, development, and cell death [1, 2]. They ultimately lead to severe diseases such as ribosomopathies [3], developmental disorders [4], and cancers [5].
Then, mRNAs are translated into polypeptide chains in ribosomes, a ribonucleoprotein complex consisted of ribosomal proteins (RPs) and the 5S, 5.8S, 18S, and 28S rRNAs [6]. Given the high energy consumption during the translation from nucleic acid sequences to amino acid sequences, it is subject to stringent controls. Translational control allows for more efficient adaptive responses to fluctuations in cellular environments, compared to upstream gene expression processes. For instance, the gene expression of RPs can be dynamically regulated by instantaneous translation in response to urgent cellular conditions [7]. In contrast, RP mRNAs undergo excessive production through transcriptional regulation, are stored as inactive messenger ribonucleoprotein particles, and are poised for immediate translation [8]. Dysregulation in translation can lead to abnormal proliferation, cell survival, and immune response, consequently contributing to the development of cancers.
The role of RNAPs and ribosomes in human longevity has been underscored by a Mendelian randomization study [9]. Over time, significant research efforts have been dedicated to examining the subunits of RNAPs and ribosomes. The perspective on ribosomes has evolved from being viewed as passive and indiscriminate structures to dynamic macromolecular complexes with specialized cellular functions. As the functions of ribosome subunits have been uncovered, we illuminate the intricate roles of individual genes and their expression regulations [10]. In this article, we briefly review the structure and function of constituent proteins of human RNAP I, RNAP II, RNAP III, and ribosomes as key players for gene expression processes. We explore the characterization of these proteins and their corresponding genes, focusing particularly on their expression quantitative trait loci (eQTL) identified for association with complex human traits. Fundamental terminology for this review is summarized in Table 1.
| Term |
Explanation |
| Expression quantitative trait locus (eQLT) | A genomic locus that explains partial genetic variability in gene expression. |
| Expression gene (eGene) | Target gene of eQTL. |
| Genome-wide association study (GWAS) | A genetic investigation to identify genetic nucleotide sequence variants associated with complex traits or diseases across the entire genome in population(s). This results in GWAS signals as independent genomic loci with significant association. |
| Ribosome | A cellular organelle responsible for translation, decoding mRNA sequences into amino acid sequences. It consists of ribosomal proteins, structural parts involved in ribosome assembly, and rRNAs, ribozymes involved in catalytic processes. |
| RNA polymerase (RNAP) | An enzyme for synthesizing RNA from a DNA template during transcription. In eukaryotes, distinct RNAPs transcribe specific types of RNAs. RNAP I transcribes 45S rRNA, RNAP II transcribes mRNAs, long noncoding RNAs, and small nuclear RNAs, and RNAP III transcribes 5S rRNAs and tRNAs. |
| Transcription | The process of copying DNA into RNA as the initial step in gene expression. |
| Translation | The process of synthesizing proteins from mRNA molecules after transcription and RNA processing. This intricate process is carried out by the translation machinery, primarily composed of ribosomes, tRNAs, and various translation factors. |
| Ribonucleic acid (RNA) | A molecule essential for various biological processes such as protein synthesis and gene regulation. RNA is classified into two types: coding RNA, which includes mRNA involved in protein synthesis, and non-coding RNA, which does not encode proteins but plays crucial roles in various cellular processes, including gene regulation. |
| Kozak sequence | A conserved sequence motif surrounding the start codon in eukaryotic mRNA that facilitates efficient translation initiation. |
| Complex trait | Phenotypes characterized by influences from multiple genetic and environmental factors, often displaying a continuous distribution within a population rather than adhering to simple Mendelian inheritance patterns. |
| Experimental Factor Ontology (EFO) | A structured vocabulary and ontology designed to describe experimental variables in biological and biomedical research, which is available in European Bioinformatics Institute (EBI) databases. |
| Bonferroni correction | An adjustment for multiple testing, typically applied by dividing the significance threshold by the number of independent tests conducted. |
| Mendelian randomization | A method employing genetic variants as instrumental variables to infer causality between an exposure and an outcome in observational studies. |
| Genetic factor | A hereditary component, such as a gene or allele, that influences an individual’s characteristics or predisposition to diseases. |
This section deals with the structure and function of RNAPs in
Saccharomyces cerevisiae, better known as baker’s yeast, which is
considered to be representative of eukaryotes and has contributed the most to our
knowledge. A caution is, however, warranted with the names of human genes in
later sections of this review because the nomenclature of many polymerase genes
and proteins within and between yeast and humans is unusual and often confusing.
In eukaryotes, the three RNAPs, RNAP I, RNAP II, and RNAP III, are complex
enzymes composed of multiple subunits that contain core and common subunits. It
is conceivable that common regulators controlling the levels of these shared
subunits effectively coordinate the functional levels of the three RNAPs. In
particular, the five out of the shared components, corresponding to prokaryotic
core RNAP composed of catalytic (
| Structural Classification |
RNAP II | RNAP I | RNAP III | Bacterial RNAP |
Function | |||||||||
| Yeast | Human | Yeast | Human | Yeast | Human | |||||||||
| Protein | Gene | Protein | Gene | Protein | Gene | Protein | Gene | Protein | Gene | Protein | Gene | |||
| Core and common subunits | Rpb3 | RPB3 | RPB3 | POLR2C | AC40 | RPC40 | RPAC1 | POLR1C | AC40 | RPC40 | RPC40 | RPAC1 | assembly | |
| Rpb11 | RPB11 | RPB11 | POLR2J | AC19 | RPC19 | RPAC2 | POLR1D | AC19 | RPC19 | RPC19 | RPAC2 | assembly | ||
| Rpb2 | RPB2 | RPB2 | POLR2B | A135 | RPA135 | RPA2 | POLR1B | A128 | RPC128 | RPC2 | POLR3B | catalysis | ||
| Rpb1 | RPO21 | RPB1 | POLR2A | A190 | RPA190 | RPA1 | POLR1A | A160 | RPC160 | RPC1 | POLR2A | catalysis | ||
| Rpb6 | RPO26 | RPABC2 | POLR2F | Rpb6 | RPO26 | RPABC2 | POLR2F | Rpb6 | RPO26 | RPABC2 | POLR2F | ω | auxiliary | |
| Rpb5 | RPB5 | RPABC1 | POLR2E | Rpb5 | RPB5 | RPABC1 | POLR2E | Rpb5 | RPB5 | RPABC1 | POLR2E | |||
| Rpb8 | RPB8 | RPABC3 | POLR2H | Rpb8 | RPB8 | RPABC3 | POLR2H | Rpb8 | RPB8 | RPABC3 | POLR2H | |||
| Rpb10 | RPB10 | RPABC5 | POLR2L | Rpb10 | RPB10 | RPABC5 | POLR2L | Rpb10 | RPB10 | RPABC5 | POLR2L | |||
| Rpb12 | RPC10 | RPABC4 | POLR2K | Rpb12 | RPC10 | RPABC4 | POLR2K | Rpb12 | RPC10 | RPABC4 | POLR2K | |||
| Rpb9 | RPB9 | RPB9 | POLR2I | A12 |
RPA12 | RPA12 | POLR1H | C11 |
RPC11 | RPC10 | POLR3K | proofreading | ||
| Dissociable subunits | Rpb4 | RPB4 | RPB4 | POLR2D | A14 | RPA14 | - | - | C17 | RPC17 | RPC9 | CRCP | formation | |
| Rpb7 | RPB7 | RPB7 | POLR2G | A43 | RPA43 | RPA43 | POLR1F | C25 | RPC25 | RPC8 | POLR3H | formation | ||
| Independent subunits | TFIIS | DST1 | TFIIS | TCEA1 |
A12 |
RPA12 | RPA12 | POLR1H | C11 |
RPC11 | RPC10 | POLR3K | proofreading | |
| TCEA3 |
||||||||||||||
| TFIIF |
TFG1 | TFIIF |
GTF2F1 | A49 | RPA49 | RPA49 | POLR1E | C37 | RPC37 | RPC5 | POLR3E | stabilization | ||
| TFIIF |
TFG2 | TFIIF |
GTF2F2 | A34 | RPA34 | RPA34 | POLR1G | C53 | RPC53 | RPC4 | POLR3D | stabilization | ||
| TFIIF-TFIIE | TFA1 TFA2 |
TFIIF-TFIIE | GTF2E1 GTF2E2 |
- | - | - | - | C82/34/31 | RPC83 | RPC3/6/7 |
POLR3C | stabilization | ||
| RPC34 | POLR3F | |||||||||||||
| RPC31 | POLR3G |
|||||||||||||
These polymerases, however, also have distinct transcription modes evident from
their different structures of other subunits. RNAP II, which targets all
protein-coding genes, interacts with a broader array of regulatory factors,
compared to RNAP I and RNAP III. Structurally, RNAP II has fewer permanent
subunits but incorporates dissociable subunits (Rpb4 and Rpb7) and independent
initiation and elongation factors (TFIIS, TFIIF, and TFIIE) as presented in Table 2. The dissociable subunits play crucial roles as a heterodimeric stalk (Rpb4/7)
in elongation process. Furthermore, RNAP II has the capacity to execute a
suitable termination mechanism by capturing dissociated RPB3, which plays a
predominant role in regulating the 3
The intrinsic subunits of RNAP I and RNAP III help conduct speedy elongation. An example is the intrinsic dimer C37/53 of RNAP III, specifically essential along with C11 for the highly efficient termination and coupled reinitiation processes in facilitating transcription of very short genes [19]. The intrinsic subunit A12 of RNAP I plays a pivotal role in RNA cleavage, facilitating proofreading, and enabling a swift resumption of elongation following a pause [16]. The intricacies involved in the resumption of elongation after pausing in RNAP II imply a necessity for specific regulatory mechanisms. This level of regulation is not essential for the comparatively simpler and faster elongation processes observed in RNAP I and RNAP III [20].
Eukaryotic ribosome, consisting of the small (40S) and large (60S) subunits, is a complex macromolecular machine that orchestrates the translation and protein synthesis as the heart of the translation machinery by collaborating with other translational apparatus molecules such as transfer RNAs (tRNAs) and translation factors. The small subunit composed of a 18S ribosomal RNA (rRNA) and 33 RPs is bound to mRNA for decoding, and the large subunit composed of a 5S rRNA, a 28S rRNA, a 5.8S rRNA, and 49 RPs is bound to aminoacyl tRNA for catalysis. The translation activity requires ribosome biogenesis through elaborate coordination of RNAP I, II, III, and over 200 ribosome assembly factors. This highly complex process inevitably requires strict regulation and smooth communication with other cellular pathways [21]. Changes in ribosome biogenesis may impact the translation process directly, influencing global gene expression during cell growth [22], differentiation [23], and disease progression (e.g., cancer metastasis potential [24]).
RPs play an inevitable role in a wide range of ribosome biogenesis, assembly, and translation as shown in Table 3 (Ref. [10, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49]). For instance, RPL33 is responsible for ribosomal-subunit joining and ribosome biogenesis. Its missense mutation (rpl33a-G76R) alters the 60S subunit, impeding ribosomal-subunit joining. This represses translation of a master transcription factor GCN4 in yeast, corresponding to ATF4 in human, and impairs the efficient processing of 35S and 27S pre-rRNAs, decreasing all four mature rRNAs responsible for the biogenesis of both ribosomal subunits [25]. RP59 is necessary for the assembly of 40S subunit, and RPL1 and RPL16 combine with 5S rRNA to produce stabilized ribonucleoprotein, which is necessary for the assembly of 60S subunit [26, 27]. RPL12 is responsible for mediating the accurate assembly of the ribosomal stalk [28]. Some RPs (RPS0, RPS14, and RPS21) participate in the cytoplasmic rRNA processing steps for the maturation of 18S rRNA [29, 30]. RPS14 is further involved in the maturation of 43S pre-ribosomes [29]. In addition, RPL25 is essential for pre-rRNA processing [31], RPL9 is crucial for the maturation of small subunit [32], and RPS15 plays a vital role in the nuclear exit of the 40S subunit precursors [50].
| Subunit | Location | RP |
Function |
| Large subunit | Peptidyl transferase center | RPL27a | Maintenance of the stability of E site [39] |
| tRNA binding pocket | RPL10 | Regulation of nuclear exports from 60S subunit [46] | |
| Polypeptide exit tunnel | RPL35 | Recognition of peptide and insertion to the translocation channel [41] | |
| Polypeptide exit tunnel | RPL39 | Maintenance of translation accuracy [47] | |
| Polypeptide exit tunnel | RPL23 | Chaperone-assisted protein folding [40] | |
| Guanosine triphosphate hydrolase center | RPL12 | Assembly of ribosomal stalk [28] | |
| RPL1 | Maintenance of the stability of 5S rRNA and assembly of 60S subunits [27] | ||
| RPL3 | Regulation of peptidyltransferase activity and translation fidelity [43] | ||
| RPL5 | Regulation of anchoring peptidyl-tRNA to the P site [45] | ||
| RPL9 | Maturation of the small subunit [32] | ||
| RPL16 | Assembly of 60S subunits [26] | ||
| RPL24 | Regulation of polyphenylalanine synthesis through P site binding [47] | ||
| RPL25 | Pre-rRNA processing [31] | ||
| RPL33 | 35S and 27S pre-rRNAs processing [25] | ||
| RPL36a | Contacting with the 3′-end of deacylated tRNA at P site [38] | ||
| RPL41 | Regulation of peptidyltransferase activity [44] | ||
| Small subunit | Decoding center | RPS15 | Accommodation of aminoacyl-tRNA at A site [37] |
| mRNA entry tunnel | RPS5 | Regulation of translation accuracy [48] | |
| mRNA entry tunnel | RPS12 | Regulation of translation accuracy [48] | |
| mRNA entry tunnel | RPS3 | Start codon recognition and ribosome-based mRNA quality control [33] | |
| mRNA exit tunnel | RPS14 | Maturation of 43S preribosome [29] | |
| mRNA exit tunnel | RPS26 | Interaction with initiation factors and recognition of the Kozak sequence [10, 34, 35] | |
| mRNA exit tunnel | RPS28 | Maintenance of translation accuracy [42] | |
| RP59 | Assembly of the 40S subunit [26] | ||
| RPS0 | rRNA processing and maturation of 18S rRNA [30] | ||
| RPS4 | Maintenance of translation accuracy [42, 48] | ||
| RPS9 | Maintenance of translation accuracy [49] | ||
| RPS20 | Regulation of mRNA binding and subunit docking [36] | ||
| RPS21 | Maturation of the 3’ end of 18S rRNA [30] |
For translation function, RPS3 conducts a vital regulation at the preliminary
translational initiation: the conserved residues R116/117 within RPS3 stabilize
interactions between the ribosome and mRNA at the mRNA entry pore, R146 and K148
contribute to the accuracy of start codon selection; K62 functions ribosome-based
mRNA quality control, and residues at 60 to 63 ensure the proper structure of the
48S preinitiation complex [33]. RPS26 promotes a selective translation of
specific mRNAs under specific cellular environment by recognizing Kozak sequence
and interacting with initiation factors eIF3a and eIF3d [10, 34, 35]. In
particular, RPS26 is a detachable component for fine regulation of mRNAs largely
with Kozak sequence according to specific situation [51]. RPS20 is responsible
for the mRNA binding and subunit docking, and its deletion reduced mRNA binding
and decreased 70S complexes, leading to initiation defects to the small subunit
[36]. RPS15 located in the decoding center plays a pivotal role in accommodating
aminoacyl-tRNA at the A site. A high-resolution cryoelectron microscopy captured
the C-terminal tail of RPS15, interacting with the tRNAs located in A- and
P-sites in the decoding center [37]. RPL36a plays a critical role in the
elongation of peptide chains by interacting with the 3
RPL23 plays a regulatory role in protein biosynthesis and chaperone-assisted protein folding as a chaperone docking site [40], and RPL23a along with RPL35 also showed an important role during signal peptide recognition and insertion of the peptide into the translocation channel by repositioning a signal recognition particle (SRP54) [41]. In addition, RPS4, RPS13, and RPS28 contribute to translational accuracy [42], and RPL3, RPL5, and RPL41 contribute to peptidyltransferase activity [43, 44, 45]. In yeast, RPL5 further plays an important role in anchoring peptidyl-tRNA to the P-site [45]. RPS4 and RPS5 are essential for preserving the accuracy of protein translation [52]. In contrast, RPS12 enhances the translation rate at the expense of a higher error rate in protein synthesis [53].
Regulatory signals targeting genes encoding RNAPs and ribosomes were collected
from Genotype-Tissue Expression (GTEx) release v8 data
(https://gtexportal.org/home/downloads/adult-gtex/qtl). These eQTL data resulted
from various tissues, including adipose (omentum visceral adipose; the GTEx term
is in parenthesis), brain (brain cortex), colon (transverse colon), liver, muscle
(skeletal muscle), pancreas, pituitary, and small intestine (small intestine
terminal ileum). For each tissue, the file
[Tissue_name].signif_variant_gene_pairs.txt.gz was downloaded from
GTEx_Analysis_v8_eQTL.tar [54]. The eQTLs were all cis-acting,
discovered (false discovery rate (FDR)
Genes encoding RNAPs/ribosomes were selected based on the gene groups curated by the HUGO Gene Nomenclature Committee (HGNC): RNAP I, RNAP II, RNAP III (ID: 726) and ribosomes (large subunit ID: 728; small subunit ID: 729) [55]. After filtering out genes without any cis-eQTL, a total of 98 genes were examined as cis-eGenes for the RNAPs and ribosomes.
All genome-wide association study (GWAS) signals associated with human diseases
and traits were collected from the NHGRI-EBI GWAS Catalog (v1.0.2; accessed on
May 1, 2022) [56]. These signals exhibited at least suggestive association with
significance threshold of p-value
GWAS signals corresponding to eQTLs for the eGenes of the RNAPs/ribosomes were selected as GWAS–eQTL pairs based on their dbSNP IDs and genomic locations. This selection did not require individual-level or complete summary statistics, making it more practical for broad insights into all GWAS signals. The GWAS–eQTL pair indicates a genetic variant that regulate gene expression for RNAPs/ribosomes and simultaneously influence complex human traits/diseases, and thus frequent GWAS–eQTL pairs imply an importance of gene groups (e.g., ribosomal genes) for complex human traits/diseases. The figures in this review present the numbers of GWAS–eQTL pairs categorized by tissues, gene groups of the RNAPs/ribosomes, and/or EFO terms.
Equivalence tests were employed to identify differences in the proportion of each EFO term corresponding to the selected GWAS signals between a population and its parental population. For instance, the proportion of each EFO term for the eQTLs associated with eGenes encoding RPs could be compared to that of all the eQTLs associated with the eGenes encoding RNAPs/ribosomes as the parental population. Multiple testing correction was applied to the equivalence test using the Bonferroni correction with a significance threshold of 0.05 divided by the number of EFO terms.
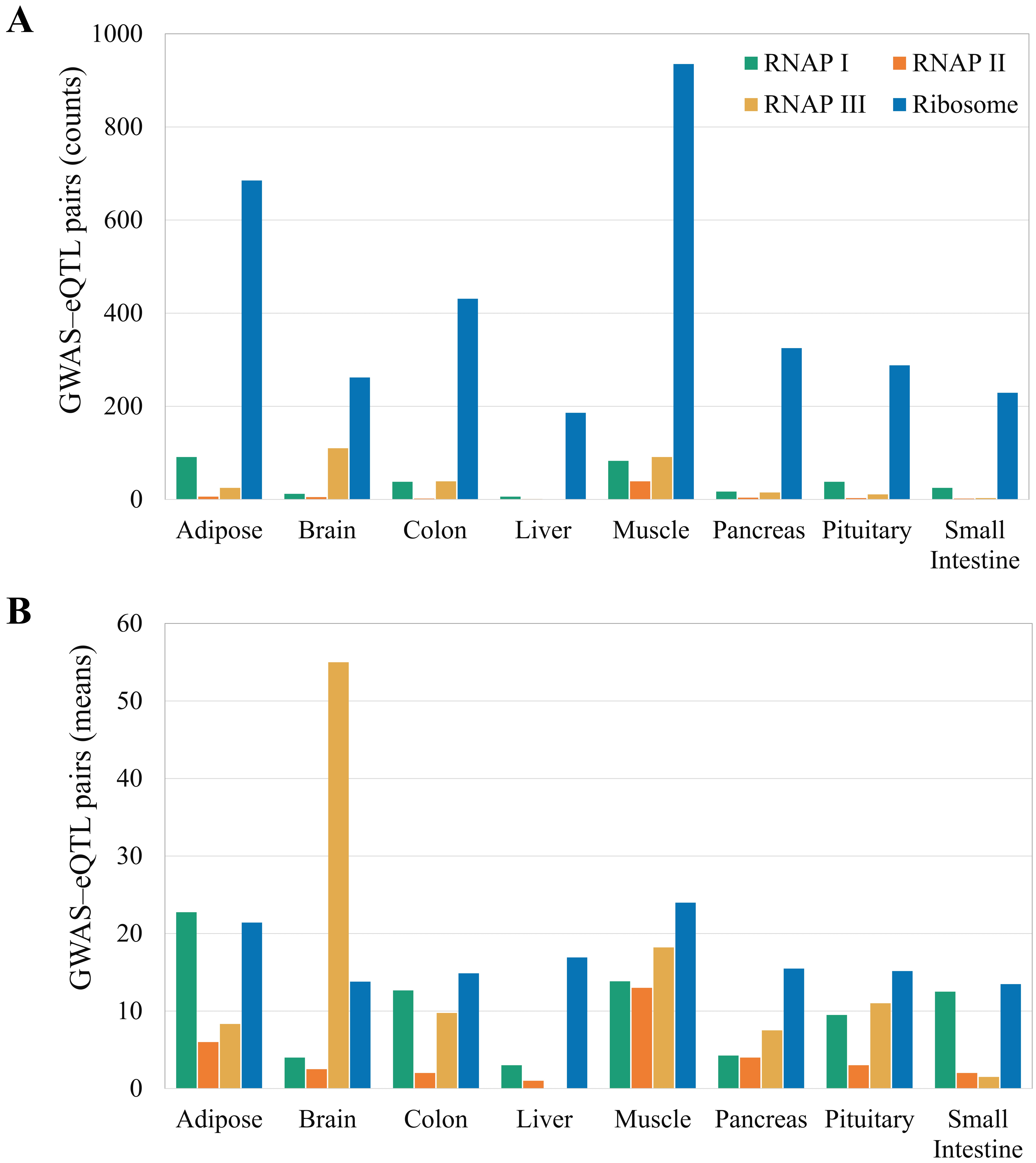
We collected 4007 pairs of GWAS signals and eQTLs (GWAS–eQTL pairs), where GWAS signals corresponded to any eQTL across eight tissues for the eGenes of RNAPs and ribosomes (Supplementary Table 1). These GWAS–eQTL pairs were discovered with 62 eGenes: 6 encoding RNAP I, 4 encoding RNAP II, 6 encoding RNAP III, and 46 encoding RP. Across tissues, the number of eGenes ranged from 12 in the liver to 53 in muscle. The highest number of identified GWAS signals (843) was observed in muscle, while the lowest (157) was in the liver.
The GWAS–eQTL pairs were most abundant for RPs and least for RNAP II across all
tissues (Fig. 1A). When considering the number of pairs per eGene, the
differences between subtypes decreased. Nevertheless, RP still exhibited the
highest number of pairs, except for brain and adipose tissues. Notably, in the
brain, RNAP III had approximately four times as many pairs per eGene as RNAP II
(55.0 vs. 13.8) (Fig. 1B). The ratio of the number of pairs for RP to the total
number of eQTLs by tissue ranged from 6.3% to 14.6% across tissues,
significantly larger than the corresponding ratios for RNAPs (p
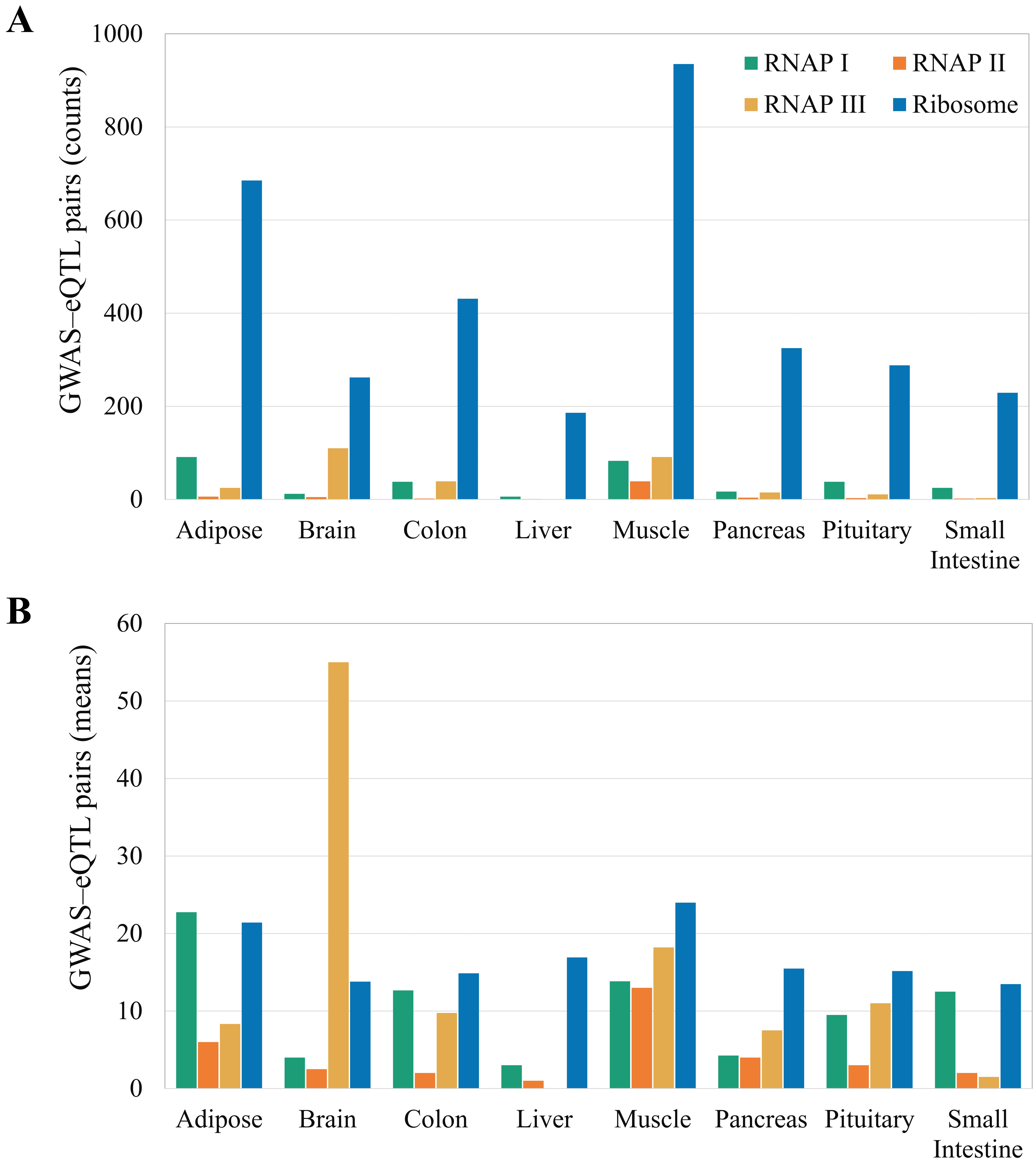 Fig. 1.
Fig. 1.Counts (A) and means (B) of GWAS–eQTL pairs by tissues. Mean was calculated as the count divided by the number of corresponding eGenes.
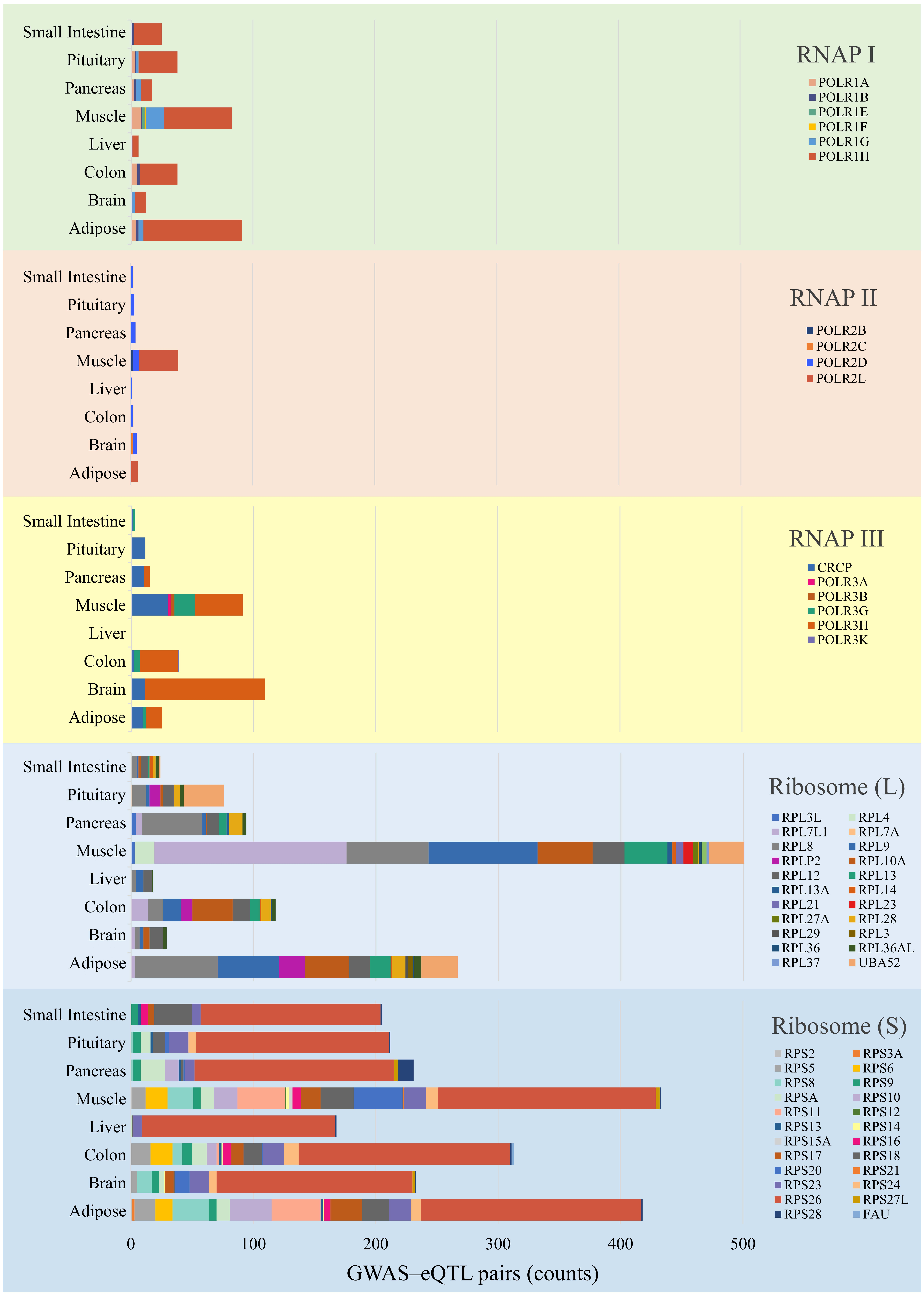
The GWAS–eQTL pairs were frequently associated with specific eGenes within RNAP I, II, III, or ribosomes, as shown in Fig. 2. For example, among RNAP I eGenes, POLR1H was dominant with 246 pairs in the eight tissues, accounting for 79.4% of the total pairs (310) associated with RNAP I eGenes. Similarly, POLR2L, POLR3H, and RPS26 had the highest frequency (61.3, 63.5, and 39.4%) in the GWAS–eQTL pairs for RNAP II, RNAP III, and RP subtypes, respectively. Notably, RPS26 exhibited a frequency high enough to be distinguished from the other eGenes.
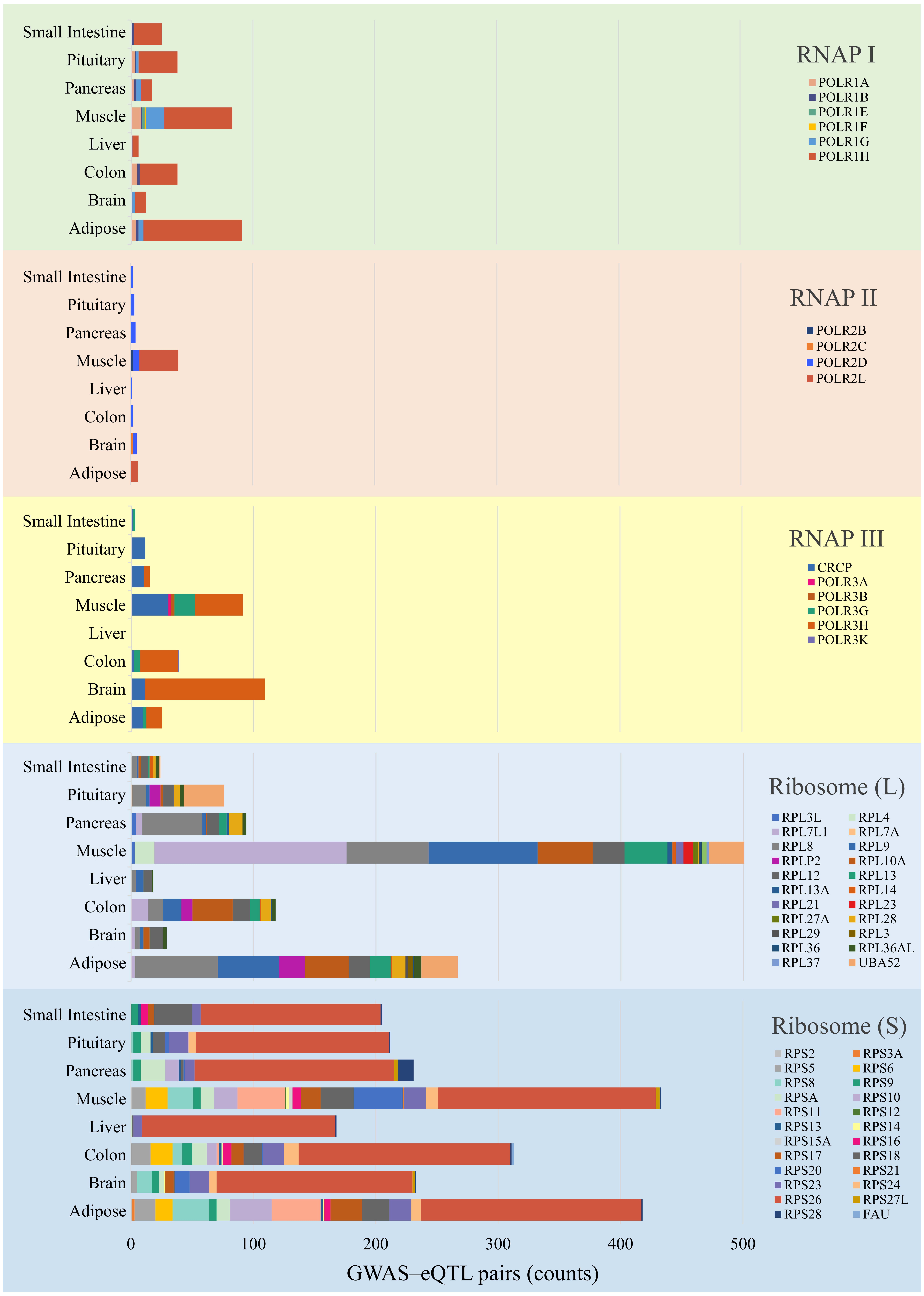 Fig. 2.
Fig. 2.Contribution of individual eGenes to the number of GWAS–eQTL pairs by tissues. The numbers of GWAS–eQTL pairs for ribosome are separately presented by small (S) and large (L) subunits.
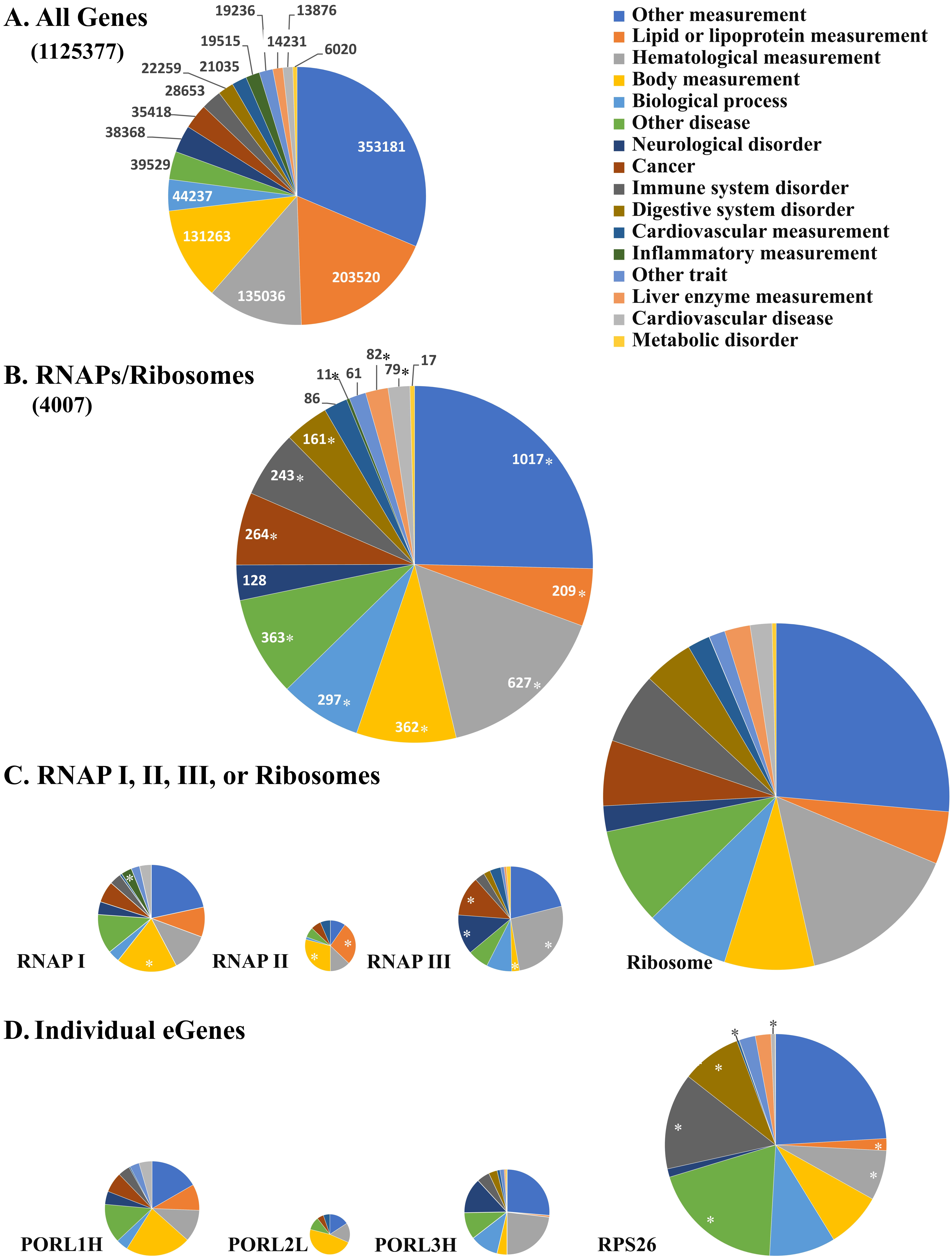
We found 512 reported phenotypes (diseases/traits) associated with eQTLs of
eGenes for RNAPs and ribosomes, mapped to 326 EFO terms and 16 parent terms.
Twelve out of 16 parent terms for eGenes for RNAPs and ribosomes had proportions
significantly different from those for all eGenes reported by GTEx (p
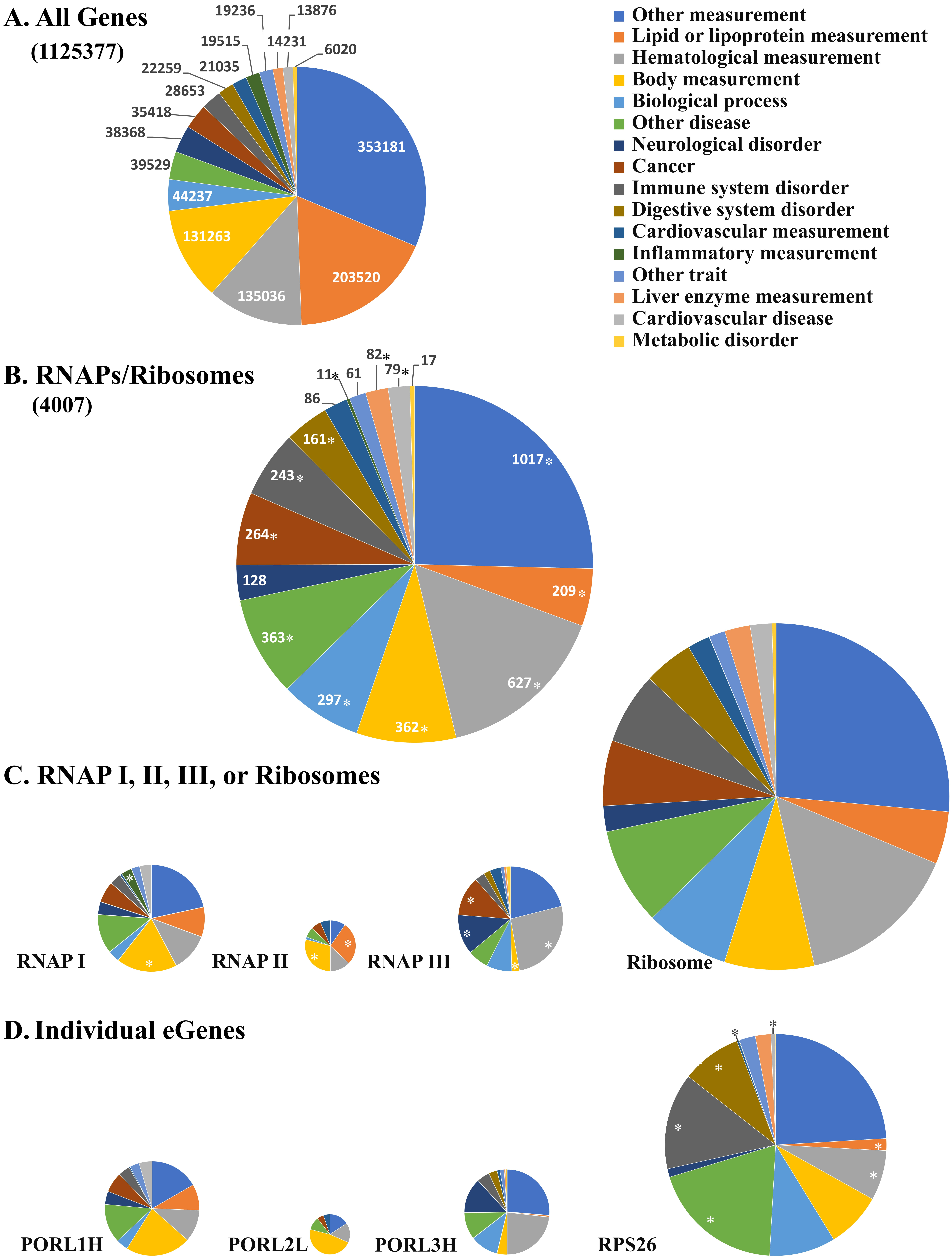 Fig. 3.
Fig. 3.Distribution of EFO parent terms for GWAS–eQTL pairs. GWAS
signal is assigned to EFO parent term according to GWAS Catalog. Proportions of
GWAS–eQTL pairs are presented for various subcategories: all eGenes available in
GTEx (A), eGenes for RNAPs/ribosomes (B), eGenes belonging to RNAP I, II, III, or
ribosomes (C), and the most frequent eGene within each of RNAP I, II, III, and
ribosomes (D). Circle size is proportional to the number of pairs across all the
pie charts except for A. Asterisk (*) in each area indicates significant difference
(p
For RNAPs/ribosomes in Fig. 3B, the three most frequent parent terms were ‘other measurement’, ‘hematological measurement’, and ‘other disease’, accounting for approximately 50% of all pairs (Fig. 3B). Their top EFO terms were ‘educational attainment’, ‘eosinophil count’, and ‘asthma’, respectively. These results were largely attributed to RNAP III and ribosome, especially two eGenes, POLR3H and RPS26, respectively.
On the other hand, RNAP I contributed the largest number of pairs (11) to the EFO term ‘white matter microstructure measurement’ within the parent term ‘other measurement’, and RNAP III contributed the largest number of pairs (7) to the EFO term ‘neuroticism measurement’. These contributions were all attributed to two eGenes, POLR1H and POLR3H, respectively.
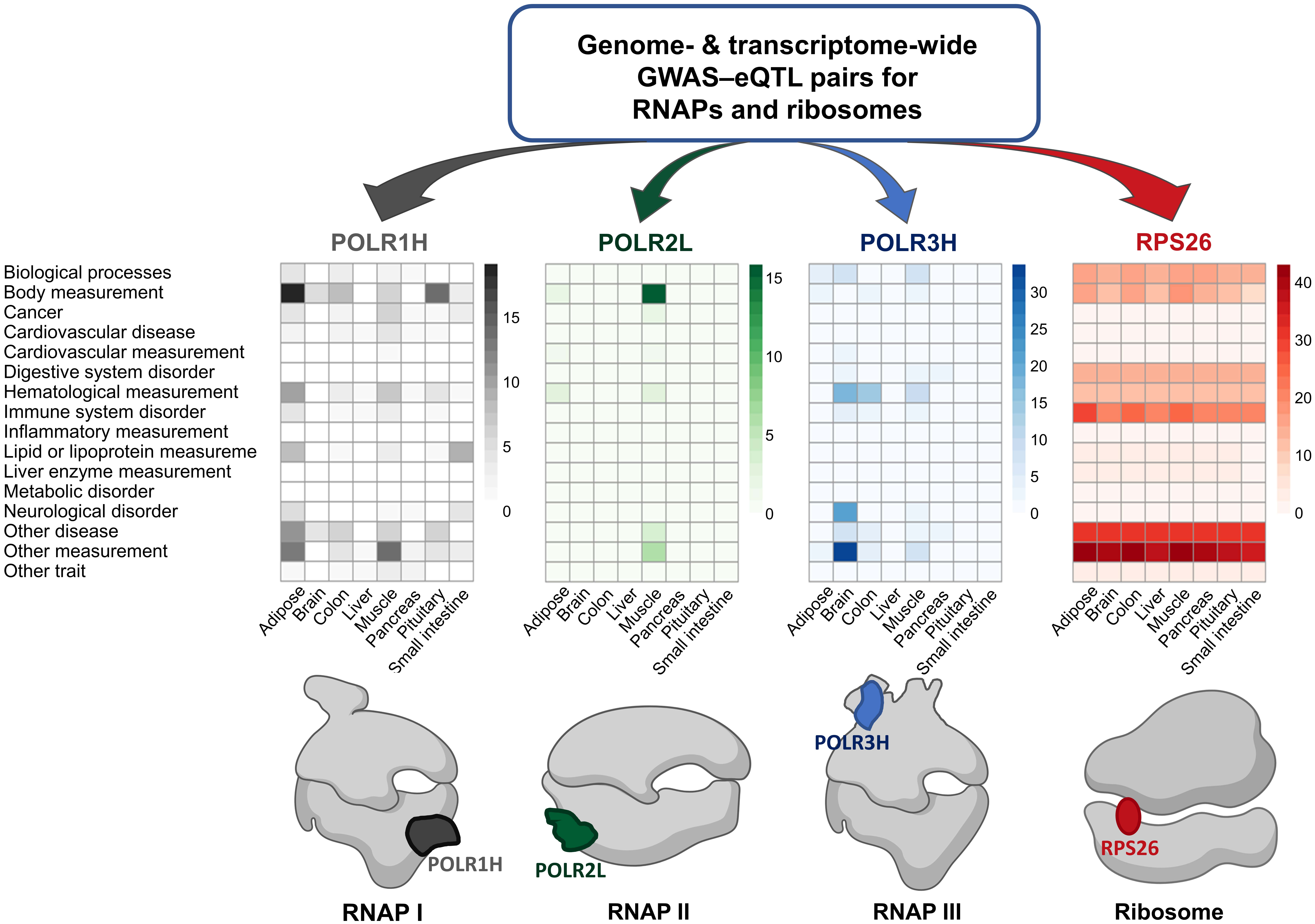
Among the remarkable eGenes within each subtype, POLR1H and POLR2L were predominantly identified in the parent term ‘body measurement’, while POLR3H and RPS26 were in ‘other measurement’ (Fig. 4). The EFO term for POLR3H included ‘neuroticism measurement’, ‘cannabis dependence measurement’, and ‘tea consumption measurement’ in the parent term ‘other measurement’, all found in brain. These eGenes showed tissue-specific pairs, except for RPS26 with ubiquitous pairs across tissues (Fig. 4).
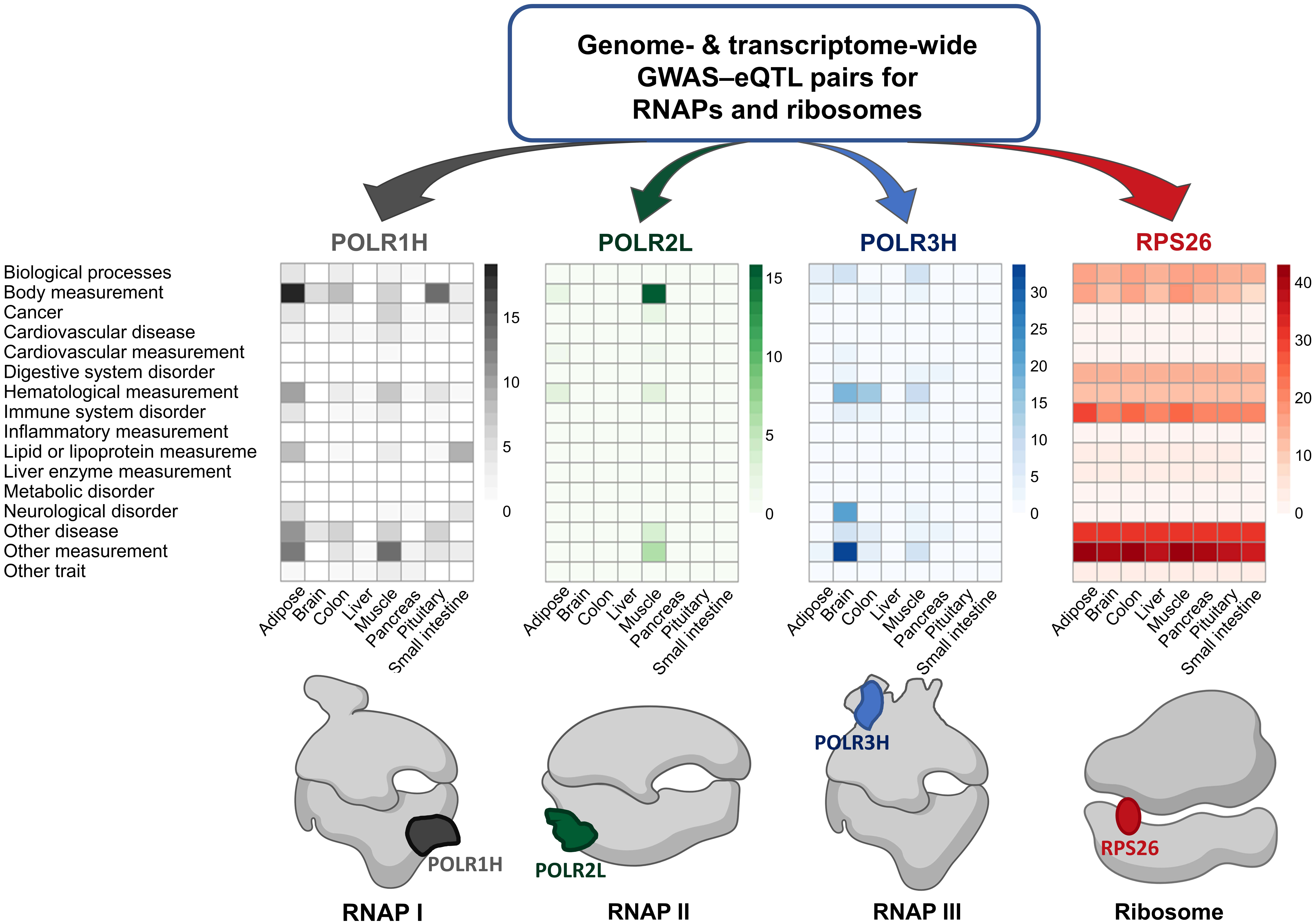 Fig. 4.
Fig. 4.Heatmap of GWAS–eQTL pairs for POLR1H, POLR2L, POLR3H, and RPS26 by EFO parent term and tissue.
Nucleotide sequence variants that regulate expression of the genes responsible for RNAPs and RPs can have profound implications in conducting gene expression. Insufficient or altered components can delay expression, cause misinterpretations during expression, or, in some cases, a complete halt in the process, resulting in deficient or undesirable proteins. The nucleotide variants are, therefore, likely to be associated with a variety of complex traits and diseases, highlighting the pivotal role of a properly functioning RNAPs/ribosomes in maintaining cellular health.
With the genes responsible for RNAPs/ribosomes, we uncovered 12 EFO parent terms of phenotypes associated with their regulatory variants. They differed in proportion from those with all the available genes. Among them, 8 EFO parent terms for RNAPs/ribosomes showed a larger portion than those for nominal genes. Notably, terms such as ‘hematological measurement’, ‘cancer’, and ‘immune system disorder’ were critically associated with protein synthesis that is closely coupled to RNAPs and ribosomes. This result concurred with previous studies in which global protein synthesis plays a role in both quiescence and differentiation of hematopoietic stem cell [58], colorectal cancer [59], hepatocellular carcinoma [60], systemic lupus erythematosus [61], and ankylosing spondylitis [62].
The differences in EFO proportions between RNAPs/ribosomes and nominal genes were considerably attributed to ribosomes. GWAS–eQTL pairs for ribosomes accounted for 83.4% of the total pairs for RNAPs/ribosomes, highlighting the significant cellular effort devoted to ribosome production, which encompasses more than half of all transcription and translation processes [63]. We hypothesize that numerous eQTL might yield ribosomes of insufficient quality and/or quantity, resulting in unfavorable translation, which could impact disease susceptibility. Heterogeneity in ribosomes results in distinct interactions with specific mRNAs, determining translation priorities in particular cell types or in response to specific environmental cues [64], exemplified by the ribosome-mediated response with RPS26 under stress [10]. The accumulation of ribosomes with altered protein stoichiometry [65] or mutations in maturing ribosomes with quality control bypass [66] may predispose human cells to cancer. In addition, a limited number of ribosomes can induce ribosome competition among cellular mRNAs, thereby altering the translation efficiency of subsets of mRNAs [67]. The undesirable translation by ribosome variability could largely influence the three parent terms ‘hematological measurement’, ‘cancer’, and ‘immune system disorder’. Hematological measurements, including corpuscular hemoglobin, hemoglobin level, and red cell distribution width, have demonstrated that highest rates of protein synthesis occur in erythroid lineage commitment [59]. Reduced ribosome levels in hematopoietic cells can affect the translation of a specific subset of mRNAs, particularly GATA1, a master regulator of hematopoiesis, impairing erythroid lineage commitment [68]. Conversely, increased RPs may regulate the p53 pathway via Mdm2 and Mdm4, inducing apoptosis and suppressing cell proliferation [69]. Enrichment analysis revealed ribosomal protein-synthetic pathways associated with GWAS signals for autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus [70].
Unlike ribosomes, we found that EFO proportion for each of RNAP I, II, and III differed from those for RNAPs/ribosomes, showing the largest difference in the portion of ‘cancer’ for RNAP III. We hypothesize that cancer susceptibility could be driven by RNAPs/ribosomes, especially RNAP III, which directly plays a pivotal role in translation from nucleic acid sequences to amino acid sequences. Dysregulation of RNAP III has been consistently observed in various cancers, including prostate cancer, lung cancer, hepatocellular carcinoma and breast cancer [71, 72, 73]. RNAP III can alter the abundance and availability of specific tRNAs in cancer cells [74], enhancing translation efficiency of EXOSC2 and GRIPAP1 genes with codons corresponding to tRNAs [75].
Individual genes with many GWAS–eQTL pairs possess some characteristics in
structure and/or function of RNAPs/ribosomes. RPS26, with the largest
number of pairs among RP eGenes, is located in the mRNA exit channel in ribosomes
and contributes to mRNA-specific translation by recognizing the Kozak sequence.
Ribosome lacking RPS26 could decrease the translation of essential mRNA with the
Kozak sequence while increasing the translation of mRNA with a long 5
For RNAP III, we found the largest GWAS–eQTL pairs in the brain among tissues,
whereas the other subtypes of RNAPs/ribosomes showed a high correlation
(r
This study showed the most abundant pairs (310 pairs) associated with eGenes
belonging to RNAP I, and this abundance was largely attributed to the eGene
(POLR1H) encoding PRA12 (246 pairs). PRA12 contributes to RNAP I passage
through nucleosomes, conveyed with RNA cleavage, enzyme backtracking and
proofreading, and transcription termination [81, 82, 83]. Depletion of its
proofreading function for rRNA transcripts may result in a reduction in the
overall fidelity of transcription. A recent study has confirmed that deletion of
PRA12 reduces transcription fidelity, accompanied by many G
A small number of RNAP II-related pairs might have been caused by a small number
of eGenes (Supplementary Table 1). This is largely because of its unique
structural organization with minimal subunits to efficiently transcribe a wide
range of genes with distinct regulatory requirements. Unlike other RNAPs, RNAP II
achieves flexibility through a combination of fewer permanent subunits and the
presence of dissociable subunits (RPB4/7). Additionally, it utilizes independent
initiation (TFIIF and TFIIE) and elongation (TFIIS) factors, whereas other RNAPs
contain built-in equivalent factors. Notably, the subunits of RNAP II are most
likely observed in other polymerases. For instance, POLR2L, the most
frequent eGene, encodes the RPABC5 protein, an essential subunit of all RNAPs for
assembly and coordination [89]. The second most eGene was POLR2D,
encoding RPB4, which constitutes RPB4/7, integral throughout the gene expression
process. This forms a stalk module of RNAP II, playing a crucial role in
transcription initiation and elongation, co-transcriptional mRNA splicing, and
3
Highlighted above are efforts to understand the functions and characteristics of individual subunits within RNAPs and ribosomes, particularly in relation to complex traits. Alongside addressing priority subunits, there is a growing interest in subunit-specific functions. For example, Rpb3 can directly bind to tissue-specific transcription factors, such as myogenin in skeletal muscles [94] and ATF4 in fibroblasts [95]. Notably, the N-terminus of Rpb3 exhibits selective inhibition in proliferating hepatocellular carcinoma cells that overexpress Rpb3 [96]. Additionally, Rpb9 has been recognized as a driver gene within a regulatory network associated with atherosclerosis, specifically targeting arterial wall tissue, as revealed by GWAS [97]. Understanding the individual genes encoding subunits of RNAPs and ribosomes, along with their specific functions, would greatly aid in comprehending the etiology, pathogenesis, and treatment of diseases.
This review suggests that differences in gene expression related to alleles of genes encoding RNAPs and ribosomes could potentially influence susceptibility to complex human diseases. However, it emphasizes caution regarding the underlying mechanisms of disease pathogenesis. While some data are sourced from GWAS involving patients, an equal proportion comes from analyzing gene expression patterns in healthy individuals with genetic variants. Experimental investigations are imperative to validate and elucidate the role of these genes and their variants in disease susceptibility, particularly in the context of patients or relevant animal models.
This study provides insightful characteristics of the regulatory factors for the RNAP I, II, III, and ribosomes that can influence human complex traits. These regulatory factors were predominantly attributed to RP, suggesting critical ribosomal perturbations in the traits. In particular, RPS26 was the notable eGene with the largest and most widespread impact across all tissues. Among RNAP III genes, POLR3H, with the largest regulatory factors, was highlighted to have brain-dominant regulatory factors associated with neurological disorders. Many regulatory factors implicate RNAP I and RNAP II genes with remarkable functions. While POLR1H encodes a built-in transcription factor, POLR2D encodes dissociable stalk module.
These results suggest the critical impact of genes and proteins responsible for RNAPs and RPs on complex human traits. The emphasis is on RPS26, POLR1H, POLR2D, and POLR3H, highlighting their significance. This information contributes to a better understanding of RNAPs/ribosomes and their regulatory factors, serving as potential prognostic and therapeutic targets in precision medicine.
Conceptualization, CL; formal analysis, JR; writing—original draft preparation, JR; writing—review and editing, CL; visualization, JR and CL; supervision, CL. Both authors have read and agreed to the published version of the manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors would like to thank two anonymous reviewers for their insightful comments on the first version of the manuscript.
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2021R1A6A1A10044154).
The authors declare no conflict of interest. Given his role as Editorial Board member, Chaeyoung Lee had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Nobuo Shimamoto and Yudong Cai.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.