

 , Trilochan Mohapatra 2,*
, Trilochan Mohapatra 2,*1 Division of Biochemistry, ICAR-Indian Agricultural Research Institute, 110012 New Delhi, India
2 Protection of Plant Varieties and Farmers' Rights Authority, 110012 New Delhi, India
Abstract
Genetic information in eukaryotic organisms is stored, replicated, transcribed, and inherited through the nucleus of a cell. Epigenetic modifications in the genetic material, including DNA methylation, histone modification, changes in non-coding RNA (ncRNA) biogenesis, and chromatin architecture play important roles in determining the genomic landscape and regulating gene expression. Genome architecture (structural features of chromatin, affected by epigenetic modifications) is a major driver of genomic functions/activities. Segregation of euchromatin (transcriptionally active) from heterochromatin (transcriptionally repressed chromosome) and positioning of genes in specific nuclear space in eukaryotic cells emphasise non-randomness in the organization of the genetic information. Not only does the base sequence of a gene carry the genetic information but the covalent modifications of bases, three-dimensional positioning of the genome, and chromatin loops are vital for switching on/off the gene and regulating its expression during growth/environmental stress. The epigenetic dynamics depend on the activities of writers and erasers under changing environmental conditions. The discovery of non-coding RNAs (one of the players in de novo methylation of DNA), increased DNA methylation protein (guide for the DNA demethylase), and methylation monitoring sequence (that helps keep a balance between DNA demethylation and methylation) have been some of the new developments in the era of epigenomics. To respond to environmental stimuli, plants depend on modulating gene expression through different mechanisms including biochemical, molecular, genetic, and epigenetic alterations. Studies on plants might provide better insights into epigenetic stress memory and molecular bases of adaptability to enable (epi)genome editing of crops for climate resilience and sustainable agriculture in the present era of multifaceted climate change.
Keywords
- epigenetic modification
- 5-methylcytosine
- histone modification
- genome architecture
- abiotic stress
- epigenetic memory
Unlike animals, plants are continuously exposed to a variety of environmental stresses. Hence, several strategies (morphological, physiological, biochemical, molecular, genetic, and epigenetic) have been evolved by plants to cope with environmental stresses [1, 2]. Our understanding of the stress sensing, signaling, and responses of plants has considerably increased over the past few decades. Growing evidence suggests that chromatin remodeling, histone modifications, DNA methylation, and non-coding RNAs play important roles in epigenetic regulations of gene expression under abiotic stresses [3, 4]. Evidence also suggests that DNA not only carries the genetic information required for the expression of a trait but epigenetic modifications on nitrogenous bases of DNA modulate chromatin architecture to control the expression of gene/trait [5]. The genome is a high-order organization of genetic information [6], which is modulated by epigenetic modifications. During the last two decades, the genome and the epigenome of eukaryotic organisms have been extensively studied to better understand the gene regulatory mechanisms. It is becoming clear that the genetic information and functions determined by a genome can be modulated under changing environmental conditions through epigenetic modulations leading to alterations in 3D chromatin organization in the nucleus [7, 8]. Since the epigenetic state of the genetic material (chromatin) varies with the changing environmental conditions (depending on the activity of the epigenetic machinery), the transfer of a gene/trait from one species to another would not only require the transfer of the gene but also the epigenetic milieu for the gene to express appropriately [9]. Therefore, understanding the epigenetic setup of a gene of interest in the donor plant becomes necessary to ensure the effective transfer of the gene/trait to the recipient.
Recent advances in high-throughput genome and epigenome analyses have provided unprecedented opportunities to generate genomic and epigenomic maps in unraveling the (epi)genomic landscape at single-base resolution. However, there are certain challenges in presenting the whole epigenome, particularly under changing environmental conditions in different tissues of an organism. The task becomes further complicated in compiling the unified information for methylome, histone modifications, and ncRNA biogenesis, as these epigenomic components vary differently with the changing environmental conditions in different cells/tissues. Generally, the epigenetic changes observed in a regular analysis present the average changes of a population of cells in the sample tissue; hence, epigenomic analysis at the single-cell level needs to be captured/performed [10, 11]. Thus, challenges in epigenomics include the required technological advancements to generate reliable/consistent data necessary for understanding the stable, reversible, and heritable components of the epigenome.
Modifications in DNA bases (at the 5th carbon of cytosine residue and/or N
Accessibility of a genomic region to transcriptional machinery is also modulated by post-translational modifications in the histone proteins and chromatin-remodeling complex that regulates nucleosome assembly and spacing [19]. Many amino acid residues, mostly at the N-terminal tail protruding out of the core histone, are subjected to covalent post-translational modifications. The rate of transcription of a gene is affected by different sites/degrees of histone modifications (e.g., H3K27ac, H3K9me2, H3K9me3, and H3K27me3), which influence the association between DNA and histone proteins, thus affecting the chromatin accessibility [20, 21, 22]. In addition, histone modifications are recognized by proteins like ATP-dependent chromatin remodelers which affect local chromatin status and regulate the expression of the gene [19, 23]. H3K9me2, established by SUVH4/5/6, is required for maintaining DNA methylation at heterochromatic regions in Arabidopsis [24, 25]. Ubiquitination of histone, when occurs at the C-terminal tail of H2A, causes transcriptional repression; whereas ubiquitination at the C-terminal tail of H2B causes transcriptional activation [26]. Moreover, histone variants have also been reported to influence nucleosome stability and interaction with the mRNA processing factors [27]. Several studies report that such epigenetic marks are involved in flowering, seed development, nitrogen fixation, and abiotic/biotic stress responses [26, 28, 29]. Roles of the important components like ncRNA (triggering de novo DNA methylation), Increased DNA Methylation (IDM) protein (involved targeted DNA demethylation), and methylation-sensing genetic element (maintains DNA methylation/demethylation homeostasis) in epigenomic regulation of genes/traits are being deciphered [25, 30].
Genome in the eukaryotes is not randomly placed in the nucleus, but it is packed into a high-order chromatin structure, which plays important roles in its functions. Understanding nuclear genome organization is attracting significant attention because processes like DNA replication/transcription, genome stability/integrity, etc. (necessary for appropriate growth/development, and stress tolerance) are regulated through chromatin structures. Eukaryotic genome organization can be studied at three levels namely (1) linear genome (nucleotide sequence), (2) epigenomic alterations (modified DNA bases, histone modifications), and (3) three-dimensional (3D) structure of the genome (depicting the arrangements of chromatins/chromosomes in the eukaryotic nucleus) [11, 31]. Chromatins are dynamic during cyclic compaction/de-compaction through cell division, cellular differentiation/developmental processes, as well as defense responses, which regulate/fine-tune the expression of genes [32, 33]. Environmental fluctuations necessitate modulations in cellular processes by switching on/off the gene in response to stress [11, 34]. Transient chromatin compaction (e.g., loosely-packed euchromatin or more compact heterochromatin) and modulation in chromatin architecture under environmental stress have been demonstrated in animals as well as plants [34, 35, 36, 37]. Environmental stresses are reported to cause alterations in chromatin architecture, improving accessibility of the stress-associated genes to transcriptional machinery through chromatin remodeling (shifting/removal of histones) [38], post-translational modifications of histones [19, 39] or replacing the histone variants [40].
In addition to the genetic factors, gene expression is also controlled by the combined actions of other regulatory mechanisms that alter chromatin architecture including DNA base modifications [5, 11], histone modifications [41, 42], chromatin remodeling [43, 44], etc. Different chromatin remodelers like CHD, ISWI, INO80, and Switch/Sucrose non-fermenting (SWI/SNF) act on chromatin architecture under different environmental conditions to alter the transcriptionally inactive to transcriptionally active chromatin state. Alterations in chromatin architecture at the promoter region are more crucial in affecting the expression of the gene [45, 46]. Developmental/environmental stimuli alter the epigenetic landscape/chromatin architecture that modulates the expression of the gene(s) necessary for proper growth and to cope with stress [11]. Some of these epigenetic marks/transcriptional repressors cooperate with chromatin remodelers to alter chromatin structure and fine-tune the expression of the gene(s). Some of these epigenetic modifications may get transmitted through the cell division/reproductive cycle and help to cope with the reoccurring stress [47]. However, identification/validation of the stress-induced heritable epigenetic mark would be necessary for utilizing it in stress tolerance improvement programs.
Initially considered to be a host defensive mechanism in prokaryotes, DNA base modifications, mainly cytosine methylation, have now been recognized to play vital roles in the regulation of gene expression in eukaryotes. In the last two decades, several epigenetic changes occurring during the developmental processes/abiotic stresses in animals and plant genomes have been reported [2, 48]. In most cases, methylation of promoter has been reported to repress the rate of transcription of the gene, mainly because of the formation of a repressive-chromatin structure affecting the binding of transcription factor on acquiring its bonding site by a methylated-DNA binding protein [49]. However, hypermethylation of promoter not causing transcriptional repression has also been reported [50], indicating that transcriptional responses of promoter methylation are context-specific. Reversal of the epigenetic modification is required for restating the gene activity, particularly in responses to environmental stress.
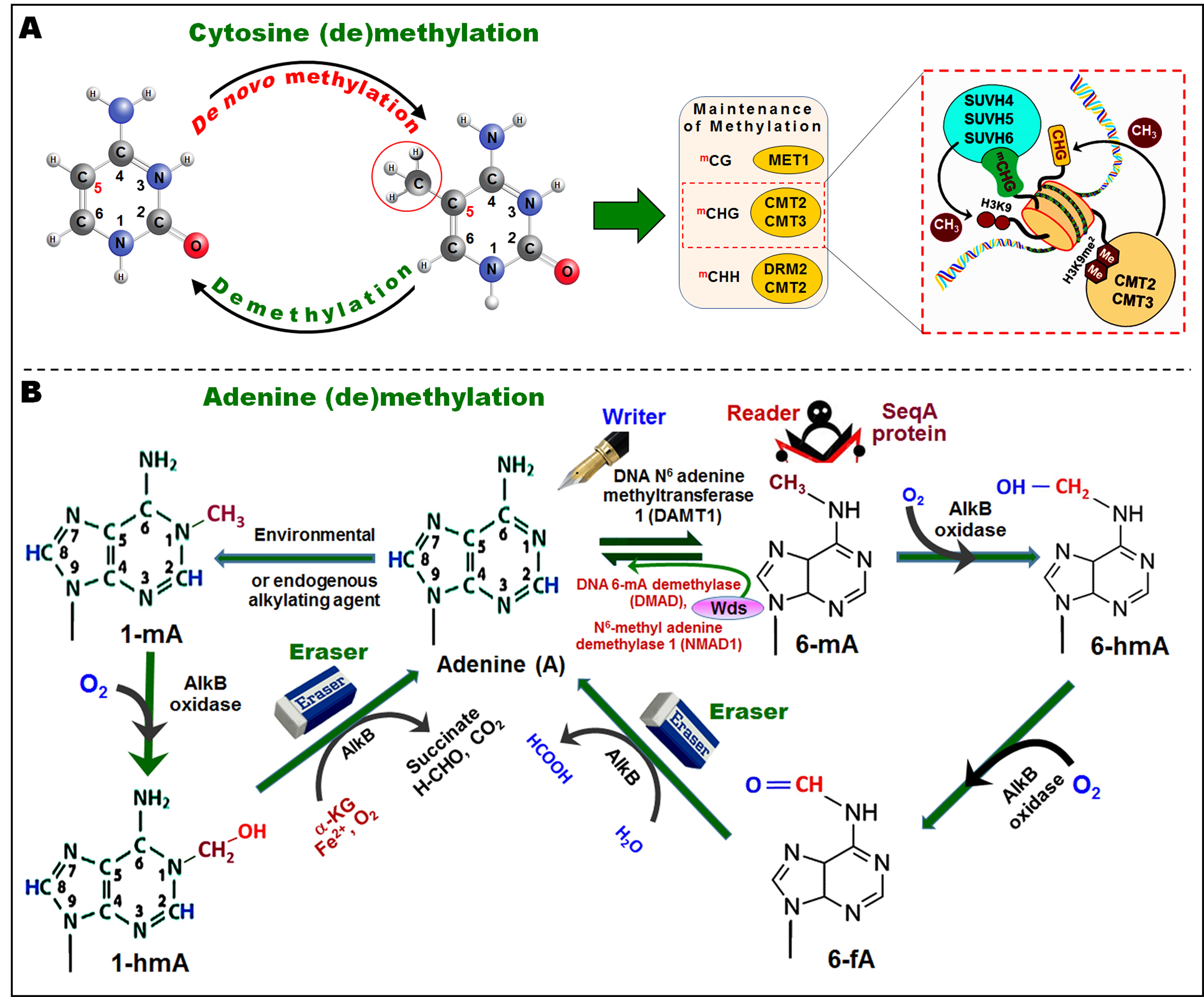
Only some of the genes have been reported to be silenced due to methylation of promoter in Arabidopsis, which indicates that DNA base modification is not the only epigenetic regulatory mechanism. About one-third of the genes in Arabidopsis were reported to be methylated at gene-body [51]. Gene-body methylation appears to be a common phenomenon in plants, as it can affect up to 60% of the genes in some species [52]. Though the function of gene-body methylation in plants is still controversial, recent findings suggest that it occurs under environmental stresses and is correlated with the fitness/adaptability of the plant [52, 53]. Generally, transposable/repetitive elements are heavily methylated in all the cytosine (CG. CHG, and CHH) contexts (Fig. 1A), but gene-body methylation meagrely occurs in non-CG contexts [51, 54, 55]. Gene-body methylation (occurring in the coding region between transcription start and termination sites) [56] and methylation in different cytosine contexts of transposable element (TE) or repetitive elements present in the introns of a gene might control mRNA processing. Epigenetic modulation of retrotransposon in the homeotic gene was reported to affect alternative splicing resulting in premature termination of transcription [57].
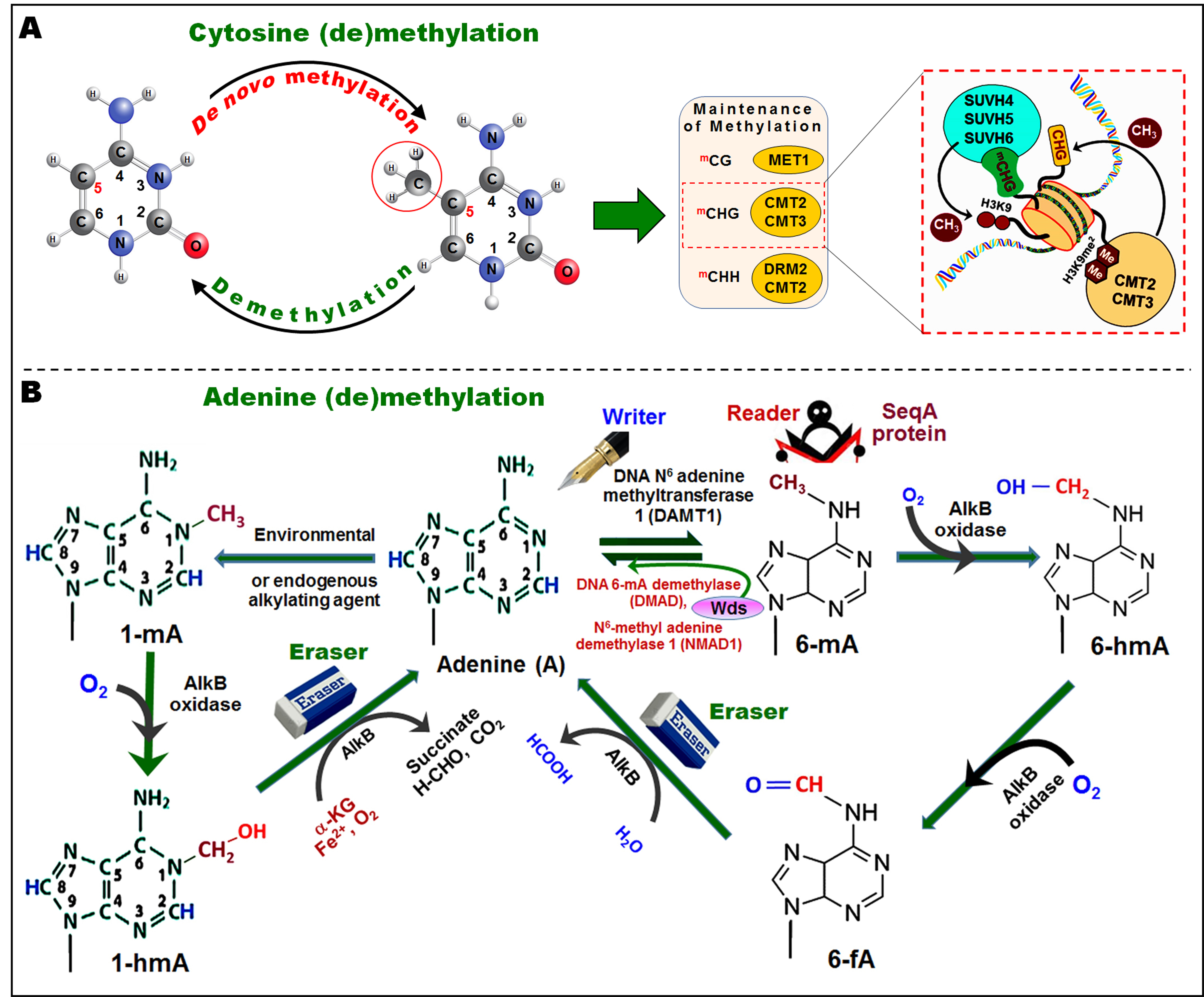 Fig. 1.
Fig. 1.DNA base methylation in plant. (A) De novo methylation
of DNA in CG, CHG, and CHH contexts. After DNA replication, methylation in CG
context is performed by methyltransferase 1 (MET1). Methylation in the CHG
context is performed by chromomethylase 2 (CMT2) or CMT3, while methylation in
the CHH context is performed by CMT2 or DRM2 through the RdDM pathway.
Methylation in the CHG context attracts binding of H3K9-specific suppressor of
variegation 3-9 homolog 4 (SUVH4), SUVH5, SUVH6 that generate dimethylated-H3K9
(H3K9me2), which promotes CMT2 and CMT3. (B) Adenine gets methylated by the
addition of the CH
Similar to cytosine methylation in DNA, an adenine can also get methylated due
to the addition of a CH
However, oxidation of methyl group (by demethylase, e.g., AlkB dioxygenase) of
6-mA results in its conversion to N
Changes in DNA methylation have been detected in several prokaryotic and eukaryotic organisms. Modifications in DNA bases in context- and region-specific manner are catalyzed by specific enzymes. In plants, heterochromatic regions are generally enriched with 5-mCs, mainly in the repetitive sequences and TEs. The dynamics of DNA base modification depend on the revocability of the processes, depending on the activity of enzymes involved in the processes that switch on/off the gene. The complex epigenetic changes are being discovered in different organisms and possible combinations/interactions among the epigenetic marks (epimarks) indicate that epigenetic codons are more complex than they are being thought [5]. Four bifunctional DNA glycosylases including Demeter (DME), Demeter-like protein 2 (DML2), DML3, and Repressor of silencing 1 (Ros1) have been known in Arabidopsis [67] that can remove methylation in any sequence context [68, 69, 70]. An AT-rich TE is favourably demethylated by DME, leading to changes in the expression of nearby genes [71, 72, 73]. Demethylation of TE by ROS1 affects transposon activity and silences the nearby genes [74]. ROS1 is also involved in demethylation of the RdDM-independent regions [75, 76]. The genomic regions demethylated by ROS1 are characterized by lowered H3K27me and/or H3K9me2 but increased H3K18Ac and/or H3K27me3 marks [74]. Some of the target sites (methylated DNA sequences) of ROS1 are created by the binding of a histone acetyltransferase “increased DNA methylation 1” (IDM1), which acetylates H3 at the site deprived of H3K4me2 and H3K4me3 [77]. Interestingly, a 39 bp “methylation monitoring sequence” (MEMS) is present in the promoter of ROS1 that functions as a sensor of MET1 and RdDM activities [5]. Moreover, the promoter of ROS1 also contains a transposon (Helitron) upstream of MEMS, which binds to cytosine methylation factors and thus makes the promoter responsive to the methylation level. Thus, like a thermostat of a machine, MEMS acts as a “methylstat” that senses and maintains the ROS1-dependent methylation level in a genomic region [78, 79]. The presence of such “methylstat” is essential for cytosine methylation dynamics and it has been reported not only in plants but also in animal systems [80, 81].
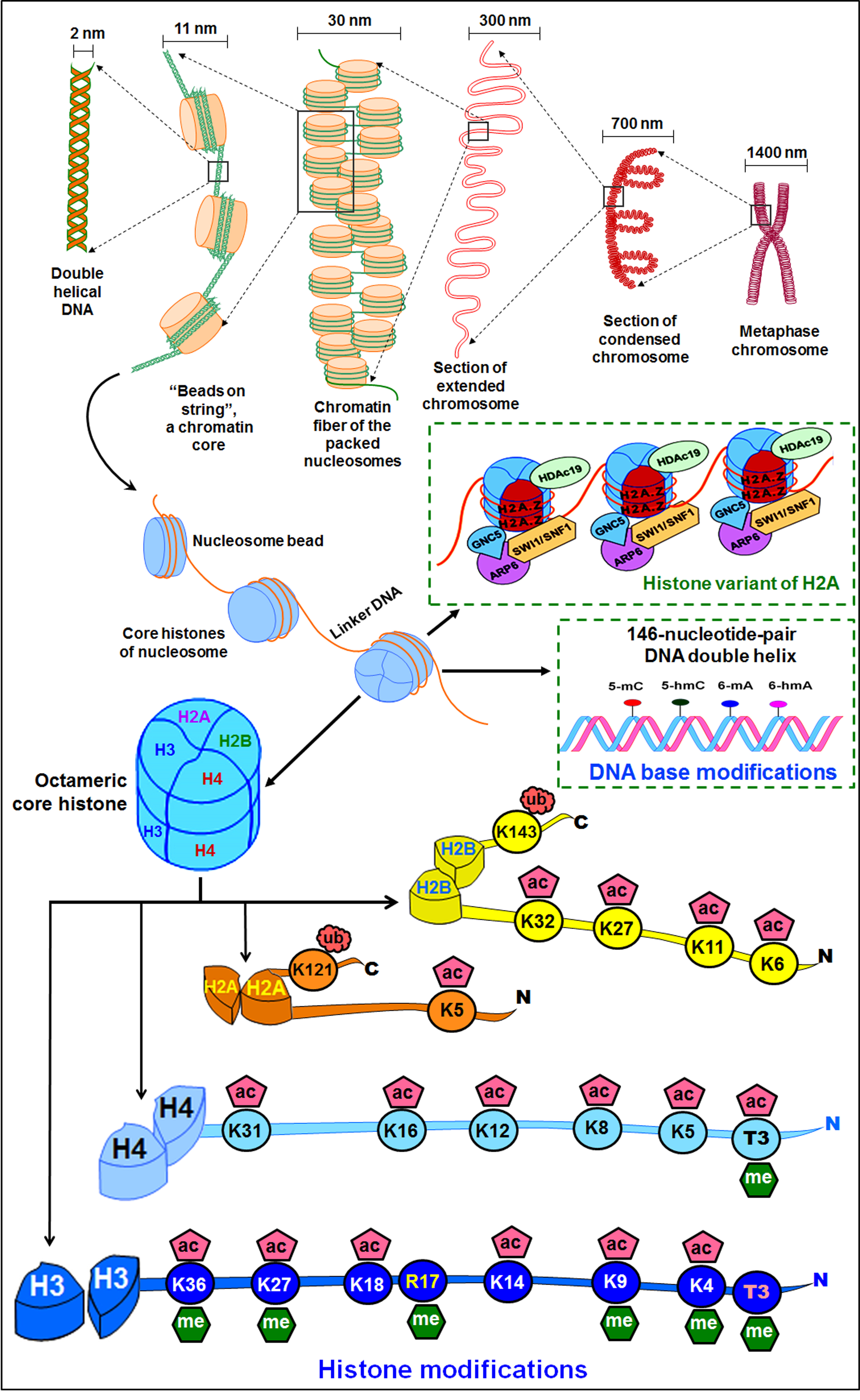
A histone octamer (two copies of each of the H2A, H2B, H3, and H4) wrapped around by a stretch of DNA (146 bp) forms a nucleosome (Fig. 2). The amino acid residues mostly at the protruding N-terminal tail, mainly of histone H3, are subjected to covalent post-translational modifications including acetylation, methylation, phosphorylation, ubiquitination, and SUMOylation, which play important roles in regulating gene expression during growth, development, and exposure to environmental stresses [82]. The post-translational modification of histone proteins, generally methylation and acetylation of histone 3 (H3) at lysine-4 (i.e., H3K4), lysine-9 (H3K9), and lysine-27 (H3K27) are some of the most common histone marks associated with controlling gene expression [83]. Acetylation of H3, such as lys-9 (H3K9ac) and lys-27 (H3K27ac), correlates with transcriptional activation of a gene [84]. Similarly, trimethylation of H3 at lys-9 (H3K9me3) and lys-27 (H3K27me3) have varying effects based on the site of methylation. While H3K9me3 and H3K27me3 have been known to cause transcriptional suppression, H3K4me3 is associated with the transcriptional activation of a gene [85, 86]. The involvements of H3K4me2, H3K4me3, H3K9ac, H4K5ac, H4K8ac, and H4K12ac in the environmental stress tolerance of plants are becoming evident now [87].
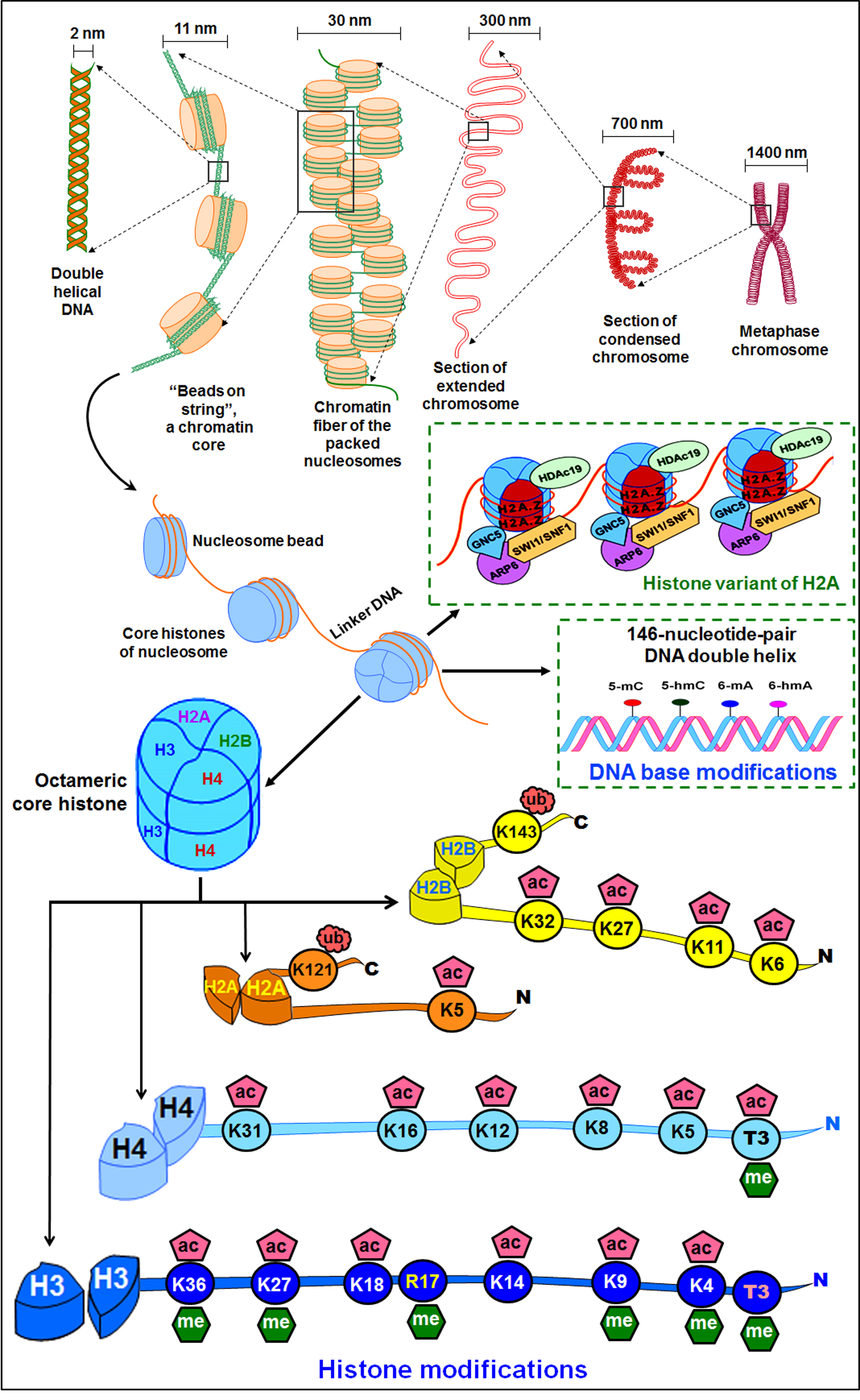 Fig. 2.
Fig. 2.Organization of the genome and epigenetic modifications. Packaging of chromosomal DNA in the nuclei through wrapping of DNA double helix on a histone octamer. The positively-charged histone proteins adhere with the negatively-charged DNA, forming a nucleosome. The nucleosomes fold into 30 nm chromatin fiber, forming loops of 300 nm, which coils to form 700 nm condensed chromatid of ~1400 nm Metaphase chromosome. DNA base modifications as well as the histone variants affect the genome functions and regulate gene expression. Modifications at the tail of core histone (H2A, H2B, H3, H4) proteins cause chromatin remodeling that affects the expression of the gene under abiotic stress.
Acetylation of a lysine residue in histone is dynamically controlled by the
action of enzymes histone acetyltransferase (HAT) and histone deacetylase (HDAC).
Acetyl-CoA is a cofactor of HAT in catalysing the transfer of an acetyl group to
the
Though modification of DNA base(s) and amino acid(s) at the tail of histones alter the chromatin architecture, the complexity of regulation of gene expression increases due to the cross-talk between different epigenetic modifications, which might help to fine-tune the gene expression (Fig. 2). The cross-talk may occur between the modified DNA base and the histone modification or between the histone modifications. The effects of cross-talk between histone modifications might be (i) competitive antagonism, particularly when more than one modifications target the same site (as in the case of lysine where acetylation, methylation, and/or ubiquitination might occur), (ii) cooperative, or (iii) disruptive [82]. Similarly, there might be cooperation between DNA methylation and histone modification as it was reported for the binding of UHRF1 protein to methylated (H3K9me3) nucleosome in HeLa cell, which is significantly enhanced when the nucleosome is methylated at CG site [91]. Moreover, DNA methylation can also inhibit protein binding to modified histone as it has been reported for the binding of KDM2A, which binds only to the nucleosome having H3K9me3 but not methylated at the DNA level in HeLa cell.
In addition to the modifications of canonical histone proteins, the incorporation of a histone variant (at different genomic regions, affecting the nucleosome stability and interaction with mRNA processing factors) as well as the chromatin-remodeling complex (regulating nucleosome assembly and spacing) also affect the nucleosome functions [19, 27, 92, 93, 94]. Histone variants are characterized by changes in a few amino acids which are encoded by separate genes. They not only affect nucleosome packaging but also modify the chromatin properties [95]. Evidence suggests that histone variants have effects on epigenetic state/transcription, chromosomal segregation, and repair of DNA damage [96]. All the core histones (except for the histone H4) and linker H1 show variants in plants. However, the focus has been on variants of H2A and H3. Among these, H2A.Z and H3.3 are evolutionarily conserved in the eukaryotes. Moreover, some tissue-specific and lineage-specific variants like H2A.W (functioning in flowering plants) and H3.10 (exists in the sperm cells of Arabidopsis only) are known [95]. Variant histone proteins exhibit distinct patterns of chromatin deposition. For example, the variants H2A.X and H2A.Z are concentrated in the euchromatin, while H2A.W is located mainly in the heterochromatic region. Furthermore, combinations of histone variants form thousands of different types of nucleosomes conferring extended potential for epigenetic codons in regulating various biological processes in plants, particularly under environmental stress. Variants of H2B are relatively less understood. One of the H2B variants (H2B.S) has been reported in sperm cells as well as in mature embryonic cells [95]. Three main variants of H3 have been known in plants including H3.1, H3.3, and centromeric H3 (CenH3) variants. The H3 variants (H3.1 and H3.3) are distinguished mainly by the amino acids present at four different locations namely 31st (Ala vs. Thr), 41st (Phe vs. Tyr), 87th (Ser vs. His), and 90th (Ala vs. Leu). These four different amino acids in H3.1 facilitate the recruitment of PRC2, which ensures the silencing of some of the development-related genes. Although H3.3 is predominant at the transcription end sites and reported to be associated with activation histone modification marks like H3K4me3, H3K9me3, and H3K36me3, it specifically affects transcription of genes associated with abiotic and biotic stresses. CenH3 is located mainly in the centromere for its formation and kinetochore assembly, particularly in the mitosis G2 phase. In recent years, manipulation of CenH3 has been exploited in haploid induction (HI) which can create true-breeding lines in a shorter period to accelerate the pace of plant breeding [97]. In addition to the above-mentioned variants of H3, some other variants of H3 like H3.10 and H3.15 are known in Arabidopsis. While H3.10 plays important roles in epigenetic reprogramming in sperm/spermatocytes, H3.15 plays essential roles in callus formation in plants [98, 99].
Research findings indicate that H2A.Z plays important roles in photo-morphogenesis and thermal morphogenesis in plants. While the accumulation of H2A.Z is essential for photo-morphogenesis, it plays an inhibitory role in thermal-morphogenesis. In rice, phosphate deficiency was reported to reduce H2A.Z accumulation on gene bodies of stress-responsive genes to facilitate the expression of the genes [100]. Transcription of drought-responsive genes was reported to be negatively correlated with the accumulation of H2A.Z in the gene body in Arabidopsis [101]. Similarly, heat stress affects flowering time in plants by altering the deposition of H2A.Z with species-specific effects [102]. H3.3 is involved in regulating flowering and seed germination as it is essential for transcriptional regulation of genes during germination [103].
Modification of histones (like H3K4me3, H3K9ac, H4K5ac, H4K8ac, and H4K12ac) in the promoter region of transcription factor (e.g., WRKY) gene has been implied to activate the expression of defensive gene(s) [104], suggesting their roles in epigenetic (histone modification) stress memory in plants. Application of acibenzolar S-methyl (a salicylic acid analog) was associated with histone modifications like H3K4me2, H3K4me3, H3K9ac H4K5ac, H4K8ac, and H4K12ac at the promoter of some of the defense associated genes [105]. Exogenous application of acetic acid was reported to promote jasmonic acid (JA) synthesis and enrichment of histone H4 acetylation, which primes the JA signaling pathway and enhanced drought tolerance in plants (Arabidopsis, rice, rapeseed, maize, and wheat) [106]. Methyl jasmonate (MeJA) was reported to increase the expression of defense-associated genes (OsBBPI and OsPOX) and modulated histone modification as well as DNA methylation. Thus, covalent (yet reversible) modification of histone interacts with DNA base modification and plays important roles in regulating gene expression. Nucleosome remodeling was reported to be implicated with heat stress memory in Arabidopsis [107]. FORGETTER1 (FGT1), encoding for a helicase, interacts with chromatin remodeling complexes (like SWI/SNF chromatin remodeler BRM) and functions as a factor required for heat stress memory. Studies also suggest that cold stress causes histone modifications (methylation and acetylation) at cold-responsive genes that can be manipulated for enhanced cold tolerance in plants [44, 108].
Non-coding RNAs (ncRNAs) are the regulatory RNAs that have significant impacts
on the expression of coding genes. Based on the size, the regulatory RNAs are
grouped into two types: (i) small non-coding RNAs (sncRNAs,
Long non-coding RNA (lncRNA) is another important group of ncRNAs having
The circRNAs are evolutionarily conserved abundant ncRNAs in plants having a loop-like (covalently closed) structure [126]. It has also been reported to be produced as an intermediate/by-product of mRNA splicing [127] or from other ncRNAs by RNA editing enzymes [128]. In rice, the majority of circRNAs are flanked by non-GT/AG splicing signals, which suggests that plants also have an alternative mechanism for their biogenesis [129]. The circRNA interacts with miRNA as a sponge and it can regulate the gene expression negatively or positively [130]. Although circRNAs are derived from protein-coding genes, they are not translated into proteins; hence, classified as non-coding RNAs [131]. As a competitor, the lncRNA can inhibit the attachment of DNA-binding protein (e.g., TF) to the target site. The lncRNA inhibits binding of DNMT1 to the target DNA sequence and thus affects DNA methylation [132]. Moreover, lncRNA can reinforce the DNA methylation process by recruiting epigenetic modifiers to the target site [133]. In mammals, lncRNA has also been reported to act as a precursor of miRNA by the action of Dicers [134].
In animals, the inheritance of epigenetic changes over the generations requires their transit through germline cells without being erased by methylation surveillance mechanisms [135]. Like animals, plants do not own germline cells, instead, they produce gametes during sexual reproduction. Therefore, some of the epigenetic modifications attained during the vegetative phase of plant growth might be inherited through mitotic and meiotic cell divisions [136, 137]. Hence, plants can remember the strategies used to fight against stressful conditions and it might be used by the plant to respond quickly to the stress on reoccurrence of the stress [138]. Plants have been reported to memorize defensive strategies against stress and trigger responses quickly on the recurrence of stress [139]. A part of the epigenetic changes induced by abiotic stress may be mitotically heritable (within the generation), which can last for several days (short-term memory) or even for the rest of plant life (long-term memory). However, abiotic stress can also induce certain epigenetic changes in plants that may show transgenerational heritability, at least to one non-stressed progeny (transgenerational memory). Although the basics of stress memories in plants have not been clear to date, reports on biochemical/metabolic, transcriptomic and/or epigenetic alterations indicate their role in short- as well as long-term memories [140, 141, 142].
A transcription activation mark (H3K4me3) was reported to play an important role in transcriptional memory [143]. To identify the heritable epigenetic marks/components associated with a trait of interest continuous efforts are being made so that they can be utilized in epigenome editing for crop improvement towards the development of climate-resilient crop varieties. However, the genome of many crop plants is considerably large and complex mainly because of their polyploid nature and occurrence of repetitive elements. How epigenetic changes superimpose the multiple copies of a gene in conferring genetic plasticity may provide some clues in developing tailor-made crop varieties to cope with multiple stresses being faced due to global climate change. Epigenetic engineering might be a potential way to achieve the desired adaptive plasticity without altering the underlying DNA sequence. However, stable inheritance of such epiallele would be essential to provide adaptive fitness/adaptability to the plant.
Under cold stress, acetylation of histone H3 by histone acetyltransferase (HAC1) was reported to promote the activation of COR genes which is involved in cold-stress memory [144]. Similarly, sRNA was reported to play a role in drought stress memory in Arabidopsis [136]. Up to 70% of the stress-induced epigenetic alterations have been reported to revert to the original state after withdrawal of the stress, but one-third of the changes might get inherited as epigenetic stress memory [9, 140, 145]. Some of the salt stress-induced modulations in the methylation of DNA were reported to be transmitted over the generations in Arabidopsis thaliana, particularly through female gametes [146]. SDC gene, coding for the suppressor of DRM1, DRM2, and CMT3, was reported to get silenced by methylation at the promoter of the gene; however, the reoccurring heat stress was reported to activate SDC [147].
Assessment of the contribution of a heritable epimark in the inheritance of a phenotypic alteration has been a challenging task because many of the epigenetic changes and DNA sequence polymorphisms co-segregate with the altered gene expression. However, evidence indicates that some of the heritable epiallelic changes in plants can be associated with a trait of interest which can be utilized in crop improvement programs. Molecular analyses indicate that variations in methylation in an epiRIL are stable across the generations. Since epiRILs are identical for DNA sequences but differ for the epigenetic marks, the phenotypic differences among them can be attributed to epigenetic variations. Thus, epiRILs can be valuable assets for epigenetic studies in plants. Epigenetic changes occurring in response to environmental stress accumulate in the first generation, but only stable/heritable components might get inherited that might be involved in the adaptability of the plant.
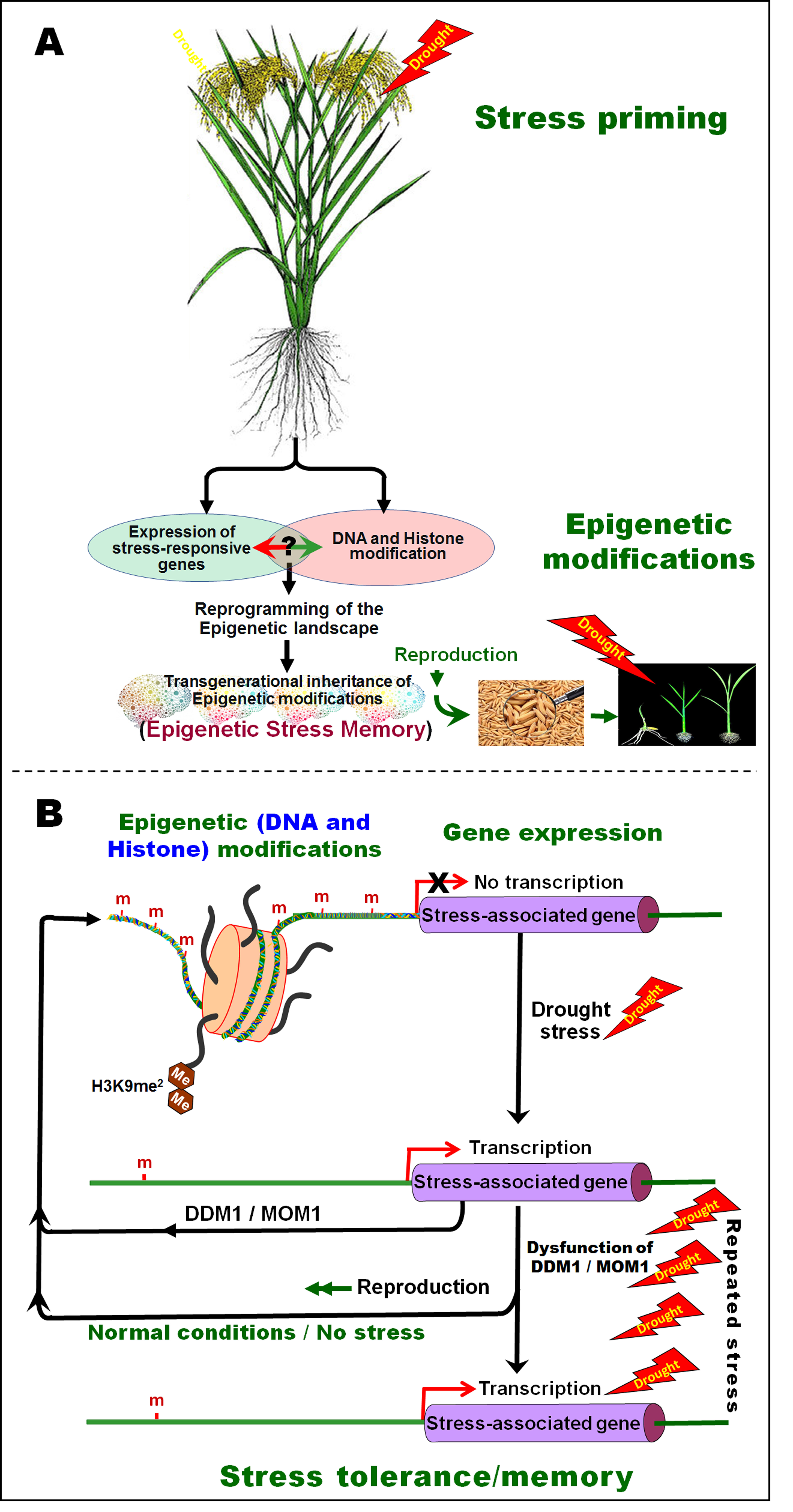
Repeated exposure of rice to drought stress for several generations was reported to improve adaptability through epi-mutations and transmission of the altered DNA methylation to the progenies [140] (Fig. 3A). In Arabidopsis, Morpheus Molecule 1 (MOM1) and DDM1 were reported to be involved in the removal of stress-induced epigenetic marks under stress-free environment. In a double mutant for ddm1–mom1, the stress-induced epigenetic marks were reported to be inherited by the progeny. However, a single mutant (either ddm1 or mom1) failed to inherit the stress-induced epigenetic mark [148]. These indicate that DDM1 and MOM1 block the inheritance of stress-induced epimark (Fig. 3B). Remembering an episode of stress and responding more efficiently to subsequent occurrences of stress constitute the way plants adapt to environmental changes. However, transgenerational epigenetic stress memory needs to be meiotically stable and heritable to at least two stress-free generations [149].
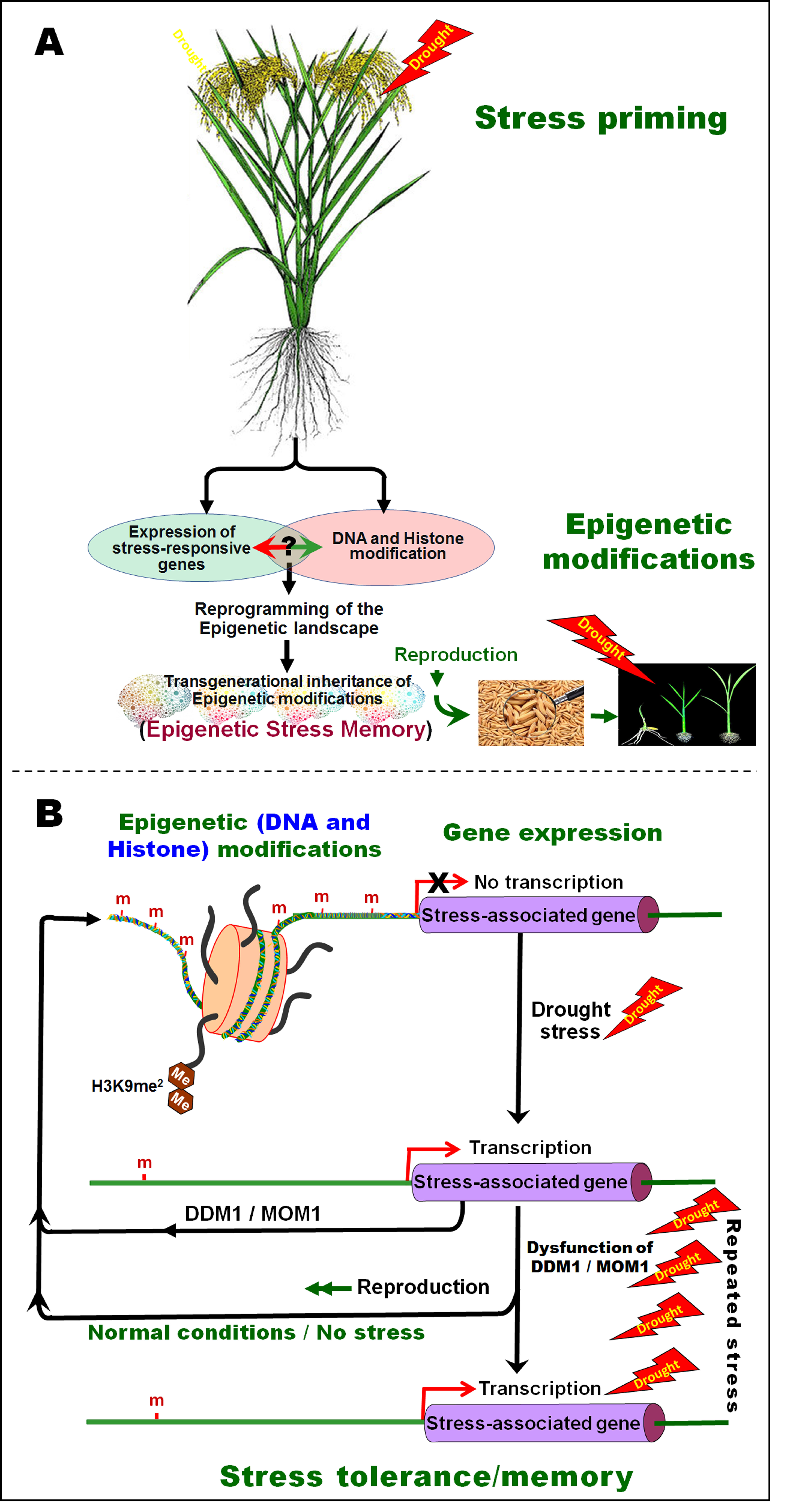 Fig. 3.
Fig. 3.Possible role of epigenetic changes in stress tolerance/memory in plants. (A) Abiotic stress alters the expression of stress-responsive genes through different epigenetic modifications. In the stress-primed plants, the epigenetic landscape is altered and a part of the epigenetic modifications gets inherited, which serves as epigenetic stress memory. (B) Stress-induced epigenetic modifications cause differential expression of the stress-responsive genes. Under normal conditions (after removal of stress), Morpheus Molecule 1 (MOM1) and DDM1 play important roles in the removal of the stress-induced epigenetic marks. However, dysfunction of MOM1 and DDM1 results in transmission of the stress-induced epigenetic marks over the generation, particularly on repeated exposure to stress.
Although considerable progress has been made in epigenomics of gene regulation in animals, the enzymes involved in DNA methylation and histone modification in plants are still being characterized. Dynamic chromatin architecture affects the accessibility of genes to transcription machinery, thus modulating the interpretation of the genetic information encoded in the DNA sequence. To date, only a little is known about the epigenetic modulators and their functions during developmental processes and environmental stress in plants. Moreover, little is known about the possible interactions between epigenetic marks. Different types of epigenetic modifications, including DNA methylation, histone modification, histone variants, and nucleosome occupancy, are being reported to be implicated in stress memory [150, 151]. Recent technological advancements like super-resolution technology and single-cell omics approaches might help in understanding the phenomenon of stress memory [11]. In addition, an assay for transposase-accessible chromatin sequencing (ATAC-seq) [152] would help decipher the interactions between DNA base methylation and histone modifications and their role in stress memory.
Furthermore, epitranscriptomic modifications [RNA base modifications, like
5-methylcytosine (m
Sequencing, assembly, annotation, and functional validation of the biological function(s) of protein-coding as well as non-coding/regulatory genes have been some of the revolutionary research during the last four decades. On understanding the biological functions of genes/proteins, it is obvious now that the DNA sequence information alone is not sufficient enough to depict/decide the expression of a gene/trait. Efforts are also being made to explore the epigenetic and 3D/4D genomic mechanisms involved in the plasticity of an organism. Interestingly, the nucleotide sequence variation and rate of meiotic recombination are correlated with 3D genomic structures. Topologically associating domains (TADs) were reported to show more single-nucleotide polymorphism, increased rate of recombination, and structural variations (SVs) compared to that observed in the inter-TAD regions [156]. Accumulating evidence suggests the involvement of epigenetic modifications such as DNA base (6-mA) modification [65] and histone modification (H3K4me3) mark [157] in stress memory and adaptation of plants. In many cases, stress memory is reported to be reset just after one stress-free generation. Though it is suggested that transgenerational stress memory over several generations might be disadvantageous to the plant [148], it might be beneficial for the adaptability of plants, particularly under fluctuating environmental conditions. 3D genomic study at the single-cell level using live-cell imaging techniques paves the way to discover the gene regulatory mechanisms needed for the development of climate-smart crops. Combined studies on genome, epigenome, epitranscriptome, and epiproteome (multi-omics analyses) and their effects/contributions to stress memory would greatly improve our understanding of keeping stress memory in plants.
SK and TM conceived the review. SK prepared the initial draft. SK and TM revised the manuscript and approved the final draft. Both authors contributed to the article and approved the final version. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Epigenomics research on abiotic stress tolerance is being carried out with financial support from the National Agricultural Science Fund (NASF/ABP-70161/2018-19) and Extramural Research Grant [18(3)/2018-O&P, NASF/EMR/(OG)/06/2021] from the Indian Council of Agricultural Research, Government of India, New Delhi, India.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.