
 , Yoshiaki Sota 2, Hideo Matsuda 1,*
, Yoshiaki Sota 2, Hideo Matsuda 1,*
1 Graduate School of Information Science and Technology, Osaka University, 565-0871 Suita, Osaka, Japan
2 Graduate School of Medicine, Osaka University, 565-0871 Suita, Osaka, Japan
Abstract
Fusion genes are important biomarkers in cancer research because their expression can produce abnormal proteins with oncogenic properties. Long-read RNA sequencing (long-read RNA-seq), which can sequence full-length mRNA transcripts, facilitates the detection of such fusion genes. Several tools have been proposed for detecting fusion genes in long-read RNA-seq datasets derived from cancer cells. However, the high sequencing error rate in long-read RNA-seq makes fusion gene detection challenging.
To address this issue, additional steps were incorporated into the fusion detection tool to improve detection accuracy. These steps include anchoring breakpoints to exon boundaries, realigning unaligned regions, and clustering breakpoints. To evaluate the accuracy of our tool in detecting fusion genes, we compared its detection accuracy with two representative existing tools, JAFFAL and FusionSeeker.
Our tool outperformed the two existing tools in detecting fusion genes, as demonstrated in long-read RNA-seq datasets. We also identified potentially novel fusion genes consistently detected across multiple tools or datasets.
The application of our tool to the detection of fusion genes in long-read RNA-seq datasets from two different cancer cell lines demonstrated the detection effectiveness of this tool.
Keywords
- fusion gene
- long-read sequencing
- RNA sequencing
Fusion genes are hybrid genes formed when two genes merge due to structural mutations in the genome, such as chromosomal translocations, inversions, or deletions. The abnormal gene fusion results in the production of a hybrid protein that may have altered or abnormal function, potentially driving cancer development, such as chronic myeloid leukemia [1], breast cancer [2], and prostate cancer [3]. Furthermore, fusion genes have been used as biomarkers for molecular targeted drugs, such as BCR::ABL1 [4, 5] and EML4::ALK [6]. Fusion genes have also been used as biomarkers for cancer diagnosis, such as breast cancer [7] and ovarian cancer [8]. Accurate detection of fusion genes is therefore of significant clinical value.
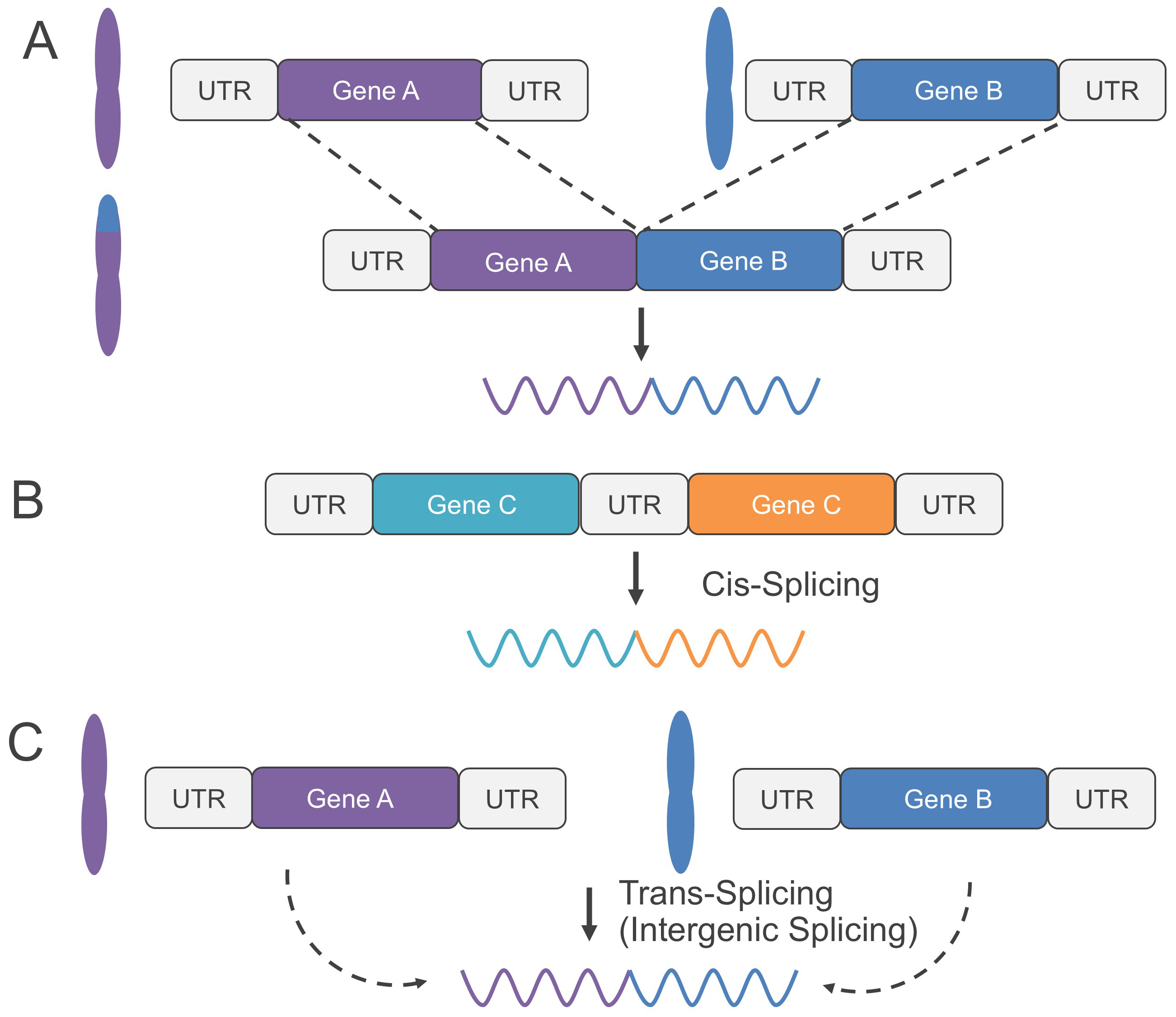
Recently, gene fusions have been classified into two categories: genomic rearrangement-dependent and genomic rearrangement-independent fusions [9]. Genomic rearrangement-dependent fusions are further divided into direct fusions formed by a single structural rearrangement event, and composite fusions produced by multiple structural rearrangements [10]. Fig. 1A illustrates a direct genomic rearrangement-dependent fusion. On the other hand, genomic rearrangement-independent fusions are further divided into cis-splicing between adjacent genes (Fig. 1B) and trans-splicing caused by transcriptional readthrough (Fig. 1C) [10]. The breakpoints of fusion genes resulting from cis-splicing and trans-splicing are significantly closer than those from genomic rearrangements [9]. Furthermore, while gene fusions typically occur in intronic regions, the splice site is conserved. As a result, a fusion transcript is generated where the breakpoint is located at the beginning or end of an exon [11].
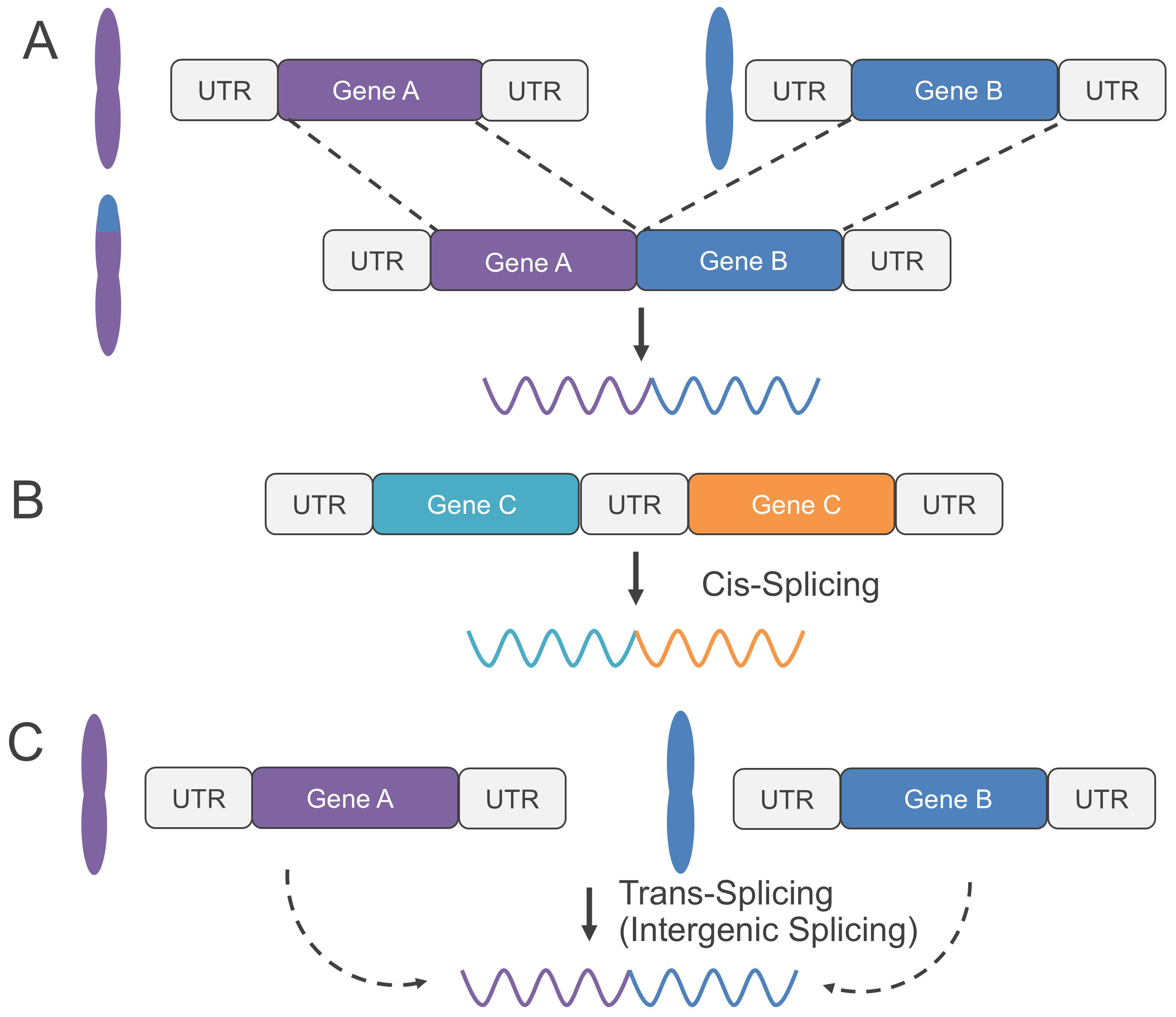 Fig. 1.
Fig. 1.
Classification scheme of gene fusions. (A) Genomic rearrangement-dependent fusion, caused by a single structural rearrangement event. (B) Genomic rearrangement-independent fusions, caused by cis-splicing between adjacent genes. (C) Genomic rearrangement-independent fusions, caused by trans-splicing (transcriptional readthrough in intergenic splicing). UTR, untranslated region.
Transcriptome sequencing (RNA-seq) has been frequently used to target actively expressed fusion genes in cancer cells. There are two types of RNA-seq: short read and long read. Short-read RNA-seq fragments mRNA transcripts into short reads of around 150 bp (base pairs) and deciphers the fragmented sequences. Thus, the detection of fusion genes in the repeat region is considered to be limited [10].
Long-read RNA-seq can directly sequence the full length of mRNA transcripts. Several technologies, such as Oxford Nanopore Technologies (ONT) [12] and Pacific Bioscience (PacBio), offer long-read sequencing. They have been used to detect fusion genes. However, these technologies have a high base-calling error rate compared to short-read RNA-seq. This high error rate may lead to inaccurate alignment, making it challenging to identify the correct fusion genes and breakpoints.
Tools such as LongGF [13], JAFFAL [14], and FusionSeeker [15] have been developed for detecting fusion genes from long-read RNA-seq datasets. These tools align long reads to the reference genome and detect fusion genes by identifying reads that align to multiple genes. We will describe how those existing tools for fusion-gene detection solve the challenges and our fusion-gene detection tool improves the detection accuracy compared to the existing tools.
We used two types of cell lines and four different datasets to evaluate the performance of fusion gene detection (see Table 1). We selected the Michigan Cancer Foundation-7 (MCF7) and SKBR3 cell lines because their long-read RNA-seq datasets have been commonly used as a benchmark for fusion-gene detection, and a previously validated list of fusion genes, considered ground truth, is available [15].
| Dataset | Cell line | Sequence type | Number of sequences | Min length | Average length | Max length |
| MCF7-ONT-1 | MCF7 | Nanopore | 4,181,740 | 45 | 1281 | 15,385 |
| MCF7-ONT-2 | MCF7 | Nanopore | 10,285,910 | 5 | 691 | 5,235,818 |
| MCF7-Pac | MCF7 | PacBio Iso-Seq | 2,389,856 | 17 | 1741 | 12,174 |
| SKBR3-Pac | SKBR3 | PacBio Iso-Seq | 3,070,545 | 12 | 3552 | 50,416 |
MCF7, Michigan Cancer Foundation-7; ONT, Oxford Nanopore Technologies; Pac and PacBio, Pacific Bioscience; Iso-Seq, isoform sequencing; RNA-seq, RNA sequencing.
The reference genome and transcriptome sequence dataset we used was the GENCODE Release version 19 (https://www.gencodegenes.org/human/release_19.html) [16]. We used JAFFAL version 2.2 (https://github.com/Oshlack/JAFFA) and FusionSeeker version 1.0.1 (https://github.com/Maggi-Chen/FusionSeeker) to compare performance with our tool. We also used minimap2 version 2.17 (https://github.com/lh3/minimap2) to align long reads to the reference genome and transcriptome and BLAT (BLAST-Like Alignment Tool) version 36x2 (https://github.com/djhshih/blat) to align gap regions to the reference genome. JAFFAL and FusionSeeker also used the GENCODE dataset as their references. The annotation of the gene set was carried out on genome assembly GRCh37 (hg19) available at: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/.
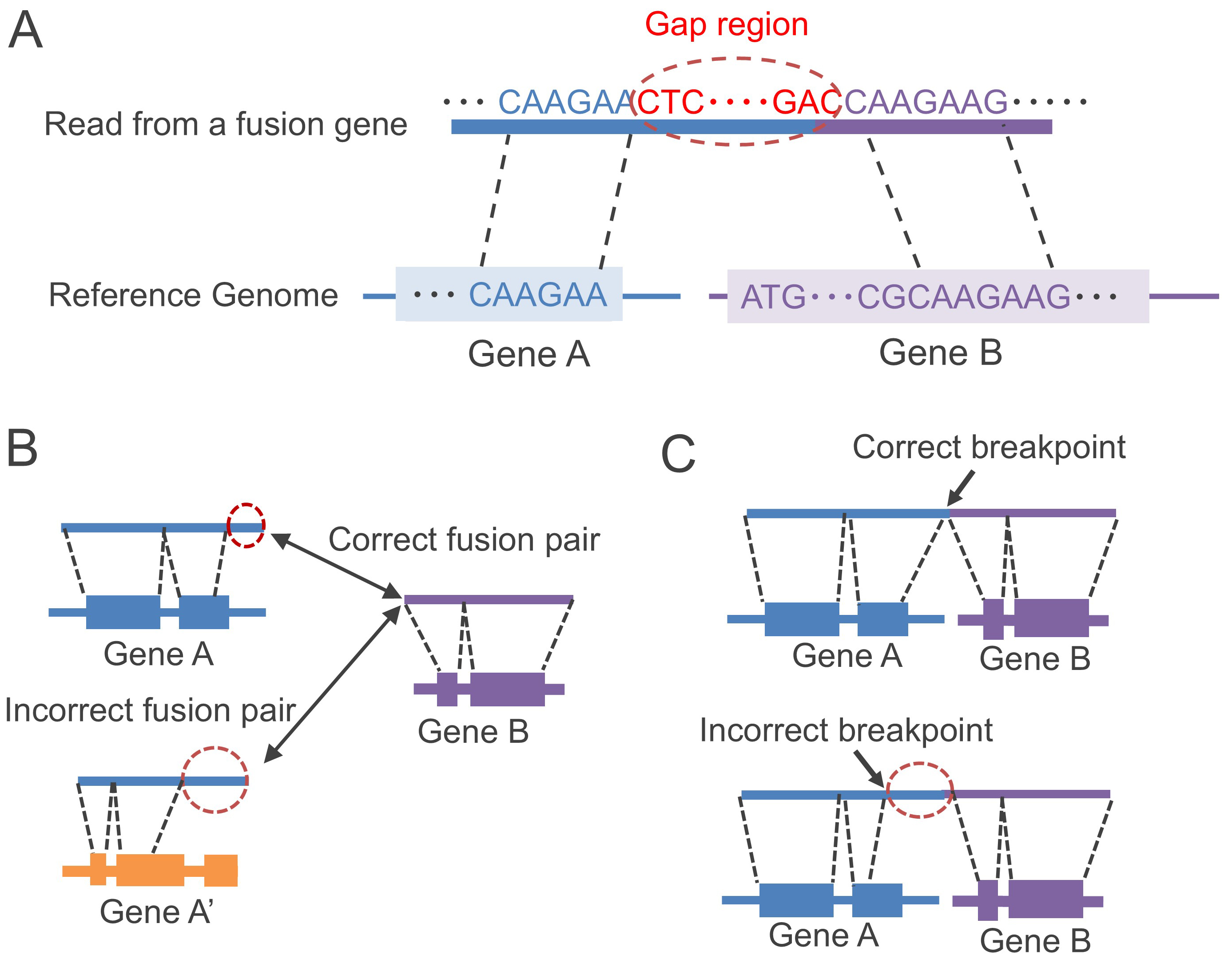
JAFFAL and FusionSeeker have reported superior accuracy compared to LongGF [14, 15]. Thus, we focused on these two tools for performance comparison. JAFFAL and FusionSeeker face two main challenges: (1) detecting the correct fusion genes, and (2) identifying the correct breakpoints (as shown in Fig. 2).
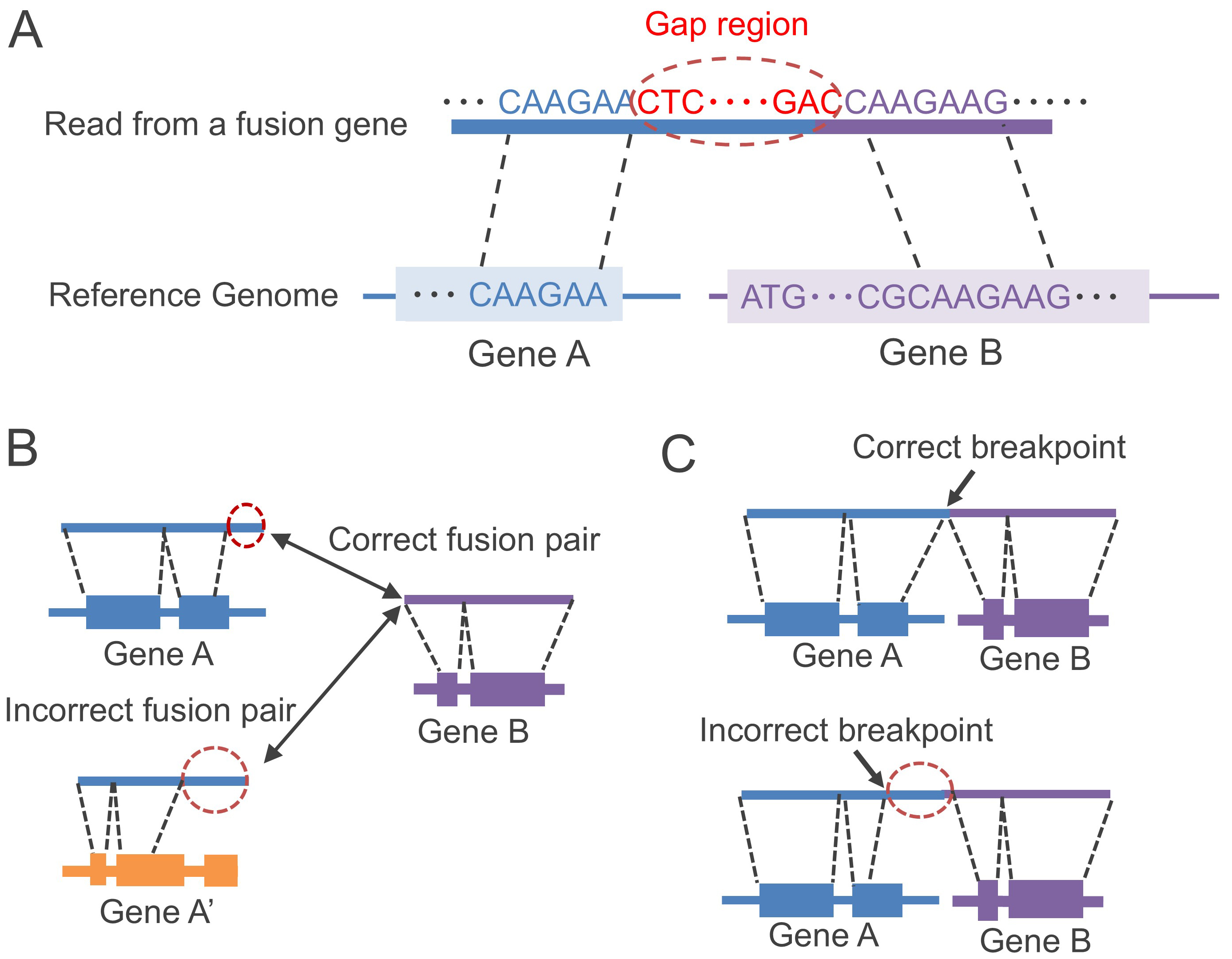 Fig. 2.
Fig. 2.
Challenges in gene fusion detection in long-read RNA-seq. (A) Some sequence regions, named gap regions, are not aligned to the reference genome due to base-calling errors in long-read RNA-seq. (B) There is a possibility of alignment with incorrect genes. (C) Incorrect positions of breakpoints may be detected due to the existence of the gap regions (indicated by red dashed circles).
JAFFAL aligns long reads to both the reference transcript and genome using minimap2 [17] and filters both alignment results by applying constraints. When certain regions of the read near the breakpoint (gap regions as shown in Fig. 2A) fail to align with the genome, possibly due to base-calling errors, it can lead to incorrect alignment with unrelated genes because of partially aligned regions (Fig. 2B), or inaccurate identification of breakpoint positions caused by the gap regions (Fig. 2C). To solve the problem of breakpoint position identification, JAFFAL compares the alignments against annotated genes in the reference genome and realigns breakpoints to exon boundaries [11]. JAFFAL also employs a restriction rule that excludes reads with large gap regions between aligned multiple genes from the analysis. However, the length of the gap region tends to be large in some gene fusion events (such as cis-splicing, as shown in Fig. 1B). In those cases, JAFFAL may not be able to detect fusion genes with large gap regions. Furthermore, either one read of a fusion gene pair can be a short fragment sequence. While minimap2, the alignment tool used in JAFFAL, is robust against sequence noise, it has limitations in accurately aligning short fragment sequences to the genome.
FusionSeeker is a tool that utilizes density-based clustering to accurately identify breakpoints that may contain misalignments. The clustering-based refining process is highly effective, especially given the challenges of achieving precise alignment with the reference genome in long-read RNA-seq. FusionSeeker primarily focuses on identifying isoforms of detected fusion genes and requires a minimum of three supporting reads to generate a consensus sequence.
Previously, we proposed FLBEA (Full-Length and Both-Ends Alignment) to address these challenges [18]. This tool generates fragment sequences from both ends of long reads and aligns them to the genome. The long reads are identified as fusion genes when their fragment sequences are aligned with two different genes. This approach improves precision by incorporating fragment sequences, as relying solely on long reads for fusion gene detection often results in false positives. It is important to note that FLBEA does not directly address the issue of gap regions shown in Fig. 2A.
FLBEA was able to detect fusion genes with higher performance than JAFFAL when the base-calling error rate was low in long reads [18]. However, when the error rate was high (more than 10%), the fragment sequences were not precisely aligned to the reference genome because the fragment sequences were shorter than the original long reads and the accuracy of the alignments was more influenced by the base-calling errors.
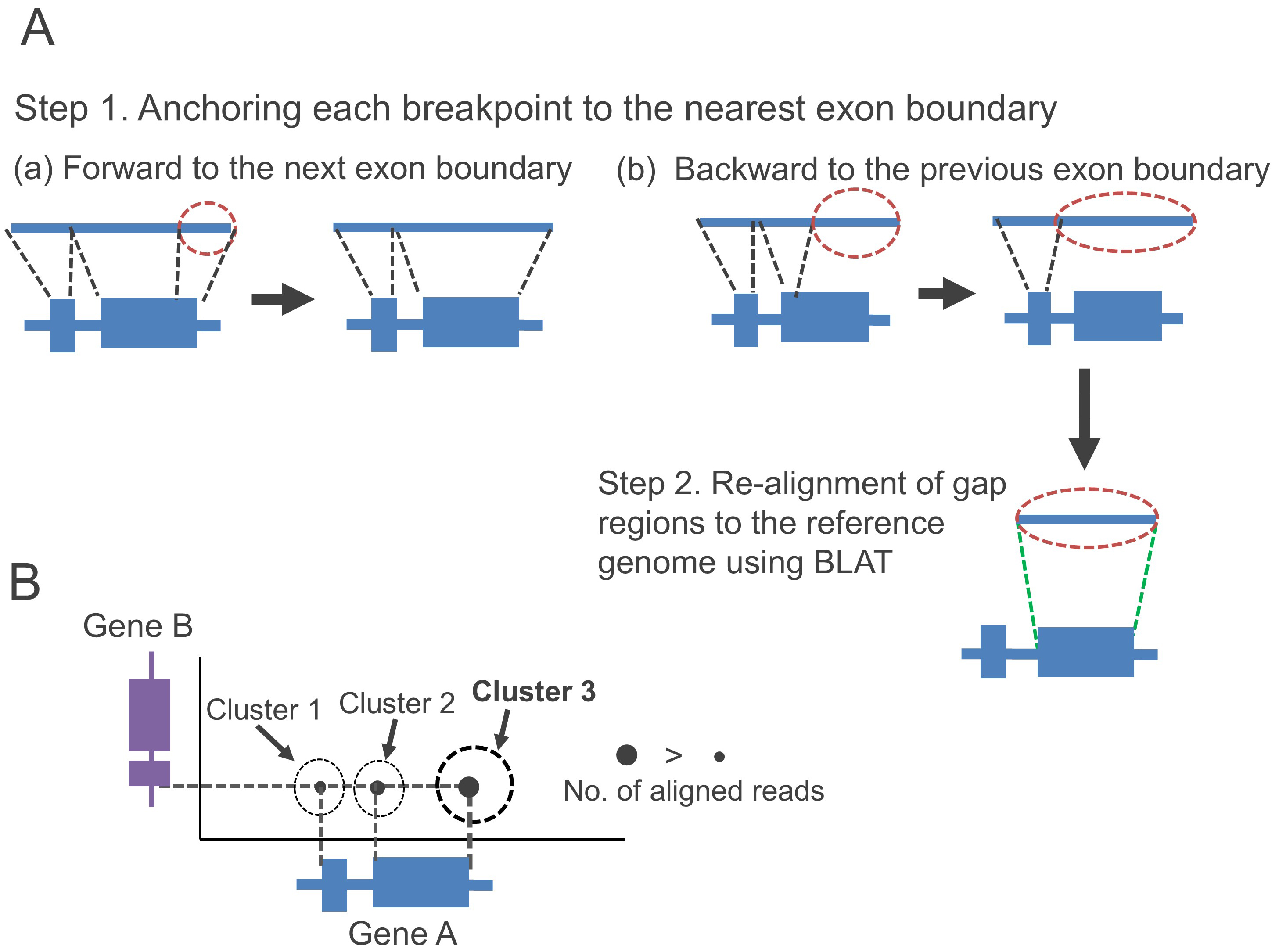
We recently proposed a fusion gene detection tool, FUGAREC (Fusion Detection with Gap Re-alignment) version 1.0 (https://github.com/Hideo-Matsuda/FUGAREC) [19]. FUGAREC aims to solve the challenges shown in Fig. 2 by the following three steps: (1) Anchors breakpoints to exon boundaries (Step 1 in Fig. 3A); (2) To align gap regions that cannot be anchored to exon boundaries (Step 2 in Fig. 3A), we use BLAT [20], instead of minimap2; and (3) Reads are clustered every 1000 bp based on the genomic position of their breakpoints. When multiple fusion reads are within a cluster, the read with the highest number of supporting reads is selected (Fig. 3B).
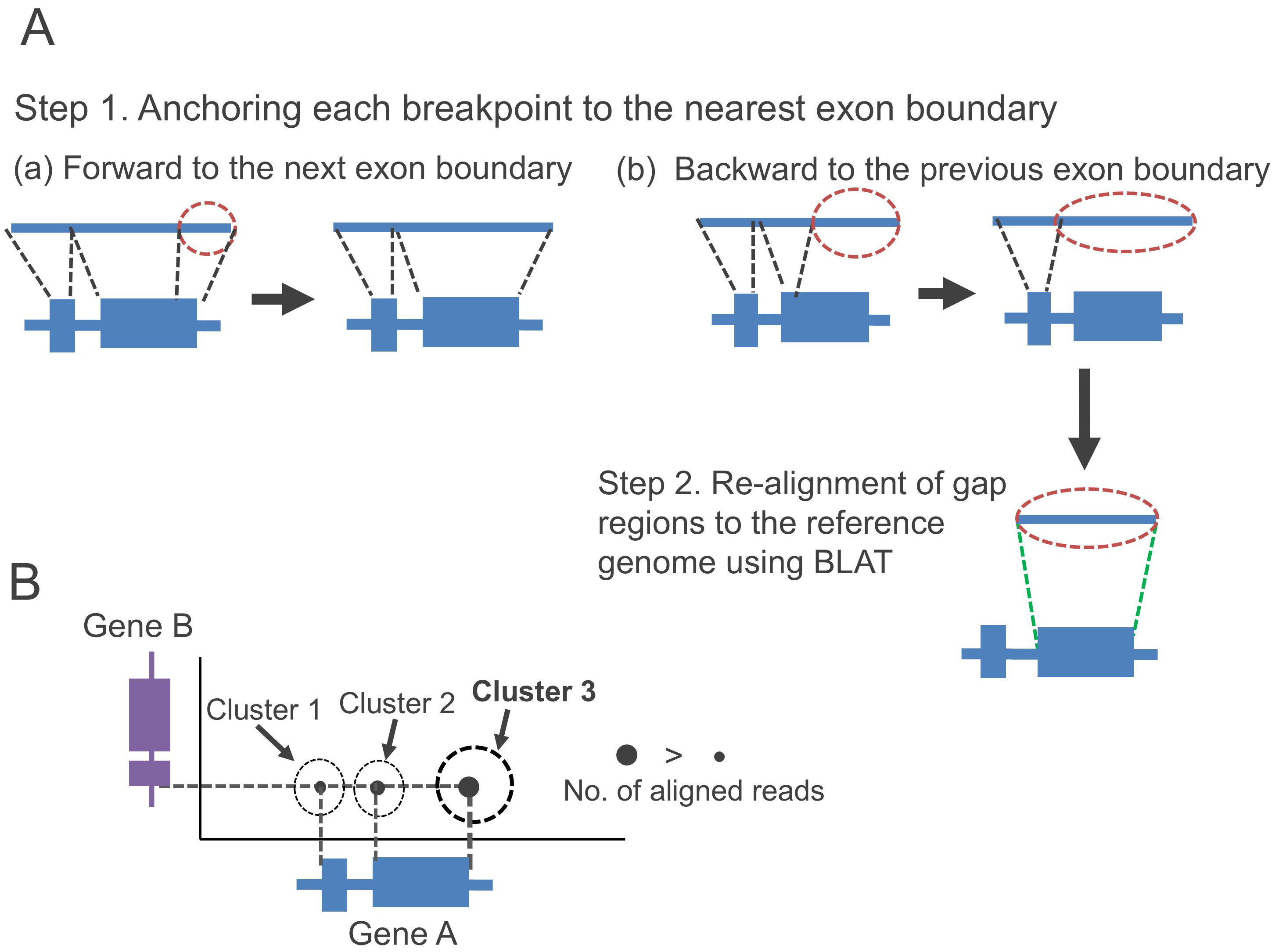 Fig. 3.
Fig. 3.
Solutions for gene fusion detection problems. (A) Anchoring each breakpoint to the nearest exon boundary between forward and backward boundaries (Step 1) and re-alignment of the gap regions to the genome using an alternative alignment tool, BLAST-Like Alignment Tool (BLAT), for (B) identification of breakpoint positions by clustering aligned boundary positions and selecting the most conserved positions.
In Step (1), the breakpoint anchoring is an extension of the JAFFAL method [14] to solve the problem of the gap regions (Fig. 2A). While JAFFAL anchors breakpoints to the exon boundaries only when the breakpoints are located within 50 bp of the exon boundaries, FUGAREC does not set any distance limit from the exon boundaries. Instead, it selects only the nearest boundary positions for anchoring breakpoints. Thus, FUGAREC may be able to detect fusion genes with large gap regions. FusionSeeker does not anchor breakpoints to exon boundaries, but it employs a different approach as described in Step (3).
In Step (2), to solve the problem of detecting the incorrect furoin pair (Fig. 2B), FUGAREC adopts BLAT to re-align gap regions to the reference genome. Since BLAT is specifically designed for aligning short sequences to the reference genome and only FUGAREC realigns the gap regions using BLAT, it may more accurately identify the correct fusion pairs compared to JAFFAL and FusionSeeker.
Step (3) aims to solve the problem of incorrect breakpoints (Fig. 2C). JAFFAL also performs clustering of reads on the genomic position of their breakpoints, which will be either the one preserving exon boundaries, or the one with the highest read support with their breakpoints ranging within 50 bp. FUGAREC achieves clustering of reads within wider range intervals, 1000 bp. In addition, FusionSeeker employs a different approach that performs clustering of supporting reads and a partial order alignment of the reads using bsalign [21] to generate a consensus sequence for each fusion gene. Although the approach can contribute to identifying correct breakpoints, it requires a minimum of three supporting reads. As a result, FusionSeeker may not detect fusion genes with low expression levels. FUGAREC does not create consensus sequences and thus can detect fusion genes even with one or two supporting reads.
Since FUGAREC always outperformed FLBEA in performance comparison using simulated data [19], we do not use FLBEA for further analysis in this paper. An earlier version of this paper [22] reported preliminary results of the fusion gene detection from the same long-read RNA-seq datasets shown in Table 1 using FUGAREC. In this paper, we have largely extended the paper [22] by adding the mechanism of gene fusion formation and discussing the possibility of detection of novel fusion genes.
Fusion genes supported by two or more reads were considered for detection. These
were evaluated as true positives (TP) if the detected genes appeared in the lists
of the previously validated gene fusions [15], and as false positives (FP)
otherwise. False negatives (FN) are known fusion genes that were not detected.
Subsequently, the performance of fusion gene detection was evaluated based on
precision, recall, and F1 score are described as precision = TP/(TP + FP), recall
= TP/(TP + FN), and F1 = 2
To evaluate the performance of our tool in detecting fusion genes, we compared it with JAFFAL and FusionSeeker using the same data. JAFFAL was executed using the configuration file JAFFAL.groovy. FusionSeeker was run with the -datatype nanopore option for Nanopore sequencing data and the -datatype iso-seq option for PacBio sequencing data. FUGAREC was run using minimap2 version 2.17 with the -x map-ont option and using BLAT version 36x2 with the options: -out=blast8 -minScore=10 -stepSize=5.
We evaluated the performance of FUGAREC compared to those of JAFFAL and FusionSeeker as shown in Tables 2,3. FUGAREC achieved the highest precision among the three tools across all long-read datasets. FusionSeeker had the highest recall in the SKBR3 dataset, but it was lower than FUGAREC in the other datasets. FUGAREC demonstrated the highest F1 score across all datasets.
| TP | FP | FN | |||||||
| Dataset | F | J | S | F | J | S | F | J | S |
| MCF7-ONT-1 | 22 | 22 | 19 | 26 | 50 | 93 | 12 | 12 | 15 |
| MCF7-ONT-2 | 21 | 20 | 17 | 19 | 19 | 52 | 13 | 14 | 17 |
| MCF7-Pac | 23 | 25 | 19 | 121 | 149 | 177 | 11 | 9 | 15 |
| SKBR3-Pac | 12 | 12 | 14 | 3 | 21 | 40 | 16 | 16 | 14 |
The numbers of true positives (TP), false positives (FP), and false negatives (FN) for each tool are listed. The highest number in TP and the lowest numbers in FP and FN for each tool is shown in bold. FUGAREC, Fusion Detection with Gap Re-alignment.
| Precision (%) | Recall (%) | F1 (%) | |||||||
| Dataset | F | J | S | F | J | S | F | J | S |
| MCF7-ONT-1 | 45.8 | 30.6 | 17.0 | 64.7 | 64.7 | 55.9 | 53.7 | 41.5 | 26.0 |
| MCF7-ONT-2 | 52.5 | 51.3 | 24.6 | 61.8 | 58.8 | 50.0 | 56.8 | 54.8 | 33.0 |
| MCF7-Pac | 16.0 | 14.4 | 9.7 | 67.6 | 73.5 | 55.9 | 25.8 | 24.0 | 16.5 |
| SKBR3-Pac | 80.0 | 36.4 | 25.9 | 42.9 | 42.9 | 50.0 | 55.8 | 39.3 | 34.1 |
Precision, Recall, and F1 scores for each tool are listed. The highest score for each tool in the three categories is shown in bold.
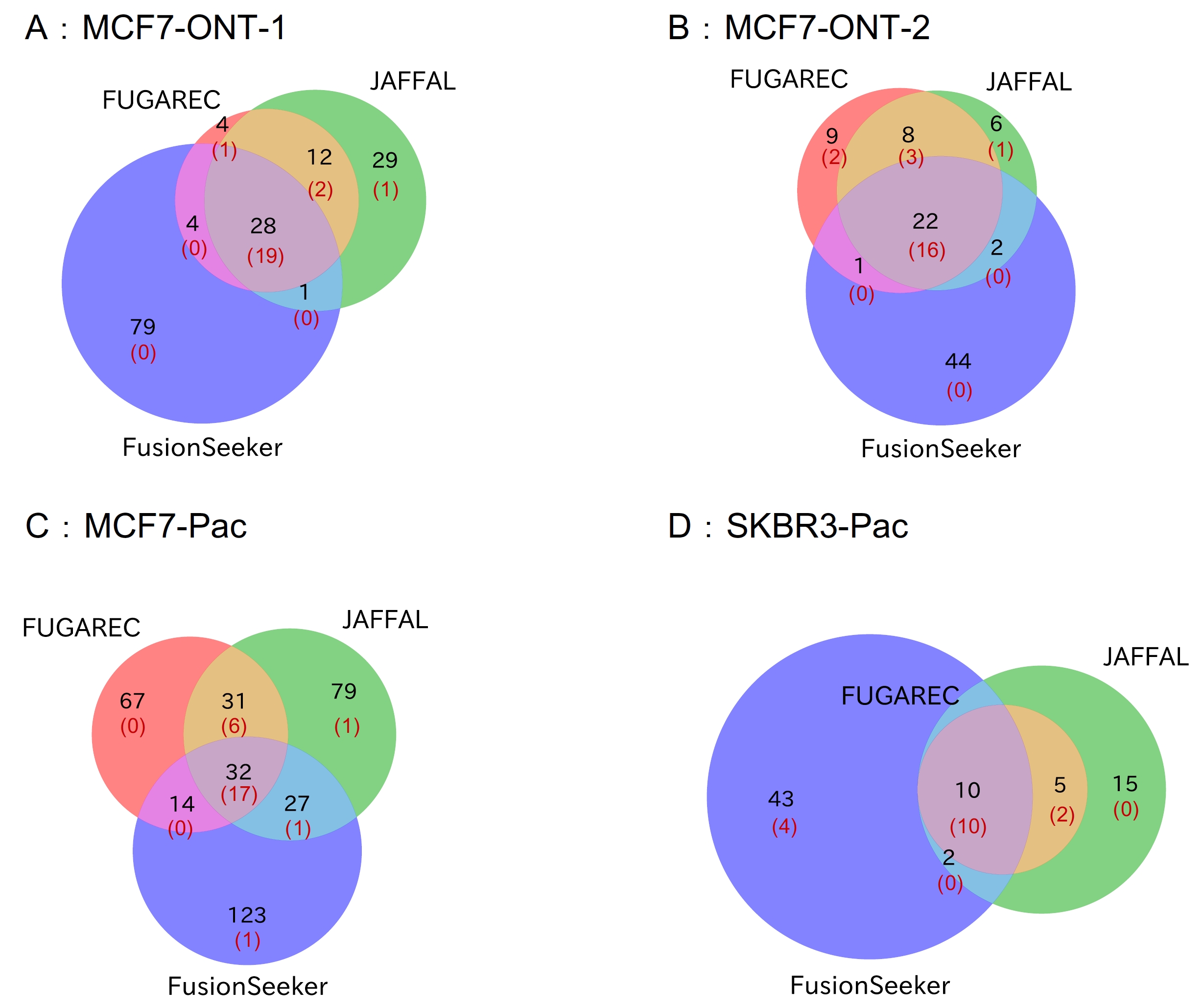
The overlap in the list of fusion genes detected by each tool is shown in Fig. 4. FUGAREC uniquely detected 4, 9, 67, and 0 fusion genes in the MCF7-ONT-1, MCF7-ONT-2, MCF7-Pacific Bioscience (Pac), and SKBR3-Pac datasets, respectively. Of these, 1, 2, 0, and 0 genes, respectively, matched the previously validated fusion genes [15].
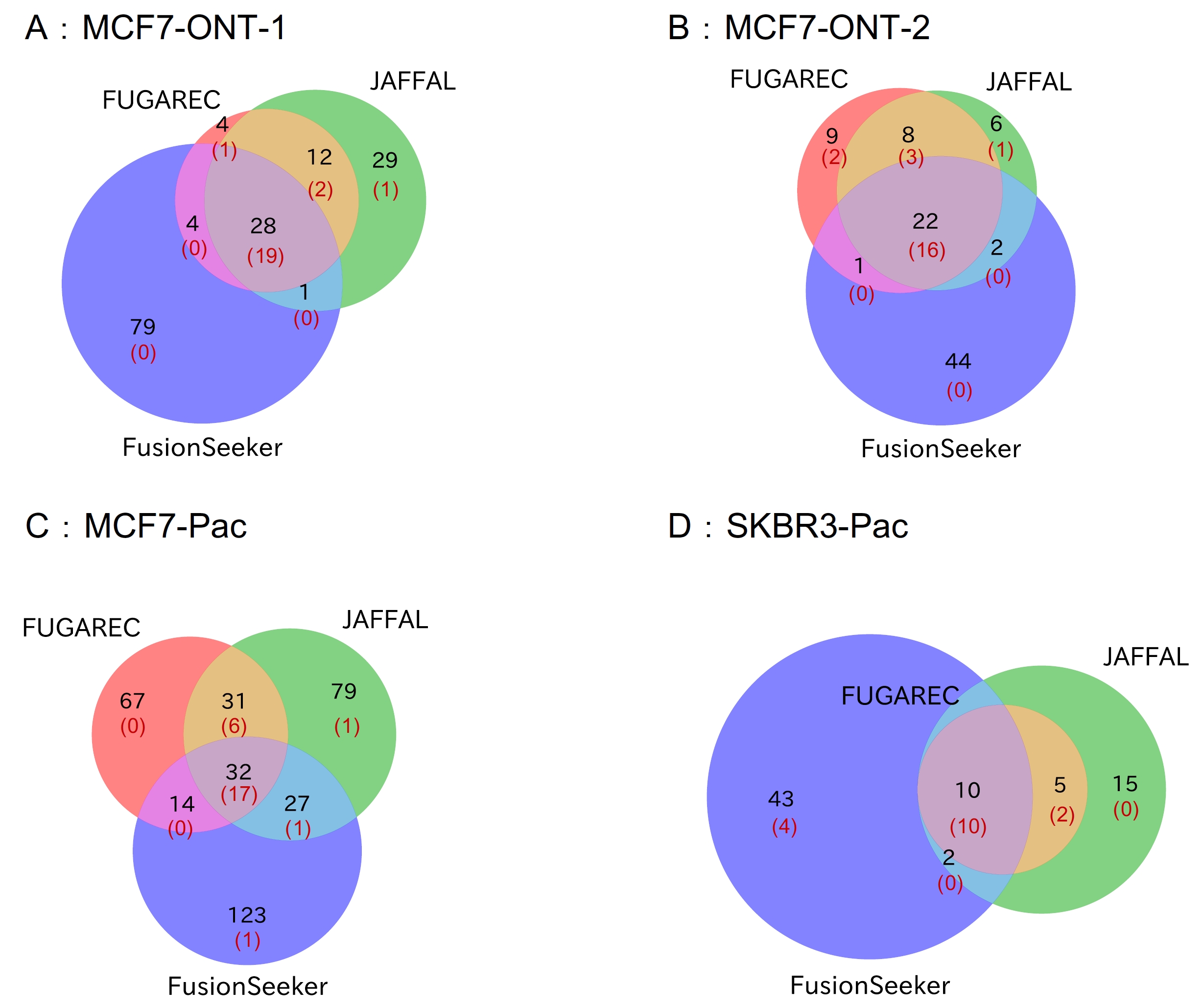 Fig. 4.
Fig. 4.
The overlap in the list of fusion genes detected by each tool in the long-read datasets. Venn diagrams of fusion gene lists by the three tools in datasets of MCF7-ONT-1 (A), MCF7-ONT-2 (B), MCF7-Pac (C), and SKBR3-Pac (D). Black: number of genes detected. Red: The number of genes matched the previously validated fusion genes.
We assume that the gene pairs detected by all three tools (JAFFAL, FusionSeeker, and FUGAREC) are fusion gene candidates. The number of candidates detected by the three tools and not included in the previously validated fusion-gene list is 9, 6, 15, and 0 for the four datasets, respectively. The numbers varied widely across the datasets, and it might not be a good criterion for novel fusion genes due to the difference between the three tools.
Additionally, we counted the number of gene pairs detected in at least two of the four datasets using FUGAREC as fusion gene candidates. The candidates that were not included in the known fusion gene list are presented in Table 4.
| MCF7-ONT-1 | MCF7-ONT-2 | MCF7-Pac | SKBR3-Pac | |
| AC099850.1::VMP1 | 1 | 1 | 1 | 0 |
| AC005808.3::NCOA3 | 1 | 1 | 0 | 0 |
| ATP9A::RP11-347D21.2 | 1 | 1 | 0 | 0 |
| BCAS3::PARD6B | 0 | 1 | 1 | 0 |
| CDKN2B-AS1::MTAP | 1 | 1 | 0 | 0 |
| EIF4E2::GIGYF2 | 0 | 1 | 1 | 0 |
| GLCCI1::RPA3-AS1 | 1 | 1 | 0 | 0 |
| GSN::SMIM22 | 1 | 1 | 0 | 0 |
| RP11-446N19.1::RP11-96H19.1 | 1 | 0 | 1 | 0 |
| RP11-977B10.2::SLC16A7 | 1 | 1 | 0 | 0 |
In this study, we evaluated the performance of our fusion detection tool, FUGAREC, in comparison with the existing tools, JAFFAL and FusionSeeker, in terms of accurately identifying fusion genes and the correct breakpoint. As shown in Fig. 4, FUGAREC and JAFFAL detected more overlapping fusion genes than FusionSeeker, likely because both FUGAREC and JAFFAL use similar approaches of anchoring breakpoints followed by breakpoint clustering. Furthermore, when comparing the number of false positives, FUGAREC and JAFFAL had fewer false positives than FusionSeeker. This suggests that the breakpoint clustering may contribute to reducing false positives.
The computational time of FUGAREC appears to increase compared to that of JAFFAL due to the additional re-alignment step of gap regions using BLAT. However, as shown in Table 2, this step typically occurs only several tens of times in a long-read RNA-seq dataset. Additionally, BLAT is a very fast tool for aligning short sequences to a genome [20]. Therefore, the slight increase in computational time is not considered a significant issue, particularly given the improvement in fusion-gene detection accuracy.
Three fusion genes, undetectable by other tools, were identified solely by FUGAREC (Fig. 4A,B). Two of these fusion genes (NAV1::GPR37L1 and PAPOL::AK7) were undetected in JAFFAL due to long gap regions (Table 5). The lengths of the gap regions identified by JAFFAL for NAV1::GPR37L1 and PAPOL::AK7 were 56 and 32, respectively; however, adjusting the breakpoints with FUGAREC reduced the lengths of the gap regions to 3. Consequently, these two fusion genes were detected by FUGAREC (Table 6).
| Gene1 | Gene2 | Gap length | |||
| Fusion gene (Gene1::Gene2) | Alignment start | Alignment end | Alignment start | Alignment end | |
| NAV1::GPR37L1 | 59 | 142 | 198 | 1383 | 56 |
| PAPOL::AK7 | 25 | 128 | 160 | 997 | 32 |
| Gene1 | Gene2 | Gap length | |||
| Fusion gene (Gene1::Gene2) | Alignment start | Alignment end | Alignment start | Alignment end | |
| NAV1::GPR37L1 | 59 | 163 | 166 | 1383 | 3 |
| PAPOL::AK7 | 25 | 134 | 137 | 997 | 3 |
Conversely, only JAFFAL could detect three fusion genes. For SLC25A24::NBPF6 and SYTL2::PICALM, one long read was partially aligned to three genes. FUGAREC detects fusion reads using only reads partially aligned to two genes. The detection of fusion reads that partially align to three or more genes is a subject for future work. FUGAREC could not detect AHCYL1::RAD51C because the supporting read was 1. JAFFAL could detect it because the supporting read was 2. AHCYL1 has exons at chr1:110546752-110547073 and chr1:110527307-110527794. FUGAREC anchored breakpoints of two fusion-derived reads to different exons. As a result, the support read was decreased.
We used the long-read RNA-seq datasets of breast cancer cell lines, MCF7 and SKBR3, since those datasets have been commonly used as a benchmark for fusion-gene detection and a previously validated list of fusion genes, considered ground truth, is available [15]. It is interesting to explore fusion genes from cancer or normal cells. Gene fusions were once thought to be unique features of cancer. However, fusion transcripts can also be found in normal cells [23]. Although FUGAREC detected fusion genes more precisely than the existing tools, the results still included many false positives (or possibly novel fusion genes). We need more ground truth data or experimental validation to analyze gene fusion in other datasets, such as human tumor cells.
In the process of detecting fusion genes using long-read RNA-seq, a significant challenge has been the inability to identify the correct fusion gene or correct breakpoint due to the presence of gap regions. To address this issue, we proposed a novel tool that integrates the anchoring of breakpoints to exon boundaries, re-alignment of gap regions, and clustering of breakpoints. Our tool demonstrated superior performance in detecting fusion genes compared to existing tools, as evidenced by our tests on two different cancer cell line datasets. Additionally, we identified potential novel fusion genes that were commonly detected across multiple tools or datasets.
However, FUGAREC faces challenges, including the detection of fusion genes from reads that partially align to three or more genes and the presence of multiple exons at close distances on the genome. To make FUGAREC applicable to clinical practice, further validation and improvement of the tool with more samples are necessary.
The two datasets of the Nanopore sequencing data of the MCF7 cell line (MCF7-ONT-1 and MCF7-ONT-2) were downloaded from NCBI Sequence Read Archive (SRA) under the accessions SRP467852 and https://github.com/GoekeLab/sg-nex-data/, respectively. The PacBio Iso-Seq sequencing data of the MCF7 and SKBR3 cell lines (MCF7-Pac and SKBR3-Pac) were downloaded from SRA under the accessions SRP055913 and SRP150606, respectively. The source code of FUGAREC and additional data are available at https://github.com/Hideo-Matsuda/FUGAREC.
KM, YS, and HM designed the research study. YS provided help and advice on fusion genes in breast cancer. KM analyzed the data. KM and HM wrote the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This work was supported in part by JSPS KAKENHI Grant Numbers JP21K19827 Japan.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.