









1 Department of General Surgery, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, 570311 Haikou, Hainan, China
Abstract
Emerging evidence indicates the essential role of cancer stem cells (CSCs) in the development and progression of various cancers, including colorectal cancer (CRC). CELF6, a member of the cytosine-uridine-guanine-binding protein (CUG-BP), Elav-like family (CELF), has been reported to be downregulated in CRC tissues. This study aims to elucidate the role and underlying mechanisms of CELF6 in CRC progression.
The expression levels and prognostic significance of CELF6, along with its association with homeobox A5 (HOXA5), were analyzed using University of Alabama at Birmingham Cancer Data Analysis Portal (UALCAN), PrognoScan, and Tumor Immune Estimation Resource (TIMER) databases. The expression of CELF6 was further assessed through quantitative real-time polymerase chain reaction (qRT-PCR), immunoblotting, and immunohistochemistry. Both in vitro and in vivo experiments were conducted to investigate the effects of CELF6 on CRC cell proliferation, stemness and tumorigenesis, and to elucidate the molecular mechanisms.
CELF6 was found to be downregulated in CRC and was associated with poor prognosis. Functional studies revealed that overexpression of CELF6 resulted in decreased CRC cell proliferation and stemness in vitro, reduced tumor growth in vivo, and induced G1 phase cell cycle arrest. Mechanistically, CELF6 regulated the expression of HOXA5 by modulating its mRNA stability. Furthermore, the knockdown of HOXA5 reversed the inhibitory effects of CELF6 on CRC cell proliferation and stemness, demonstrating that silencing HOXA5 counteracted the suppressive effects of CELF6.
This study is the first to identify CELF6 as a suppressor of stemness and a modulator of CRC progression. These findings provide new insights into the role of CELF6 in CRC and highlight its potential as a novel therapeutic target.
Keywords
- CELF6
- cancer stemness
- colorectal cancer
- HOXA5
- proliferation
Colorectal cancer (CRC) is one of the leading causes of cancer-related mortality worldwide, with 1.9 million new cases and 935,000 deaths reported in 2020 [1]. CRC encompasses both colon and rectal cancers and is characterized by considerable heterogeneity [2]. The intestine’s distinct proximal-to-distal developmental progression results in unique microbial populations and varying gene and protein expression profiles across different intestinal segments, contributing to diverse physiological functions [3]. This variability leads to differences in pathological characteristics, therapeutic approaches, and prognostic outcomes between colon and rectal cancers [4, 5]. Despite advances in surgical techniques and improvements in radiotherapy and chemotherapy regimens [6], there have been limited improvements in mortality rates and prognosis for CRC [7].
Cancer stem cells (CSCs) are recognized for their high oncogenic potential, self-renewal capacity, and ability to differentiate into multiple cell types, playing a crucial role in tumor initiation, progression, and persistence [8, 9]. It has been shown that the activation and maintenance of stemness-associated pathways are closely linked to CRC recurrence and metastasis [10].
The cytosine-uridine-guanine-binding protein (CUG-BP), Elav-like family (CELF) consists of RNA-binding proteins found in both animals and plants that regulate various post-transcriptional processes, including mRNA splicing, degradation, and translation [11]. CELF6, a member of this family, is known to bind mRNA, regulating its stability [12]. CELF6 deficiency has been associated with impaired behavioral responses and autistic-like behaviors [13, 14]. In triple-negative breast cancer, CELF6 acts as a tumor suppressor by binding to the 3′ untranslated region (UTR) of FBP1, thereby enhancing its mRNA stability and expression, which suppresses tumor cell proliferation [15]. Additionally, CELF6 stabilizes p21 mRNA, increasing p21 protein levels and inhibiting tumor cell proliferation and cell cycle progression [16]. Despite these findings, the role of CELF6 in CRC remains underexplored, and its mechanisms are not well understood. Bioinformatics studies suggest that CELF6 expression is reduced in CRC, with lower CELF6 levels correlating with poorer prognosis. Thus, further research is needed to elucidate the significance of CELF6 in CRC and its potential role in regulating CSC “stemness”.
The homeobox (HOX) gene family plays an important role in regulating various biological processes, including growth and development, cell differentiation, proliferation, apoptosis, angiogenesis, and receptor signaling [17, 18]. Homeobox A5 (HOXA5), located on human chromosome 7, participates in embryonic development and adult stem cell differentiation [19]. In addition, its overexpression has been associated with several cancers and cancer progression [20]. In CRC, re-expression of HOXA5 has been shown to inhibit tumor progression and metastasis by disrupting the CSC phenotype [21]. HOXA5 also interacts with HuR, an RNA-binding protein that enhances HOXA5 stability and expression [22]. Using Chromatin immunoprecipitation sequencing (ChIP-seq) technology, CELF6 has been identified as a potential regulator of HOXA5, binding to its 3′ UTR region [23]. Our study further corroborates the positive association between CELF6 and HOXA5 expression in CRC through online database analyses. We hypothesize that CELF6 regulates CRC progression through its interaction with HOXA5.
The University of Alabama at Birmingham Cancer Data Analysis Portal (UALCAN)
database (http://ualcan.path.uab.edu) [24, 25] was utilized to compare the
expression levels of CELF6 between colon adenocarcinoma (COAD) tissues and paired
malignant non-transformed tissues. The prognostic significance of CELF6
expression in CRC was assessed using the PrognoScan database
(http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html) [26]. A significance
threshold of p
Tissue samples were collected from sixty CRC patients, along with matching paracancerous normal tissues. Immediately following excision, all tissue samples were preserved at –80 °C. The study received approval from the Ethics Committee of Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University (Approval No. Med-Eth-Re [2023] 33). Written informed consent was obtained from the patients or their families/legal guardians.
Five human CRC cell lines (HT29, HCT116, LoVo, SW48, and HCT15) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The normal colonic epithelial cell line NCM460 was sourced from the BeNa Culture Collection (Beijing, China). All cell lines were authenticated by short tandem repeat (STR) profiling and were confirmed to be free of mycoplasma contamination. Cells were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and maintained at 37 °C in a 5% CO2 incubator.
The CELF6 cDNA sequence, cloned into the pcDNA vector, and a control vector were provided by GenePharma (Shanghai, China). Additionally, short hairpin RNAs (shRNAs) targeting CELF6 and HOXA5 were also supplied (shNC: 5′-TTCTCCGAACGTGTCACGTAA-3′; shCELF6-1#: 5′-GCCCAGATTTACTTCTTTCAA-3′; shCELF6-2#: 5′-CATACAGACATTCCTGCCCTT-3′; shHOXA5: 5′-GCCATTATAGCGCCTGTATAA-3′). The HCT15 and SW48 cell lines were transfected with these constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Total RNA was extracted from tissue samples and cultured cells using TRIzol reagent (Invitrogen, USA). The RNA was then reverse-transcribed into complementary DNA (cDNA) using the TaqMan™ Reverse Transcription Reagent (Invitrogen, USA). qRT-qPCR was performed using the ABI 7500 Fast Real-Time PCR System. The following primers were used: CELF6, forward: 5′-CCCATCGGGGTCAATGGATTC-3′, reverse: 5′-GCCCGTTATTGTAGAGCGTGT-3′; HOXA5, forward: 5′-AACTCATTTTGCGGTCGCTAT-3′, reverse: 5′-TCCCTGAATTGCTCGCTCAC-3′; GAPDH, forward: 5′-GGAGCGAGATCCCTCCAAAAT-3′, reverse: 5′-GGCTGTTGTCATACTTCTCATGG-3′.
Total protein was extracted from cells and tissues using
radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, Shanghai,
China), and the protein samples were electrophoresized and transferred to
polyvinylidene fluoride (PVDF) membranes at 4 °C. The membranes were
then blocked with 5% skimmed milk at room temperature. Primary antibodies were
applied at 4 °C: anti-CELF6 (ab173282; Abcam, Shanghai, China; 1/1000),
anti-HOXA5 (ab140636; Abcam, China; 1/1000), anti-cyclin-dependent kinase 1
(CDK1) (ab133327; Abcam, China; 1/10,000), anti-cyclin D1 (ab16663; Abcam, China;
1/1000), anti-cyclin B1 (ab32053; Abcam, China; 1/50,000), anti-p27kip1
(ab32034; Abcam, China; 1/5000), anti-Nanog (ab109250; Abcam, China; 1/2000),
anti-OCT4 (ab200834; Abcam, China; 1/10,000), anti-SRY-box transcription factor 2
(SOX2) (ab92494; Abcam, China; 1/1000), anti-CD133 (ab222782; Abcam, China;
1/2000), and anti-
Tissue sections were fixed in 4% formaldehyde, embedded in paraffin, and subjected to antigen retrieval. Sections (5 µm) were incubated with the following primary antibodies at room temperature: anti-CELF6 rabbit monoclonal antibody (Abcam, China; ab173282; 1/50 dilution), anti-HOXA5 rabbit polyclonal antibody (Invitrogen, USA; PA5-69008; 5.0 µg/mL dilution), anti-SOX2 rabbit monoclonal antibody (Abcam, China; ab92494; 1/1000 dilution), and anti-CD133 rabbit monoclonal antibody (Abcam, China; ab222782; 1/1000 dilution). Biotinylated secondary antibodies were then applied, and the sections were visualized using a microscope after staining with 3,3’-diaminobenzidine (DAB) chromogenic agents.
Cell viability was assessed using the cell counting kit-8 (CCK-8) assay (Beyotime Biotechnology, Shanghai, China). Cells were plated into 96-well plates at a density of 2000 cells per well and incubated for 0, 24, 48, or 72 hours. Following incubation, 10 µL of CCK-8 solution was added to each well, and absorbance was measured at 450 nm using a microplate reader. For the colony formation assay, the cells were seeded at a density of 1000 cells per well in 6-well plates. After two weeks of incubation, colonies were fixed, stained, and counted under a microscope (Olympus, Tokyo, Japan).
Cell cycle distribution was analyzed using the Cell Cycle and Apoptosis Analysis Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Aldehyde dehydrogenase 1 (ALDH1)-PE (#65583; 1/50 dilution) and CD44-allophycocyanin (APC) (#80813; 1/160 dilution) antibodies were used to identify ALDH1- or CD44-positive cells. FCM was conducted using a flow cytometer (Beckman Coulter, Fullerton, CA, USA) after staining.
The cells were collected and prepared as a single-cell suspension in a medium supplemented with basic fibroblast growth factor (20 ng/mL), recombinant human epidermal growth factor (10 ng/mL), and B27 (2%). Sphere formation was assessed after 14 days when spheres reached a diameter of 50 µm.
The stability of HOXA5 mRNA was evaluated using an ActD assay. The cells, either transfected with CELF6 overexpression plasmids or control cells, were treated with ActD (5 µg/mL; Sigma-Aldrich, St. Louis, MO, USA). HOXA5 mRNA levels were measured by qRT-PCR at various time points (0, 2, 4, and 8 hours).
The wild-type or mutant HOXA5 3′ UTR sequences were cloned into luciferase reporter vectors. These vectors were co-transfected with CELF6 overexpression plasmids, and luciferase activity was measured using the Dual Luciferase Assay Kit (Thermo Scientific, China).
The RIP assay was performed using the Dynabeads™ Protein G Immunoprecipitation Kit (Invitrogen, USA). Cell lysates were incubated with anti-CELF6 (5 µg) or IgG-conjugated magnetic beads. The RNA-protein complexes were then isolated and analyzed by qPCR.
The cell lysates were incubated with 50 pmol of a biotin-labeled HOXA5-RNA probe in an RNA binding buffer at 4 °C for 2.5 hours. RNA pull-down assays were performed using the Pierce™ Magnetic RNA-Protein Pull-Down Kit (Thermo Scientific, China). After incubation, the complexes were captured with pre-saturated streptavidin beads for 30 minutes, washed, and then subjected to western blot analysis.
All animal experiments were approved by the Institutional Animal Care and Use
Committee of Hainan General Hospital, Hainan Affiliated Hospital of Hainan
Medical University (Approval No. Med-Eth-Re [2023] 33). CELF6 overexpressing or
control SW48 cells (5
Data are expressed as mean
The investigation began with an analysis of CELF6 expression in colon adenocarcinoma (COAD) using the UALCAN database. This analysis revealed that CELF6 expression was significantly reduced in COAD tissues compared to healthy controls (Fig. 1A). To further explore the clinical relevance of CELF6, we examined its association with survival outcomes in CRC patients through the PrognoScan database. Our analysis indicated that lower CELF6 levels were correlated with poorer survival rates, including overall survival (Cox P = 0.039360) and disease-specific survival (Cox P = 0.081047) (Fig. 1B).
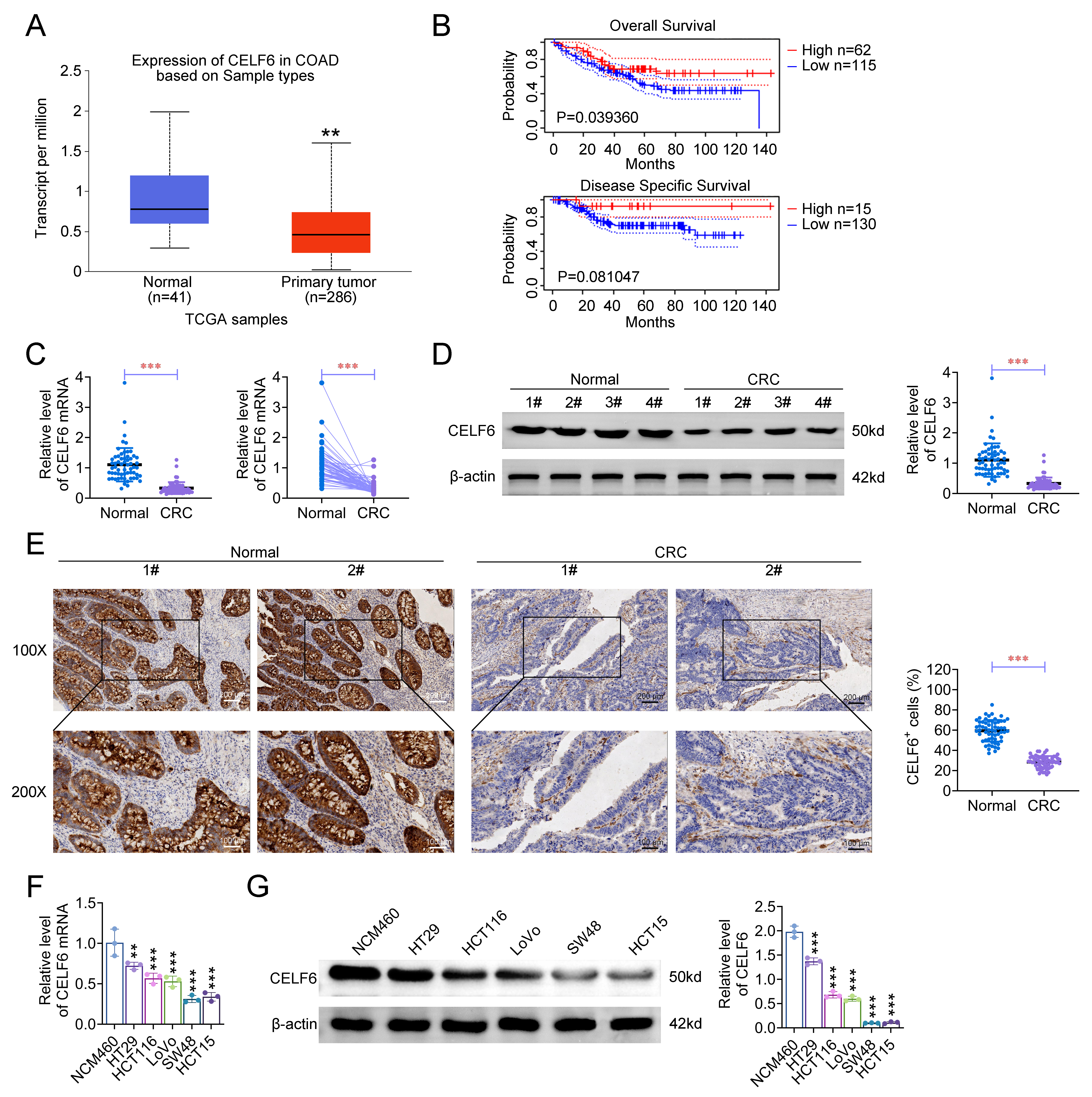 Fig. 1.
Fig. 1.
Cytosine-uridine-guanine-binding protein (CUG-BP), Elav-like
family (CELF) 6 is downregulated in colorectal cancer (CRC) tissues and cells.
(A) Analysis of CELF6 expression in colon adenocarcinoma (COAD) and adjacent
non-transformed tissues using data from The Cancer Genome Atlas (TCGA) database
accessed via UALCAN. (B) Kaplan-Meier survival curves comparing high and low
CELF6 expression levels in CRC patients, as retrieved from the PrognoScan
database. (C) quantitative real-time polymerase chain reaction (qRT-PCR) analysis
of CELF6 mRNA expression in 60 CRC tissues and corresponding adjacent
non-cancerous tissues (unpaired, left; paired, right). (D) Immunoblotting
analysis of CELF6 protein expression in four randomly selected pairs of CRC and
adjacent non-cancerous tissues. (E) Representative immunohistochemistry (IHC)
images and statistical results showing CELF6 expression in two pairs of CRC and
adjacent non-transformed tissues at 100
Next, we validated these findings in a cohort of 60 CRC tumor specimens and their matched adjacent non-cancerous tissues. qRT-PCR analysis demonstrated a significant reduction in CELF6 mRNA expression in CRC tissues compared to normal tissues (Fig. 1C). Western blotting and immunohistochemistry (IHC) further confirmed the downregulation of CELF6 protein in CRC tissues (Fig. 1D,E). Similar trends were observed in CRC cell lines, where both mRNA and protein levels of CELF6 were significantly lower compared to the normal colonic epithelial cell line NCM460 (Fig. 1F,G). These results suggest that CELF6 expression may serve as a diagnostic and prognostic biomarker for CRC.
To investigate the functional role of CELF6 in CRC, we established stable HCT15 and SW48 cell lines with either CELF6 overexpression or knockdown (Fig. 2A). Overexpression of CELF6 resulted in reduced cell viability (Fig. 2B) and decreased colony formation (Fig. 2C,D) in both cell lines. Conversely, CELF6 knockdown led to increased cell proliferation. FCM analysis revealed that CELF6 overexpression induced G1 cell cycle arrest in both HCT15 and SW48 cells, whereas CELF6 knockdown impeded this G1 arrest (Fig. 2E,F). Western blot analysis showed that CELF6 overexpression reduced the protein levels of cell cycle regulators CDK1, cyclin D1, and cyclin B1, while increasing the expression of the cyclin-dependent kinase inhibitor p27kip1. In contrast, CELF6 knockdown resulted in elevated levels of CDK1, cyclin D1, and cyclin B1, along with decreased levels of p27kip1 (Fig. 2G). These findings suggest that CELF6 modulates CRC cell proliferation by regulating cell cycle progression.
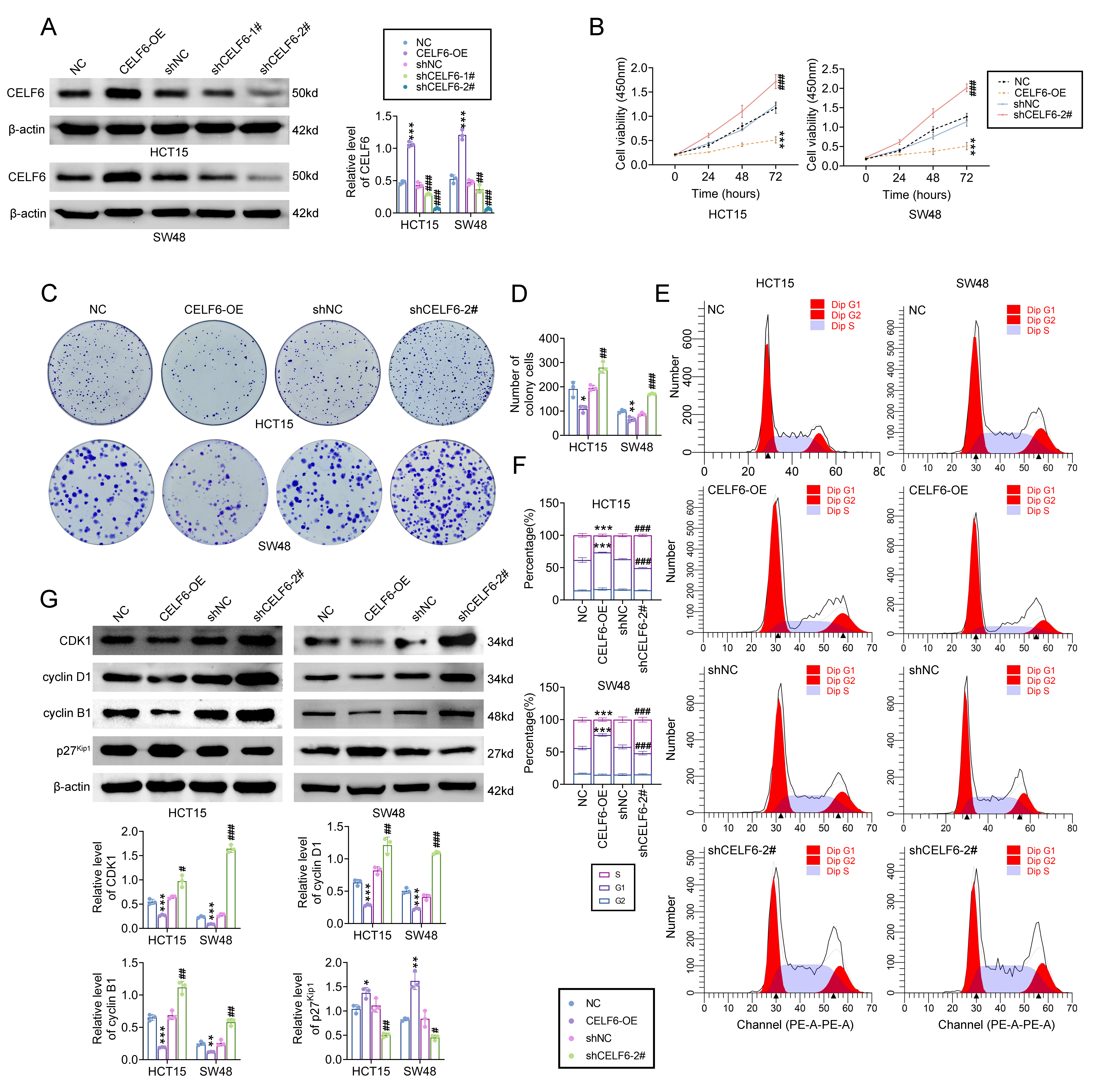 Fig. 2.
Fig. 2.
CELF6 inhibits CRC cell proliferation and cell cycle
progression. (A) Western blot analysis of CELF6 levels in HCT15 and SW48 cells
with CELF6 overexpression or knockdown. CELF6 knockdown was achieved using two
different shRNAs (shCELF6-1# and shCELF6-2#). (B) Assessment of cell
proliferation by cell counting kit-8 (CCK-8) assays in CELF6 overexpressed or
knocked-down HCT15 and SW48 cells. (C,D) Colony formation assays evaluating the
impact of CELF6 overexpression or knockdown on cell growth in HCT15 and SW48
cells. (E,F) Flow cytometry analysis of cell cycle distribution in CELF6
overexpressed or knocked-down HCT15 and SW48 cells. (G) Western blot analysis of
cell cycle proteins cyclin-dependent kinase 1 (CDK1), cyclin D1, cyclin B1, and
p27^kip1 following CELF6 overexpression or knockdown. The
results are presented as the mean
To explore the role of CELF6 in regulating stem cell characteristics in CRC, we assessed the expression of CSC markers, including Nanog, OCT4, SOX2, and CD133. Western blot analysis revealed that CELF6 overexpression significantly reduced the levels of these CSC markers, whereas CELF6 knockdown led to increased expression of these markers (Fig. 3A). Sphere formation assays demonstrated that CELF6 overexpression inhibited the sphere-forming ability of HCT15 and SW48 cells, indicating reduced stemness, while CELF6 knockdown enhanced sphere formation (Fig. 3B). Additionally, FCM analysis showed that CELF6 overexpression resulted in a significant reduction in ALDH1 and CD44 activities in HCT15 and SW48 cells. Conversely, CELF6 knockdown increased the activities of ALDH1 and CD44 (Fig. 3C). These results collectively suggest that CELF6 plays a critical role in diminishing the stemness features of CRC cells in vitro.
 Fig. 3.
Fig. 3.
CELF6 suppresses the stemness features of CRC cells. (A)
Western blot analysis of stem cell markers Nanog, OCT4, SOX2, and CD133 in HCT15
and SW48 cells with CELF6 overexpression (CELF6-OE) or knockdown (shCELF6-2#).
(B) Representative images and quantification of sphere formation assays in HCT15
and SW48 cells with CELF6 overexpression or knockdown. Scale bars, 200 µm.
(C) Flow cytometry analysis of ALDH1 and CD44 activities in HCT15 and SW48 cells
with CELF6 overexpression or knockdown. The results are presented as the mean
To investigate the role of CELF6 in CRC development, we examined its effect on HOXA5 expression. The results indicated that CELF6 positively regulates HOXA5 expression in both HCT15 and SW48 cell lines. Specifically, CELF6 overexpression resulted in a significant increase in HOXA5 expression at both the mRNA and protein levels (Fig. 4A,B). Conversely, CELF6 knockdown led to a decrease in HOXA5 expression. qRT-PCR analysis of CRC tissues confirmed reduced HOXA5 mRNA levels in tumor samples compared to adjacent non-cancerous tissues (Fig. 4C). A positive correlation between CELF6 and HOXA5 mRNA levels was also observed in clinical samples, highlighting a potential regulatory relationship (Fig. 4D). IHC analyses further supported these findings, showing downregulation of HOXA5 protein expression in CRC tissues (Fig. 4E,F). Collectively, these data suggest that HOXA5 is upregulated following CELF6 overexpression and may act as a downstream effector in the regulatory network mediated by CELF6, influencing CRC progression.
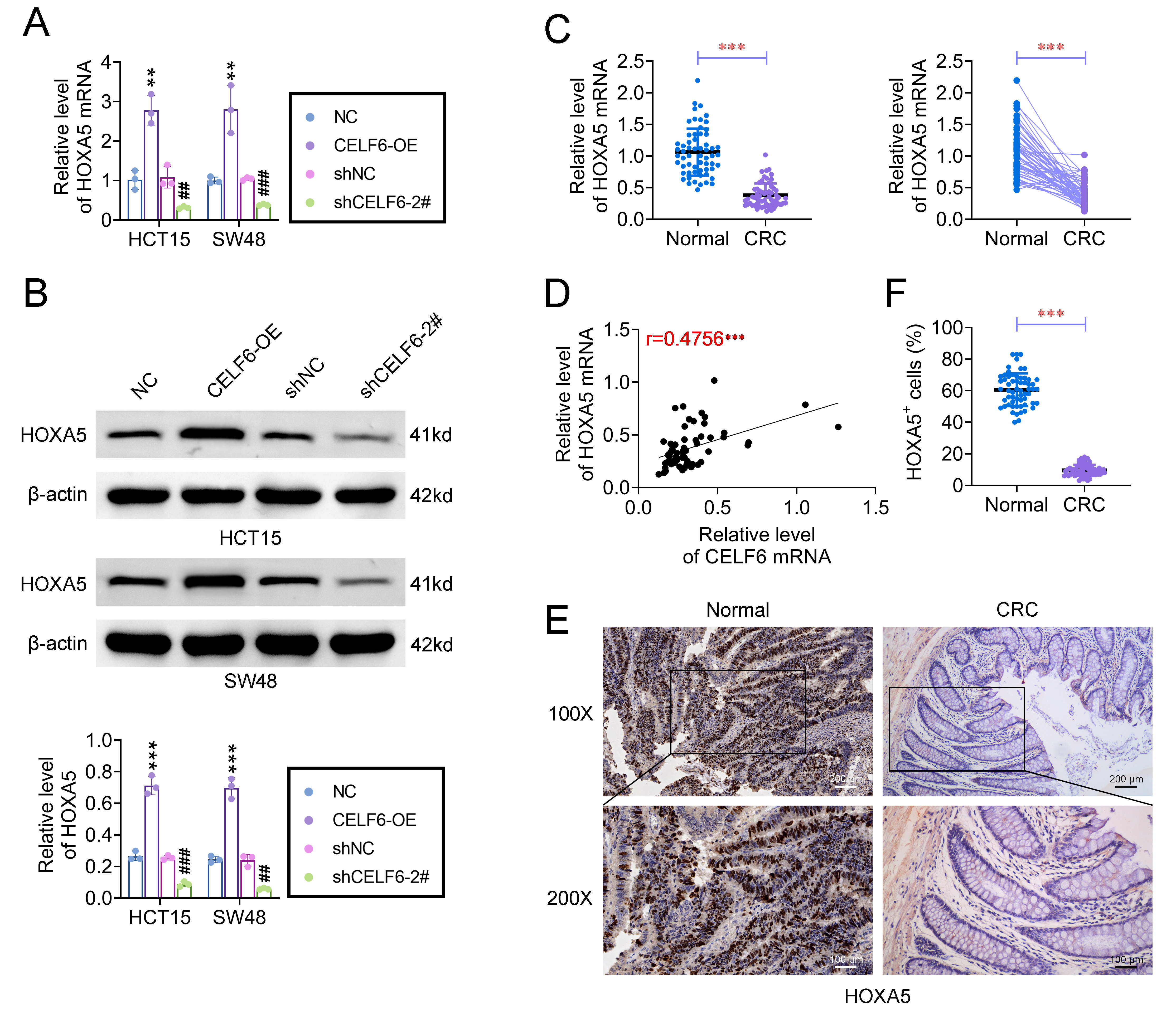 Fig. 4.
Fig. 4.
CELF6 promotes HOXA5 expression in CRC cells. (A,B) Impact of
CELF6 overexpression (CELF6-OE) or knockdown (shCELF6-2#) on HOXA5 mRNA and
protein levels assessed by qRT-PCR and western blotting. (C) Relative mRNA
expression levels of HOXA5 in 60 paired CRC tissues and adjacent non-cancerous
tissues measured by qRT-PCR. (D) Correlation analysis between HOXA5 mRNA and
CELF6 expression levels in 60 CRC samples. (E,F) Representative IHC images and
quantification of HOXA5 protein expression in CRC and adjacent non-cancerous
tissues at 100
Regulation of mRNA stability by RNA binding proteins (RBPs) is a well-established mechanism [27]. We hypothesized that CELF6 interacts with HOXA5 and influences its mRNA stability. Actinomycin D (Act D) assays demonstrated a gradual decrease in HOXA5 mRNA levels in control cells, whereas CELF6 overexpression significantly slowed the decay of HOXA5 mRNA (Fig. 5A), indicating that CELF6 stabilizes HOXA5 transcripts.
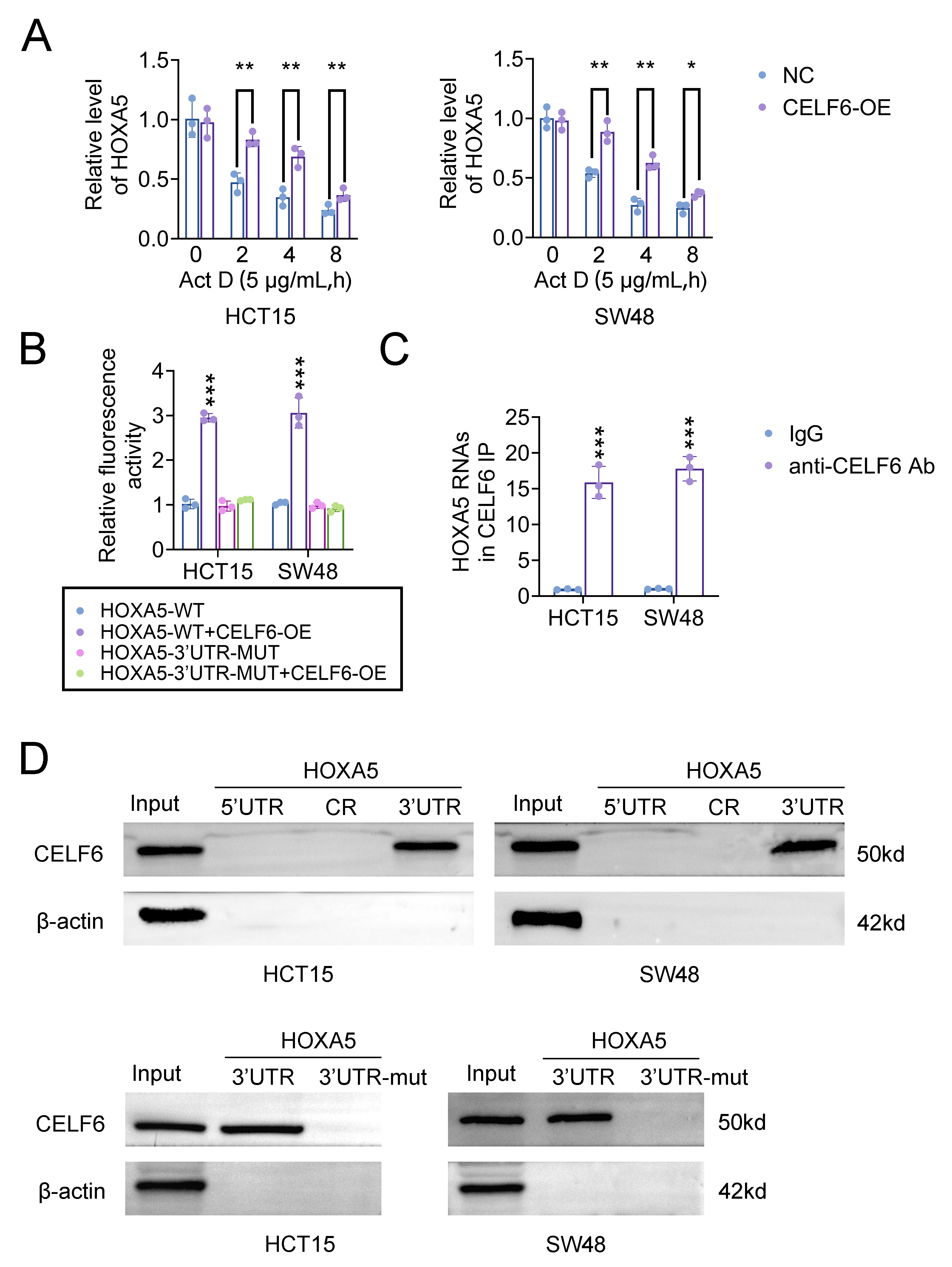 Fig. 5.
Fig. 5.
CELF6 positively modulated HOXA5 by binding to its 3′ UTR. (A) qRT-PCR analysis of HOXA5 mRNA levels in
CELF6-overexpressing and control HCT15 and SW48 cells treated with Actinomycin D
for the indicated durations. Statistical significance is indicated as *p
To investigate whether CELF6 stabilizes HOXA5 mRNA through direct interaction, we cloned the HOXA5 3′ UTR and a control vector into luciferase reporter plasmids. These plasmids were introduced into CELF6-overexpressing cell lines and their respective controls. Dual luciferase reporter gene assay showed that in CELF6-overexpressed CRC cells, the luciferase activities of the wild-type HOXA5 3′ UTR reporter were significantly increased, whereas those of the mutant reporter were not significantly altered (Fig. 5B). Further analysis using RIP assays confirmed a direct interaction between CELF6 protein and HOXA5 mRNA (Fig. 5C). To corroborate this interaction, RNA pull-down assays coupled with western blotting were performed. The results indicated that CELF6 was specifically enriched in the HOXA5 3′ UTR, with no significant enrichment in the 5′ UTR or coding region (CR) (Fig. 5D). Overall, these findings suggest that CELF6 mediates the regulation of HOXA5 mRNA stability through direct binding to its 3′ UTR.
Given that CELF6 regulates HOXA5 mRNA stability and abundance, we aimed to validate HOXA5 as a key mediator of CELF6’s antitumor effects. To investigate this, HCT15 and SW48 cells with CELF6 overexpression were transfected with shRNA targeting HOXA5 or a negative control (shNC). Western blot analysis demonstrated that CELF6 overexpression significantly counteracted the reduction of HOXA5 protein levels caused by HOXA5 knockdown in both cell lines (Fig. 6A).
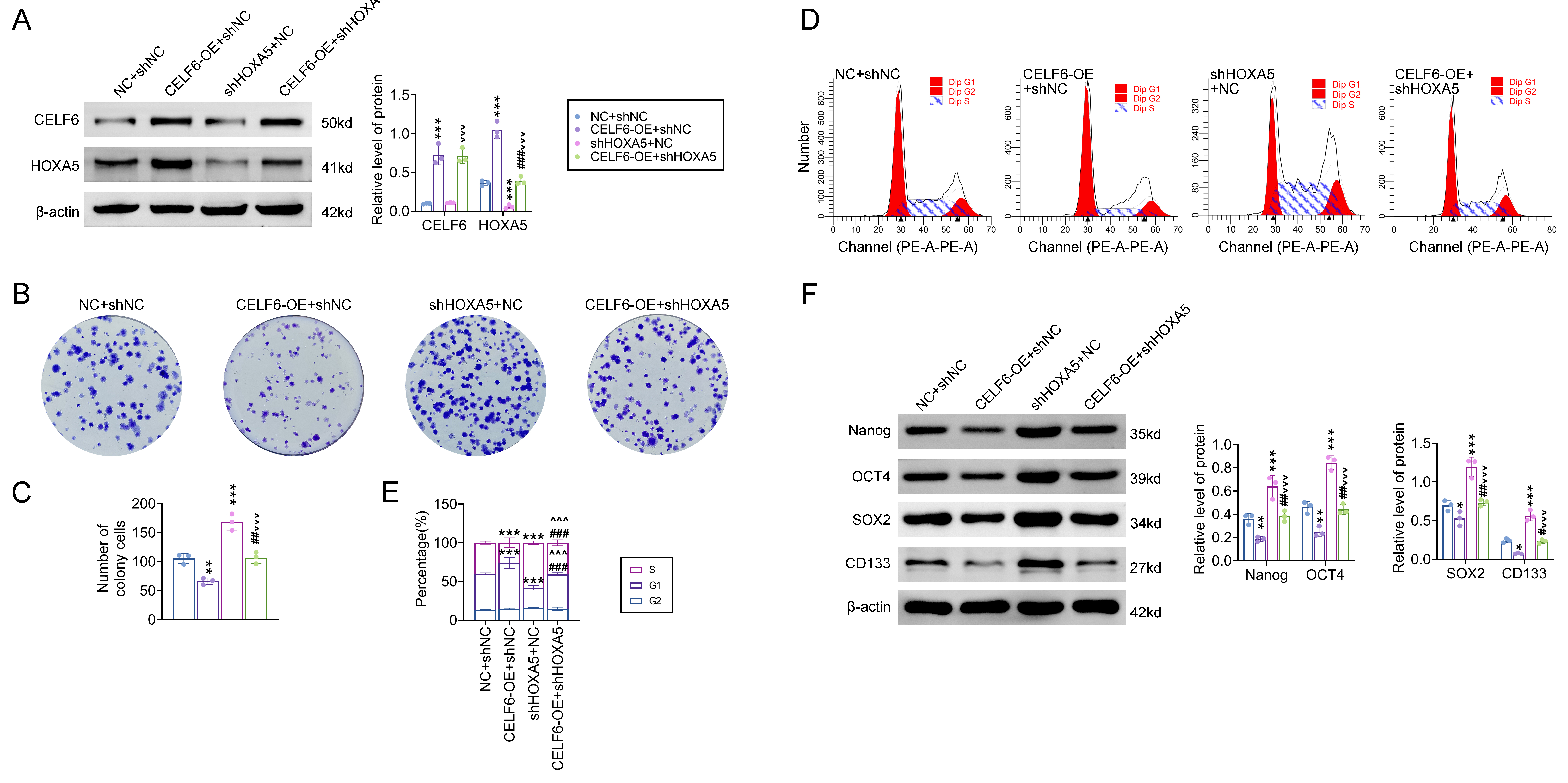 Fig. 6.
Fig. 6.
Knockdown of HOXA5 reverses the antitumor activity of CELF6.
The HOXA5 gene was knocked down in HCT15 and SW48 cells overexpressing CELF6
through transfection with shHOXA5 or shNC. (A) Western blot analysis of CELF6 and
HOXA5 protein levels in HCT15 and SW48 cells overexpressing CELF6 and transfected
with shHOXA5 or shRNA. (B,C) Colony formation assays assessing the proliferative
potential of CELF6-overexpressing cells with HOXA5 knockdown. (D,E) Flow
cytometry analysis of cell cycle distribution in CELF6-overexpressing cells with
or without HOXA5 knockdown. (F) Protein levels of Nanog, OCT4, SOX2, and CD133 in
CELF6-overexpressing cells with HOXA5 knockdown assessed by western blotting. The
results are presented as the mean
Colony formation assays revealed that HOXA5 knockdown significantly restored the proliferative capacity of CELF6-overexpressing cells (Fig. 6B,C). Furthermore, FCM analysis indicated that HOXA5 knockdown reversed the G1 cell cycle arrest induced by CELF6 overexpression (Fig. 6D,E). Additionally, the suppression of HOXA5 attenuated the reduction in stemness markers Nanog, OCT4, SOX2, and CD133 caused by CELF6 overexpression (Fig. 6F). Collectively, these results suggest that HOXA5 plays an essential role in mediating CELF6’s effects on CRC cell aggressiveness.
To investigate the impact of CELF6 on tumorigenesis in vivo, we conducted xenograft experiments using SW48 cells with CELF6 overexpression. Consistent with our in vitro findings, tumors derived from CELF6-overexpressing cells grew significantly slower compared to those from control cells (Fig. 7A). Additionally, the tumor volume and weight were notably reduced in the CELF6 overexpression group (Fig. 7B,C). Additionally, qRT-PCR analysis of xenograft tumors revealed increased mRNA levels of both CELF6 and HOXA5 in the CELF6 overexpression group (Fig. 7D), suggesting that CELF6 modulates HOXA5 expression in vivo. IHC staining of tumor tissue sections further demonstrated elevated expression levels of CELF6 and HOXA5, while the stem cell markers SOX2 and CD133 showed a marked decrease in the CELF6 overexpression group (Fig. 7E,F). These results indicate that CELF6 inhibits CRC tumor growth in vivo through the regulation of HOXA5.
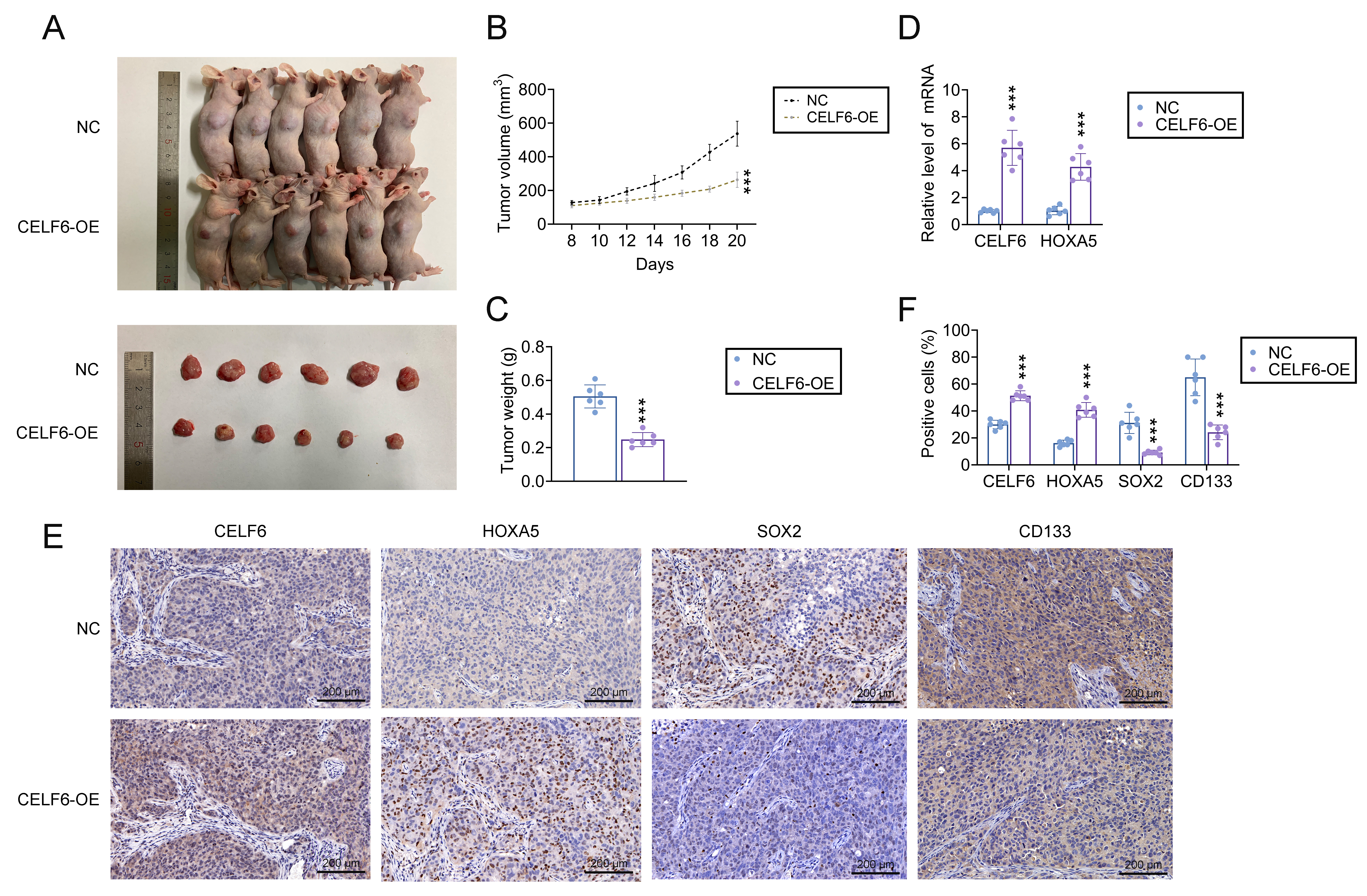 Fig. 7.
Fig. 7.
CELF6 overexpression ameliorates the CRC cell growth in
vivo. Nude mice were inoculated with either CELF6-overexpressing SW48 cells or
control cells. (A) Representative images of nude mice and tumor xenografts
derived from CELF6-overexpressing and control SW48 cells. (B,C) Measurement of
tumor size and weight in CELF6-overexpressing versus control groups. (D) qRT-PCR
analysis of CELF6 and HOXA5 expression in xenograft tumors. (E,F) Representative
IHC images (scale bar = 200 µm) and statistical analysis of CELF6, HOXA5,
SOX2, and CD133 expression in tumor sections. The results are presented as the
mean
This present study demonstrates that CELF6 expression is significantly reduced in CRC tissues and correlates with adverse clinical outcomes. CELF6 overexpression was associated with reduced CRC cell proliferation and reduced cancer stemness, possibly through the induction of HOXA5 activity. These findings suggest that CELF6 functions as a suppressor of cancer stemness in CRC, highlighting its potential as both a prognostic marker and a therapeutic candidate for CRC.
CELF6, a recently identified member of the CELF family discovered in 2004, is expressed ubiquitously in the nervous system, kidneys and testes and is known for its tumor suppressor functions through post-transcriptional regulation [28]. Its involvement in cancer was first suggested in studies related to cervical cancer susceptibility, which explored the association between CELF6 gene variations and cancer risk [29]. Additionally, CELF6’s role in stabilizing FBP1 mRNA has been shown to inhibit aggressive behaviors in triple-negative breast cancer [15]. A recent study has also identified low CELF6 expression in lung cancer tissues and its prognostic significance [30]. In CRC, CELF6 has been reported to impede proliferation and metastasis, either by stabilizing the p21 protein [16] or by inhibiting oncogenic CD44 isoforms [31]. These findings collectively support the hypothesis that restoring or inducing CELF6 expression, in conjunction with standard treatment modalities, could be beneficial [12, 16, 32]. Herein, CELF6 was found to be suppressed in CRC samples and cell lines, with lower expression levels predicting poorer prognosis in CRC patients, and consistent with previous reports indicating that CELF6 inhibits CRC cell growth through G0/G1 phase arrest [16]. In contrast, CELF1, another member of the CELF family, is highly expressed in CRC and promotes tumor cell proliferation, migration, and invasion [33]. Our in vivo xenograft studies further corroborate the inhibitory effect of CELF6 on tumorigenesis, reinforcing its potential as a therapeutic target in CRC.
CSCs are pivotal in cancer development, progression and recurrence due to their self-renewal capabilities [34, 35]. Our study highlights the role of CELF6 in regulating CSC properties in CRC. ALDH1 [36] and CD44 [37] are crucial CSC indicators, and a recent study showed that CELF6 negatively impacts CRC metastasis by modulating CD44 [31]. We found that CELF6 overexpression significantly inhibited sphere formation, a key characteristic of CSCs, which aligns with previous findings showing that CELF6 modulates CD44, an important CSC marker [31]. Our data further demonstrate that CELF6 re-expression reduced the fraction of ALDH1+/CD44+ stem cells and downregulated stem cell-related genes, including Nanog, OCT4, SOX2, and CD133. These observations suggest that CELF6 acts as a negative regulator of CRC stemness.
HOXA5, a member of the HOX gene family, is known for its role in regulating
various biological processes such as proliferation, differentiation, and
apoptosis in tumor cells [38, 39, 40]. Aberrant HOXA5 expression can disrupt normal
organ function and contribute to cancer progression. Studies have shown that
HOXA5 can counteract stemness mechanisms and inhibit pathways associated with
cancer progression. For instance, Ordóñez-Morán et al. [21]
demonstrated that HOXA5 can override the Wnt/
In our present research study, we observed a positive correlation between CELF6 and HOXA5 expression in CRC samples and demonstrated that CELF6 stabilizes HOXA5 mRNA by binding to its 3′ UTR, thus positively regulating HOXA5 levels. Our rescue experiments revealed that the knockdown of HOXA5 reversed the effects of CELF6-induced suppression of cell motility and sphere formation in CRC cells. These findings indicate that CELF6 modulates CRC cell growth and stem cell-like properties through the regulation of HOXA5 mRNA stability, as depicted in our proposed molecular model (Fig. 8).
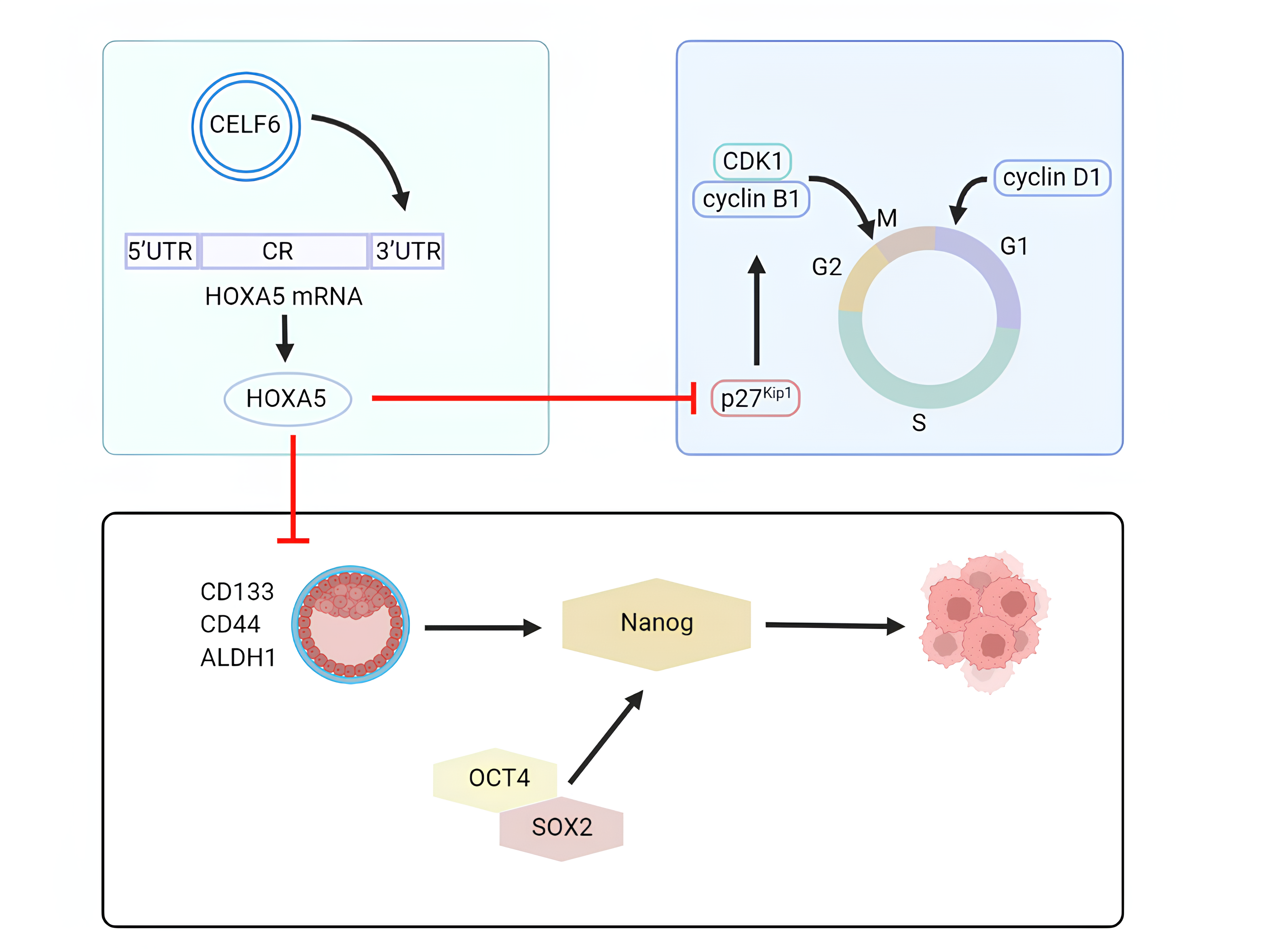 Fig. 8.
Fig. 8.
Molecular working model of CELF6 in CRC cells. Schematic diagram illustrating the antitumor roles of CELF6 in CRC, depicting its impact on cell growth arrest and stemness inhibition.
To further substantiate our findings, additional research is needed to explore the specific mechanisms by which CELF6 regulates CSCs and the precise role of the CELF6-HOXA5 axis in this context. Moreover, future studies could investigate how the CELF6-HOXA5 axis influences CSCs versus regular cancer cells and elucidate any differences in their regulatory mechanisms. Moreover, examining the role of this axis in cancer recurrence and resistance to chemotherapy might provide deeper insights into its clinical relevance, and utilizing patient-derived CRC stem cells could also offer valuable validation of our results and advance the understanding of CELF6’s potential as a therapeutic target.
In summary, our study reveals that CELF6 expression is significantly reduced in CRC tissues and is associated with poor clinical outcomes. We have elucidated a critical regulatory pathway involving CELF6 and HOXA5 that plays a key role in modulating cancer stemness. Specifically, CELF6 overexpression inhibits CRC cell proliferation, suppresses stem cell-like characteristics, and impedes tumor growth through stabilization and upregulation of HOXA5. These findings highlight the potential of CELF6 as a promising therapeutic target in CRC.
The databases presented in this study can be found in online repositories (UALCAN and PrognoScan databases). The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Conceptualization, Methodology, and Writing - Original Draft were performed by ZMF; Formal analysis, Resources, and Investigation were performed by XW; Formal analysis, Visualization and Data Curation were performed by ZJC; Project administration, Supervision, and Validation were performed by BCW; Validation, Supervision, and Writing - Review & Editing were performed by WWH and XL. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All procedures performed in studies involving human participants were those of the 1964 Helsinki Declaration and its later amendments for ethical research involving human subjects. A written consent was signed by the patients or their families/legal guardians. All procedures performed in studies involving human participants and all animal experiments were approved by the Ethics Committee of Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University for the use of animals (Approval No.Med-Eth-Re [2023] 33). All animal experiments are conducted in accordance with the National Institutes of Health Laboratory Animal Care and Use Guidelines.
Not applicable.
This work was supported by the Natural Science Foundation of Hainan Province (Grant No. 820RC758).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.