



 , Ahmed M. Elsayed 2, Mohamed I. Husseiny 3,*
, Ahmed M. Elsayed 2, Mohamed I. Husseiny 3,*1 School of Medicine, University of California Irvine, Irvine, CA 92617, USA
2 Division of Infectious Diseases, The Lundquist Institute at Harbor-UCLA Medical Center, Torrance, CA 90502, USA
3 Department of Translational Research & Cellular Therapeutics, Arthur Riggs Diabetes & Metabolism Research Institute, Beckman Research Institute, City of Hope National Medical Center, Duarte, CA 91010, USA
Abstract
Regulatory T-cells (Tregs) play a crucial role in maintaining immune homeostasis, ensuring a balanced immune response. Tregs primarily operate in an antigen-specific fashion, facilitated by their distinct distribution within discrete niches. Tregs have been studied extensively, from their point of origin in the thymus origin to their fate in the periphery or organs. Signals received from antigen-presenting cells (APCs) stimulate Tregs to dampen inflammation. Almost all tumors are characterized by a pathological abundance of immune suppression in their microenvironment. Conversely, the lack thereof proves detrimental to immunological disorders. Achieving a balanced expression of Tregs in relation to other immune compartments is important in establishing an effective and adaptable immune tolerance towards cancer cells and autoantigens. In the context of cancer, it is essential to decrease the frequency of Tregs to overcome tumor suppression. A lower survival rate is associated with the presence of excessive exhausted effector immune cells and an increased frequency of regulatory cells. However, when it comes to treating graft rejection and autoimmune diseases, the focus lies on immune tolerance and the transfer of Tregs. Here, we explore the complex mechanisms that Tregs use in human disease to balance effector immune cells.
Keywords
- regulatory T-cells (Tregs)
- immune imbalance
- Foxp3
- natural Treg (nTreg)
- induced Treg (iTreg)
- thymic Treg (tTreg)
- peripheral Treg (pTreg)
Immune regulation balances pro-inflammatory and anti-inflammatory immune responses. Classifications characterize the immune compartments in an effort to improve understanding of the mechanisms involved [1]. Regulatory T-cells (Tregs) are known for their powerful function in regulating the immune system and improving self-tolerance, which is pivotal in immune homeostasis [2, 3]. Tregs account for 3–10% of the peripheral CD4+ T-cell population in humans [4]. Tregs are initially observed in the thymus during embryogenesis, specifically at the 12th week of gestation, and their levels remain constant throughout pregnancy and infancy [4]. The immunosuppressive characterization of Tregs from the thymus during fetus development showed early expression of forkhead box P3 (Foxp3) and other markers associated with their function, such as glucocorticoid-induced TNFR-related protein (GITR) and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [5].
Tregs are derived from either the thymus, referred to as (tTregs), or
conventional T-cells in tissues outside the thymus, known as peripheral Tregs
cells (pTregs). The tTregs typically possess T-cell receptors (TCRs) with higher
affinity for autoantigens compared to conventional T-cells and pTregs [6]. In
both homeostatic and inflammatory conditions, the conversion of conventional
CD4+ T-cells into pTregs occurs on interaction with self-antigens or
exogenous antigens and in the presence of transforming growth factor beta
(TGF-
The development of Tregs in the thymus during the neonatal period leads to
dominant tolerance [15]. Only 2–3% of developing CD4+ single positive (SP)
thymocytes are represented by thymic Tregs. Their development is propelled by the
combination of a strong TCR stimulation and a CD28 co-stimulatory signal [15].
The progression of CD25+ Foxp3+ Tregs into a mature stage is enhanced
by a third signal mediated by
Treg development is dependent on TCR/CD28 stimulation and cytokine signaling,
both of which follow a two-step model. First, induction of CD25 occurs
in CD25– Foxp3– CD4+ SP thymocytes through moderately to slightly
strong binding on TCR signals. The outcome of this process is the generation of
CD25+ Foxp3– Treg precursors, commonly referred to as CD25+ Tregs.
The binding to the TCR signal is required to be lower than the clonal deletion
binding of SP CD4+ thymocytes [18, 19]. The CD28- CD80/86 co-stimulatory
axis plays a crucial role in Treg development, starting from their precursor
stage and facilitating the induction of Foxp3 expression [20]. The binding of TCR
signaling when associated with CD28 drives to the upregulation of tumor-necrosis
factor receptor superfamily (TNFRSF) members, which are TNFR2, GITR and OX40
[21]. Second, the maturation of CD25+ Treg through stimulatory
Identification of another Treg precursor subset, which expresses a low level of Foxp3 and lack CD25 (CD25– Foxp3low) suggests another developmental pathway [17]. When migrating from thymus, it is found that CD25+ Treg differentiates into mature Treg. These findings confirm that both CD25+ and Foxp3low Treg can be the sources of the generation of the mature Tregs. Foxp3low Tregs appear to be dependent on IL-15 for survival, and IL-2 is necessary for maturation into Tregs. The development of periphery CD25+ Treg is likely dependent on IL-2. Therefore CD25+ Tregs show a higher affinity for self-antigens than Foxp3low Tregs [17].
CD4+ Tregs were phenotypically known as CD4+ CD25+ Foxp3+
cells. Based on surface marker expression, different classifications emerged. The
first functional classification was based on the cytokine expression of Th-like
Treg subtypes [26]. In this classification, CD45RA is crucial for the division of
CD4+ Tregs. This subset is further subdivided into three subsets. First is
the resting (rTreg) or naïve Tregs with phenotype CD45RA+ Foxp3low
CD25low. The second is the effector Tregs (eTregs) with phenotype
CD45RA– Foxp3high CD25high that has strong inhibitory and
stabilizing functions. The third is non-Tregs (noTregs) with phenotype
CD45RA– Foxp3low CD25low that mainly secrete inflammatory
cytokines and promote the immune response [27]. The rTregs can suppress the
immune system as it expresses CD62L and the IL-7 receptor (CCR7), which are
indicators of naïve cells. Upon antigen stimulation, they differentiate into
eTregs, which exhibit stronger
immunosuppressive and higher proliferation capabilities. These eTregs are
short-lived and more prone to apoptosis. Unlike the earlier identified Tregs
subsets, non-Tregs are categorized as Tregs and have the ability to secrete
pro-inflammatory cytokines like
interferon-
Another classification is based on the cytokine secretion profiles and
transcription factor expression (Table 1, Ref. [27, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42]). CD4+ Treg has been classified
into four distinct subtypes. The first, Th1-like Tregs secrete IFN-
| Treg subset | Origin | Phenotype | Function | Markers for Differentiation | References |
| Natural or Naïve Tregs (nTregs) Cells | Thymus | CD4+CD25+Foxp3+ | Maintain self-tolerance and immune homeostasis by suppressing immune responses via direct cell-cell contact and secretion of inhibitory cytokines like IL-10 and TGF- |
Express CD4+, CD25+, Foxp3+, Helios+, CTLA-4+, GITR+. | [27, 30, 31, 32] |
| Induced Tregs (iTregs) | Peripheral tissues from nTregs | CD4+CD25+Foxp3+ | Induced in response to specific antigens and cytokines. Suppress immune responses and maintain tolerance. | Express CD4+, Foxp3+, CD25+, CTLA-4+, ICOS+. | [27, 31, 33, 34, 35] |
| Type 1 Regulatory T (Tr1) Cells | Peripheral tissues from CD4 T-cells | CD4+CD25‒CD49b+LAG-3+ | Suppress immune responses through the secretion of IL-10 and TGF- |
Do not express Foxp3‒ but produce high levels of IL-10, IFN- |
[33, 34] |
| T Helper 3 (Th3) Cells | Peripheral tissues from CD4 T-cells | CD4+CD25−Foxp3‒LAP+ | Regulate mucosal immunity and oral tolerance primarily through TGF- |
CD4+, TGF- |
[36, 37, 38] |
| CD8+ Regulatory T-Cells | Thymus and peripheral tissues from CD8+ T-cells | CD8+CD25+Foxp3‒ | Suppress immune responses through direct cytotoxic activity or cytokine production. They can inhibit the function of other T-cells and APCs. | Express CD25 and Foxp3 in some cases. | [39, 40] |
| Double Negative Regulatory T- (DN Tregs) Cells | Develop from CD4‒ CD8‒ T-cells in Peripheral tissues | CD3+CD4‒CD8‒TCR |
Suppress immune responses through cytokine production and direct cell-cell contact. They are involved in controlling autoimmune responses and graft-versus-host disease. | T-cells are expressing the |
[41, 42] |
CD25, IL-2 Receptor Alpha; Foxp3, Forkhead box P3; CTLA-4, Cytotoxic
T-Lymphocyte Associated Protein 4; GITR, Glucocorticoid-induced tumor necrosis
factor receptor; LAG-3, Lymphocyte Activation Gene-3; TGF-
In both classifications, Foxp3 expression was not associated with any immunosuppressive potentiality, which leads to conclusions not in line with majority of studies that have employed Foxp3 to demonstrate immunosuppressive capacity. Still, the most acceptable phenotype for CD4+ Treg isolation and detection is CD4+ CD25+ CD127low/-, because CD127low/- cells have higher suppressor potentiality than CD25+ subsets [29]. Different classes of Tregs were characterized in Table 1.
Foxp3 is essential for deciding which lineages to commit to throughout the formation of tTregs in the thymus. In addition, this mechanism is responsible for the maintenance of the extrathymic tTregs population and ensuring the continuous expression of genes that are fundamental in defining the specific characteristics of the Tregs signature [43]. Under homeostatic conditions, the majority of Tregs maintain their commitment to the tTregs lineage even after leaving the thymus. Despite this, a notable fraction (10-20%) of Foxp3+ Tregs experience a loss of Foxp3 expression, leading them to become exhausted Tregs [44]. This population mainly originates from activated conventional T-cells that exhibit temporary expression of Foxp3, as well as from pTregs [8]. Foxp3 plays a crucial role in determining the lineage commitment of Tregs. Foxp3 is not the only factor that influences the Treg gene program and suppressor activity (Fig. 1) [45].
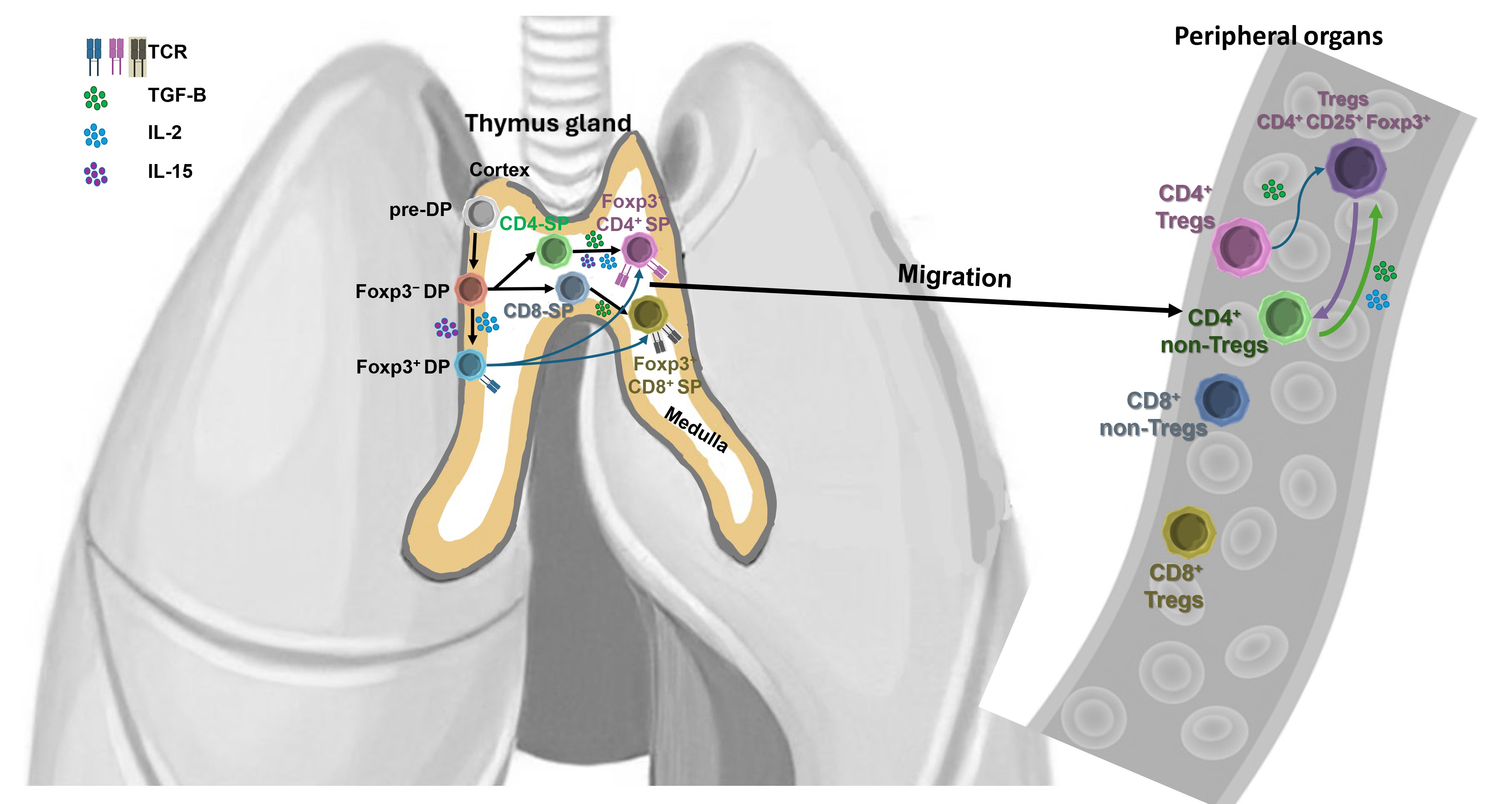 Fig. 1.
Fig. 1.
Tregs thymic development, excursion to periphery, cytokine profiling, and Foxp3. In the cortex of thymus, pre-double positive T (pre-DP)-cells are matured and Foxp3 is not yet expressed. Foxp3– DP cells are committed to be either traditional T-cells or tTregs, depending on the presence of IL-2 and IL-15. Foxp3– DP or Foxp3+ DP cells are ready to migrate to the medulla of the thymus for maturation. In the medulla, Foxp3– DP mature to become naïve CD4 single positive T-cells (CD4-SP) or CD8 single positive T-cells (CD8-SP), which then migrate to the peripheral organs. A subset of CD4-SP and CD8-SP cells are converted to serve as Tregs expressing Foxp3. When Foxp3+ DP migrate from the cortex to medulla, they become Foxp3+ CD4 T-cells or Foxp3+ CD8 T-cells. TCR, T-cell receptor.
Foxp3 is responsible for maintaining the stability of the core suppressive mechanism of Tregs, ensuring that peripheral inflammatory and non-inflammatory signals do not interfere with it [46]. Tregs driven by Foxp3 are characterized by a distinct gene signature. They express a gene set associated with an activation program that is shared with conventional T-cells [47]. Foxp3 is essential for the establishment of suppressive activity of Tregs since a frameshift mutation of Foxp3 leads to immune dysregulation and polyendocrinopathy, enteropathy X-linked (IPEX) syndrome [48]. It is possible to repair the severe autoimmunity of Treg-deficient mice and efficiently restore their suppressive function by restoring Foxp3 transcription [48]. This suggests that there is a mechanistic role of Foxp3 in the establishment of Tregs, that grants periphery Tregs suppressive activity. It also implies that there are multiple molecules and mechanisms that likely play a role in regulating the function of both tTregs and pTregs.
When non-self-antigens are recognized, thymus derived Tregs develop based on
their interactions with self-peptide-MHC complexes in the presence of
TGF-
The signaling pathway mediated by TGF-
Metabolic pathways can determine the Th17/Treg balance. Naïve T-cells rely
on oxidative phosphorylation and fatty acid oxidation or become anabolic to match
cell proliferation and growth. Activated naïve T-cells rely on mammalian
target of rapamycin (mTOR) as a critical regulator of differentiation and
function. The proper complex function of the two mTOR complexes is necessary for
glycolysis upregulation and specific effector subset differentiation. Without
this, CD4 T-cells cannot activate glycolytic machinery for effector function,
leading to a regulatory phenotype [59]. On the other hand, Tregs metabolize fatty
acids, amino acids, and glucose, besides carrying out oxidative phosphorylation.
Rapamycin enhances Foxp3 expression and expands tTregs. Therefore, faulty mTOR
activity affects the Th17/Treg balance by enhancing T-cell sensitivity to
TGF-
Tregs modify their phenotypes without compromising their suppressive function.
They express transcription factors and chemokine receptors associated with
various T-cell types, but they do not produce inflammatory cytokines due to
Foxp3-dependent repression [61]. These events enable Tregs to express a
Th-determining transcription factor and migrate to the site of inflammation.
Tregs in healthy tissues are involved in immune suppression, tissue repair, and
other non-immune activities [62]. Skeletal muscle Tregs produce amphiregulin to
aid muscle repair [63]. The molecular signature of Tregs differs with
the pathogenic condition. The transcriptomic profile of Foxp3+ Tregs from
individuals with colorectal cancers compared to cells from people without cancer.
Tregs showed upregulation of chemokine receptors -4, -1, -2, and -7, and
cytokines IFN-
Foxp3 is being now studied in the development and function of Tregs [65]. Foxp3 is the primary transcription factor accounting for Treg function [15]. Mutations in the Foxp3 gene have a detrimental effect on the function of Tregs, with a particular impact on their ability to suppress immune responses [63, 65]. Foxp3+ nTregs are highly stable and can effectively prevent autoimmune diseases in animal models [33, 34, 63]. Upon constant exposure to self-antigens and microbial antigens, their functionality becomes stable through flexibility of their highly proliferative state [66]. However, Foxp3 gene expression alone was reported insufficient to maintain Treg function. In line with this, 70% of genes were noted to vary between conventional T-cells and Foxp3+ naïve Tregs. Ikzf2 (Helios), Ikzf4 (Eos), CTLA-4, NFAT, Nr4a2, and AP-1 and TNF-receptor superfamily are examples of gene differences that are correlated with and/or were able to enhance Foxp3 transcription in Tregs [67]. At a Treg precursor stage, histone modifications occur before Foxp3 expression, and the genes reach their peaks of expression prior to Foxp3 induction [63].
The signature of specific genes affects the behavior and pathological role of
Tregs. For example, in mice with psoriasis, mutations in inhibitor of Kappa B
Kinase beta (IKBKB) disrupted the balance between pro-inflammatory (TNF and
IFN
The development of Tregs, both in the thymus (tTregs) and in the periphery
(pTregs), is triggered by the induction of Foxp3 in response to antigen exposure.
TGF-
pTregs are identified as true Tregs with the expression of canonical Treg
markers like CTLA-4, GITR, and CD103 and their correlation with IL-2 [15].
Non-immunogenic antigen delivery methods are the most effective triggers for the
induction of pTregs [71]. When tTregs are employed for in vitro assays,
the pTregs are obtained and accompanied with highly effective suppressive
function. Although TGF-
By examining different cell populations, a fascinating variation was found that sheds light on the inherent adaptability of pTregs. Helios, which is an ikaros family transcription factor, is a specific marker for tTregs. Helios is highly expressed on Foxp3+ Tregs in the thymus [73]. Approximately 70% of pTregs express Helios, which can be used to distinguish genotypically between tTregs and pTregs [74].
When stimulated with CD28, TCR signaling, or IL-2, Tregs undergo differentiation into mature or eTregs, which exhibit strong suppressive activity. These eTregs express more immunosuppressive molecules like Human Leukocyte Antigen – DR isotype (HLA-DR), CTLA-4, Helios, and T-cell immunoreceptor with Ig and ITIM domains (TIGIT), chemokine receptor 4 (CCR4), C–X–C chemokine receptor Type-4 (CXCR4), and CXCR5, to enhance infiltration of Tregs into tumor microenvironment (TME) and maturation of nTregs to become eTregs (Fig. 2) [75]. This type of infiltration of eTregs promotes immune tolerance.
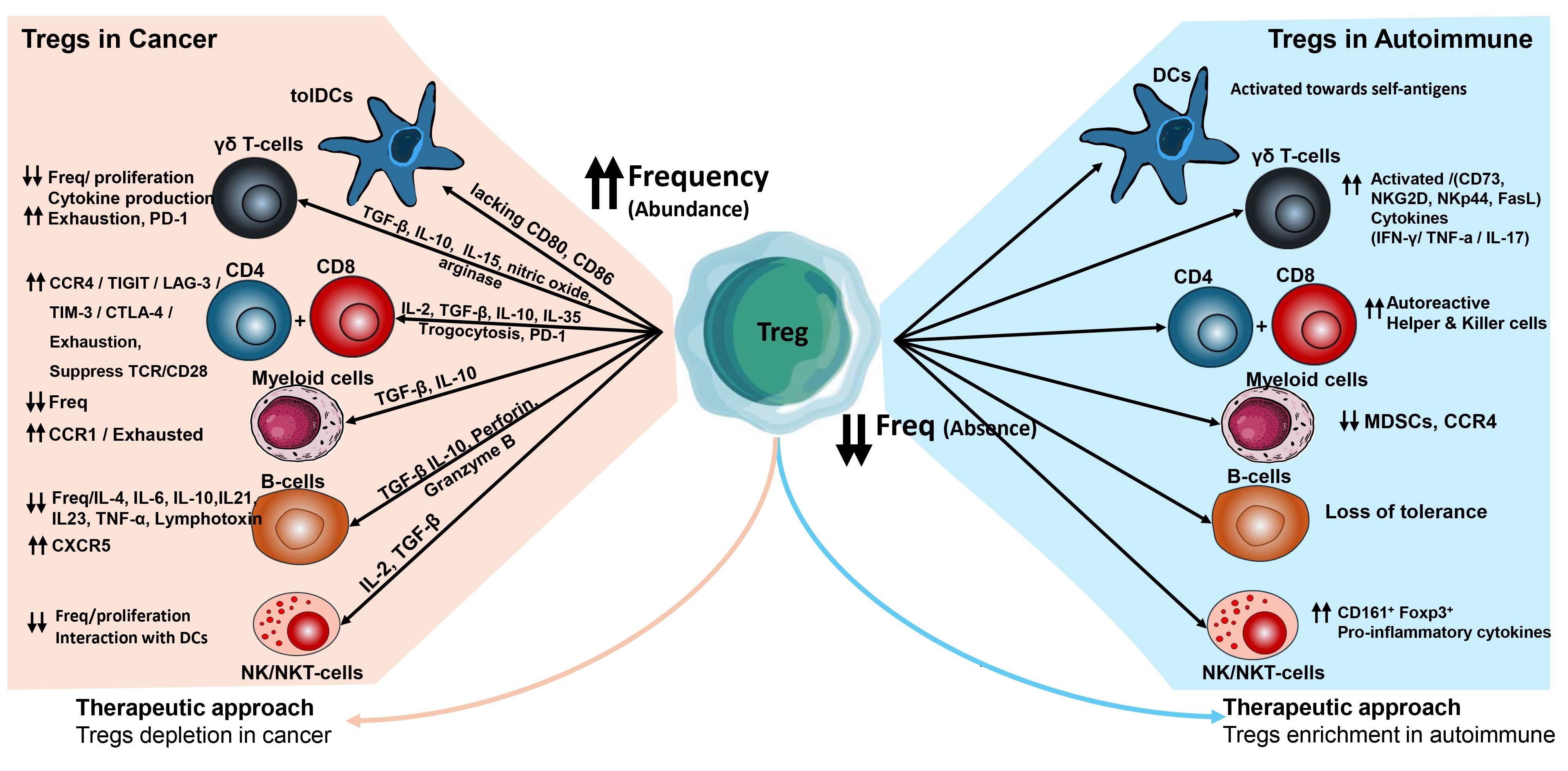 Fig. 2.
Fig. 2.
Role of Tregs in protection from autoimmune diseases
and cancer development. In cancers, Tregs induce immune suppression of other
immune subsets through cytokines, chemokines, and cell-to-cell contacts, tolDCs,
Tolerogenic dendritic cells; CCR, chemokine receptor; TIGIT, T-cell
immunoreceptor with Ig and ITIM domains; LAG-3, lymphocyte activation gene-3;
CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CXCR1, C–X–C chemokine
receptor 1; MDSCs, myeloid-derived suppressor cells; NK/NKT-cells, natural
killer; PD-1, programmed cell death protein 1; TNF-
Tregs exhibit diverse inhibitory activities for efficient immune cell control
[76] including expression of immune suppressive cytokines, such as IL-10 and
TGF-
Long-term tolerance can be achieved through adoptive transfer of Tregs and depletion of Tregs did not affect tolerance in this situation. T-cells from immuno-tolerant mice can establish long-term tolerance in recipient animals, demonstrating the long-term tolerogenic qualities of Tregs to surrounding immune cells both directly and indirectly, and ensuring the durability of their effect [33, 34].
The innate immune response is finely tuned by regulatory Tregs, which work to maintain immune homeostasis and modulate the activities of other immune cells, through suppressing effector cell functions, controlling antigen presentation, controlling cytokine production, maintaining tissue integrity, and via their interaction with innate-like T-cells [80, 81].
Tregs help prevent excessive inflammation and tissue damage that can occur
during immune responses by dampening the activity of the macrophages, dendritic
cells (DCs), and natural killer (NK) cells [81]. Tregs inhibit the maturation and
antigen-presenting function of DCs, which modulate the activation of T-cells and
other effector cells of the innate immune system. Tregs controls other immune
subsets by the production of anti-inflammatory cytokines, such as IL-10 and
TGF-
Treg can exert their anti-inflammatory and pro-tolerogenic effects by modulating
the behavior of neutrophils. During immune/inflammatory responses, neutrophils
and different Treg subtypes establish a complex crosstalk. Lipopolysaccharide
(LPS) or CD3/CD28 ligation triggers the activation of Tregs, resulting in the
expression of a range of immune suppressive pathways in neutrophils through
various mediators, alongside the promotion of their apoptosis [83]. Culturing
human neutrophils with activated Treg led to elevated levels of anti-inflammatory
molecules such as IL-10, TGF-
Tregs possess the capability to restore injured tissues through the production
of healing molecules like amphiregulin, and tissue-regulatory protein peroxisome
proliferator–activated receptor
Tregs play a crucial role in tissue repair and regeneration by regulating
inflammation and orchestrating the activity of both innate and adaptive immune
systems [83]. Following tissue injury, a symphony of immune responses is set in
motion until a new tissue is regenerated. Tregs play a role in each of the
various stages. Tregs can counteract the onset of inflammation by suppressing the
secretion of inflammatory cytokines such as IL-6, IFN-
In skeletal muscle, Tregs are linked to mesenchymal stromal cells, nerves, and IL-33 secretion. All are connected when, through calcitonin-gene-related peptide, Tregs accumulated [90]. IL-33 acts on Tregs containing the ST2 receptor encoded by the IL1rL1 gene compared to that of Tregs in lymphoid tissue. IL1rL1 is upregulated in Tregs isolated from damaged muscle [91]. Tregs in muscles have been found to express significant amounts of amphiregulin [92]. By directly interacting with satellite cells, these specialized Tregs have an impact on supporting muscle regeneration. The administration of amphiregulin normalized the muscle transcriptome during muscle repair. Amphiregulin also enhances myogenic differentiation [92]. Treg depletion in injured muscle was associated with less tissue regeneration, prolonged inflammation, and impaired production of myogenic transcription factors, macrophages polarization from M1 to M2 phenotype [93]. Tregs protect against tissue deterioration in neurodegenerative [89], cardiac [94], lung [95], autoimmune [27], atherosclerotic [86], and skin diseases [96].
Tregs have a crucial role in preventing immunopathogenic reactions to various viral, bacterial, fungal, protozoal, and helminth infections [97]. During acute infection, Tregs inhibit the accumulation of cytotoxic CD8+ T-cells. Second, Tregs produce IL-10 that promotes the maturation of memory T-cells [15]. Different Treg populations that emerge during acute infection with Listeria monocytogenes [98]. These populations have distinct effects on regulating CD8+ T-cell responses at various stages, including the priming and contraction phases [99]. Different Treg subpopulations separated from several time points of the same animal model were clonally unique, suggesting that they most likely came from different cell progenitors [100].
During chronic infection, limiting Treg number boosts immune responses mediated by cytotoxic CD8+ T-cells, leading to improved control of the infection [101]. The generation of clones of Tr1 cells that produce IL-10 was only observed in situations involving persistent infection [102, 103]. Meta-analyses compared the role of Tregs in acute versus chronic infections. Increased CD4+ Treg frequencies were noted in chronic hepatitis B virus (HBV) infection, pointing to the role of Tregs in disease progression, viral load, absence of therapy, and risk of hepatocellular carcinoma [104]. Tregs attracted CD4+ and CD8+ T-cells to the liver through chemokines CCL17 and CCL22 and reduced their inflammatory response in cases of chronic hepatitis C virus (HCV) infection, leading to the prolonged existence of pathogens [15]. On the other hand, Tregs may be functional in reducing the amount of liver damage caused by HCV [105, 106, 107].
CD4+ Tregs mediated inconclusive anti-HIV immune responses and were comparatively more abundant in the mucosa and bloodstream [108]. CD4+ Tregs decreased HIV replication in T-cells in vitro by altering ectonucleotide levels through CD39 and by transferring cAMP through gap junctions formed with conventional T-cells [109]. Blocking CD39 restored the ability of HIV-gag-stimulated CD8+ T-cells to produce cytokines [110]. By interfering with the immunological synapse, they inhibited the spreading of virus from DCs to T-cells [109]. There is a positive correlation between the frequency of CD4+ CD39+ Tregs and HIV viral load and disease progression [108]. These somewhat contradictory data may be resolved by distinguishing between acute and chronic infection [108].
An understanding of Tregs stimulated efforts to treat autoimmune disorders,
organ transplant rejection, and inflammation-related neurodegenerative diseases.
This is a result of extensive comprehension of the molecular and mechanistic
aspects of Treg biology (Fig. 2) [27]. High expression of IL-2R
The advantage of Treg therapies lies in their capacity to educate and propagate endogenous cells to exhibit suppressive activities, thereby facilitating long-term tissue protection even in the absence of survival of the infused Tregs [112]. Preclinical research showed that Tregs have the ability to prevent and reverse disease. Adoptive Treg therapy prevented GvHD in individuals after allogeneic hematopoietic stem cell transplant. Tregs were applied to r transplant- and autoimmune-related diseases [113]. Tregs were infused efficacious without adverse side effects such as systemic immunosuppression [114]. As well, Tregs were used in the treatment of solid organ transplantation [113, 115, 116, 117, 118, 119], spontaneous abortion [114], and autoimmune disease [14, 120, 121, 122, 123].
Tregs can modify their metabolic functions, including glycolysis, oxidative phosphorylation (OXPHOS), fatty acid oxidation (FAO), and amino acid metabolism, in order to meet their energy needs. Nevertheless, there is disagreement and a lack of clarity about the connections between these processes and the underlying mechanisms [124]. The metabolic program of the cell is influenced by its activity state, and varies between naïve, activated, and memory cells. To illustrate, when cells are in a resting state which needs energy to maintain survival and circulation, they rely on energy sources from OXPHOS such as ATP [125, 126]. Through the TCR and co-stimulatory CD28, effector T-cells switch from OXPHOS to glycolysis [125, 127]. Upon their activation, cells use glutaminolysis in addition to glycolysis to generate energy [125, 126]. Tregs exhibit a distinct metabolic profile compared to other T-cell subsets. Initially, they employ glycolytic metabolism for activation, migration, and proliferation. However, they subsequently undergo a metabolic shift, becoming independent of glucose and relying on the oxidation of lipids and pyruvate [128, 129].
Tregs that proliferate exhibit heightened glucose transporter 1 (GLUT1)
expression and mammalian target of rapamycin (mTOR) activity, resulting in
reduced suppressive capacity and simultaneous downregulation of Foxp3 expression
[114, 116, 127, 128, 129]. Tregs are regulated glycolytically by different mechanisms,
such as the phosphoinositide 3-kinase (PI3K)-Akt-mTOR signaling network. This
pathway enhances the glycolytic rate of Tregs and significantly influences their
differentiation and functional stability [130]. The PI3K-Akt-mTOR pathway is
regulated by many factors including AMP-activated protein kinase (AMPK),
phosphatase and tensin homolog (PTEN), and hypoxia-inducible factor 1
Tregs were generated from human CD4+ cells by inhibition of fatty acids binding proteins. This dysregulated mitochondria, decreased OXPHOS, and increased glycolytic pathways [134]. The persistence of eTregs is linked to mitochondria, as they acquire energy through FAO. The transfer of mesenchymal stem cell mitochondria to CD4+ T-cells aids in the differentiation of Tregs, providing relief from GvHD [135, 136]. Mitochondrial complex III and mitochondrial transcription factor A prevented DNA hyper-methylation to suppress Foxp3 expression [137].
The pro-migratory molecule lymphocyte function-associated antigen 1 when stimulated by its ligand, increases iglucose uptake [138]. Multiple metabolic processes rely on the participation of amino acids. Immune homeostasis and responses are regulated by the availability and metabolism of amino acids. Treg generation and function is linked to amino acid transporters, such as those responsible for branched-chain amino acids (glutamate, glutamine, and glutathione). Furthermore, the catabolism of tryptophan and arginine was noted [127]. Maintaining cholesterol balance is essential for Tregs as it impacts their lipid metabolism, biofilm and lipoprotein composition, mTOR-class 1 activation, and immune synapse formation [139]. The rise in cholesterol levels in cells interferes with mTOR signaling, leading to the promotion of Tregs. Insufficient lipids disrupt the mevalonate pathway, resulting in protein modification [140] and increases PD-1 and eTreg numbers [141].
Tregs are essential for the development of immunotherapies against cancer and
autoimmune diseases. In cancer models, Treg depletion induced an anti-tumor
immune response [142, 143]. Tregs are important players that can either
contribute or protect against diseases. This raises the question of the dual role
of these cells. Within the hepatocellular tumor microenvironment, the frequency
of
Tregs are one of the main gatekeepers of the immune system and serve as a protector in preventing and treating autoimmune diseases. In glomerulonephritis, Tregs provided protection against renal tissue injury that is linked with pathogen driving Th1 and Th17 effector cell activation [153, 154]. The incidence of most autoimmune diseases is somehow correlated with dysfunction of suppressor immune cells, mainly Tregs [155]. The mutation of the autoimmune regulator gene in Tregs leads to loss of normal immune tolerance and increased the incidence of autoimmune polyendocrine syndrome type 1 (APS-1) [155, 156]. Furthermore, a close link between Tregs malfunction and the type 1 diabetes was noted. Specifically, adoptive transfer of genetically engineered Tregs in non-obese diabetic mice limited disease [157]. Inflammatory and autoimmune diseases are now treated by adoptively transferred and genetically altered Tregs (Fig. 2) [158].
Germane to this, most autoimmune diseases and transplantation rejection emerges
from abnormal immune tolerance as well as deficiency or malfunctions of normal
existing Tregs in tissue and periphery [159, 160, 161]. Tregs induce immune suppression
to other immune subsets by crosstalk though cytokines, chemokines, and
cell-to-cell contacts, such as Tregs crosstalk towards T-cells [162], myeloid
cells [163], B-cells [164], NK cells [165], and
Ageing enhances Treg senescence and limits proliferation [175]. Tregs migrated less and did not regenerate muscle in aged animals [176]. In order individuals, Tregs had less ability heal lung damage caused by influenza [177]. Differentiation of Tregs diminished with age, which is significant when comparing the differentiation of naïve T-cells from aged mice to those of young animals [178]. Similarly, a reduction in the de novo induction of antigen specific Tregs in the aged mice was less compared to young animals [179].
Retinaldehyde dehydrogenase 2 (RALDH2) was decreased in DCs from mesenteric
lymph nodes (MLN) from older mice [15]. Additionally, CD11b– CD103+
PD-L1high DCs, characterized by elevated RALDH2, were fewer [179] in
conjunction with TGF-
The accumulation of Tregs appears to be age dependent, with middle-aged mice exhibiting Treg levels that are in between those of young and old mice [182]. For obvious reasons, Tregs are assessed in human blood samples. More aTregs (Foxp3high CD45RA–) and less rTregs (Foxp3low CD45RA+) were noted in blood from older individuals [181, 183]. Related to this, Tregs were more numerous in the skin of older subjects and could account, in part, for fewer in the circulation [184].
Technology advancements and a better understanding of Treg biology will likely drive the development of Treg-targeting therapeutics in several fascinating ways.
Precision Medicine: Treg-targeting therapies may be more effective and less
likely to cause side effects if they are customized to each individual based on
their unique immune profile and genetic background. This is becoming more
possible with advances in proteomics and genomic technology [185]. Combination Therapies: Combining Treg-targeting strategies with other
immunotherapies to increase therapeutic efficacy, such as checkpoint inhibitors
or chimeric antigen receptor (CAR)-T cell therapy [186]. The goal is to produce
synergistic effects that enhance anti-tumor or autoimmune responses. Combination
checkpoint inhibitor therapy slowed tumor growth by blocking several pathways,
such as PD-1 (Nivolumab), LAG-3 (Relatlimab), and CTLA-4 (Ipilimumab) [187]. The
FDA approved six CAR-T therapies for cancer including Kymriah (tisagenlecleucel),
Tecartus (brexucabtagene autoleucel), Yescarta (axicabtagene ciloleucel), Abecma
(idecabtagene vicleucel), Breyanzi (lisocabtagene maraleucel), and Carvykti
(ciltacabtagene autoleucel) [186]. Nanotechnology: Using nanoparticles (NPs) and nanocarriers to precisely deliver
drugs or therapeutic agents to Tregs to improve efficacy and lower systemic
toxicity [188, 189, 190]. NPs can administer monoclonal antibodies (anti-PD1). Other NP
formulations deliver small interfering RNAs to disrupt immunological checkpoints
[191, 192]. Using NPs, antigen, such as CAR-encoding DNA in vivo and
CAR-encoding mRNA, can be delivered to T-cells [193]. NPs that release
TGF- Gene Editing Technologies: Tregs can be precisely modified using tools like
CRISPR/Cas9. This could involve engineering Tregs to either enhance their
suppressive functions for autoimmune diseases or reduce their inhibitory effects
for cancer treatment. CRISPR/Cas9 can edit both primary T-cells and engineered
T-cells, including CAR-T and TCR-T, in vivo and in vitro to
regulate T-cell differentiation and activation [196]. Tregs can more effectively
detect islet-associated antigens and improve the immune-suppressive environment
using CRISPR-Cas9 to replace endogenous TCR with islet-specific TCR and stable
Foxp3 expression [197, 198]. CRISPR/Cas9-edited dual-targeted (CD19/CD22) CAR-T,
was safe and efficient in individuals with B-cell acute lymphoblastic leukemia
(B-ALL) [196]. Novel Drug Development: Novel pharmaceuticals or biological treatments
that precisely modulate Treg survival or function are being developed. These may
provide more efficient and selective modulation of Tregs. Selective Targeting: Developing therapies that target pathogenic Tregs precisely
while protecting Tregs that maintain normal immune tolerance. This may lessen
adverse effects and improve safety profiles [11].
Integrating regulatory T-cells into medicine requires careful consideration and is not straightforward. Clinical correlative studies should be considered when examining the delicate immunological balance of Tregs in their macro- and microenvironments. Varying roles of regulatory T-cells are found in many situations and diseases, in aging, between sexes, and potentially underestimated factors. Tregs crosstalk to other immune cells through complicated network mechanisms. This is necessary for a balanced immune reaction. Sometimes Tregs have a beneficial role and sometimes a harmful role. In autoimmune diseases, Tregs are not of adequate number or function mainly secondary to hyperactive immune cells recognizing self-antigens. Consequently, proinflammatory cytokines and chemokines are secreted to augment the immune reaction. Enrichment of the affected organ with autologous Tregs might restore the immune balance. In cancers, Tregs are abundant and under the control of cancer cells to maintain a balanced less severe tumor immune response. In this case, depletion of Tregs from the immune compartments of the tumor may increase cancer killing.
MS, AME, and MIH, Conceptualization, writing, and editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.