



 , Muhammad Imran Qadeer 2,3,*, Khadija Zahid 2, Elena Vladimirovna Cherepkova 4, Sayakhat Taurbekovich Olzhayev 5
, Muhammad Imran Qadeer 2,3,*, Khadija Zahid 2, Elena Vladimirovna Cherepkova 4, Sayakhat Taurbekovich Olzhayev 51 Department of Biotechnology, Kinnaird College for Women University, 54000 Lahore, Pakistan
2 Sundas Foundation Hematological and Molecular Analysis Center, 54000 Lahore, Pakistan
3 Department of Microbiology and Molecular Genetics, University of the Punjab, 54000 Lahore, Pakistan
4 All-Russian Research Institute for Civil Defence and Emergencies, 121352 Moscow, Russia
5 Department of Oncology, Kazakh-Russian Medical University, 050004 Almaty, Kazakhstan
Abstract
Immune thrombocytopenia (ITP) is an autoimmune bleeding disorder. It involves impaired production and excessive destruction of platelets. It is a complex and heterogeneous disorder with unknown pathophysiology. Both genetic and immunologic perturbations have been implicated in the disease pathogenesis. Immune dysregulations involve both the humoral and cellular immunity. Attack of anti-platelet autoantibodies has been found to be the fundamental cause of platelet destruction. Other mechanisms including T cell mediated platelet destruction, complement activation, apoptosis, and desialylation have also been found in the development of ITP. Genetic testing has revealed various predispositions including single nucleotide polymorphisms (SNPs), copy number variations (CNVs), and epigenetic changes in the immunoregulatory genes of ITP subjects. Varying methylation patterns have also been found in the immune-related genes. This review summarizes the dysregulated immune cells, immunologic cascades, altered signaling pathways, genetic mutations and epigenetic changes in ITP pathogenesis. These alterations induce autoimmune responses against the platelets resulting in complex bleeding manifestations and onset of ITP.
Graphical Abstract
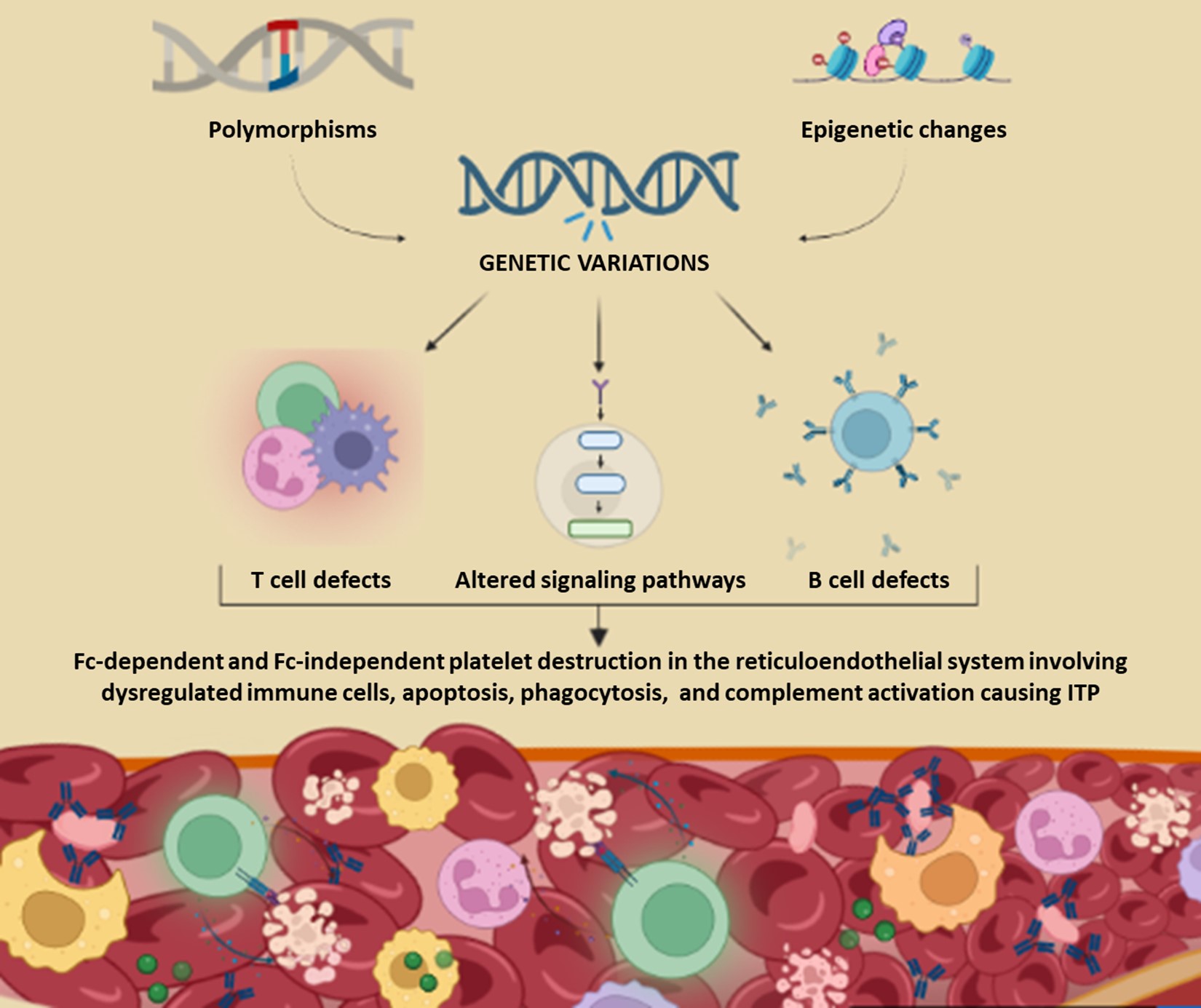
Keywords
- immune thrombocytopenia
- autoimmune
- platelets
- immunologic
- genetic
- polymorphisms
Immune thrombocytopenia (ITP) is an acquired autoimmune bleeding disorder. It
was previously referred to as “idiopathic” because of its non-evident cause of
pathogenesis [1, 2]. It is characterized by reduced platelet count, less than 100
ITP can be categorized as acute or chronic. The frequency of incidence of the disorder range from 3.3 to 3.9 per 100,000 adults and from 1.9 to 6.4 per 100,000 children per annum [4]. In adults with the age range of 18–40 years, affected individuals have a female predominance with ~2:1 affected females as compared to males. In individuals above 60 years of age, equal ratio of males and females are affected [5, 7, 8]. Out of the total ITP cases, about 80% cases are initially diagnosed as primary ITP. However, ~18–38% of the patients might have a comorbid condition or co-medication due to which the diagnosis can be changed to secondary ITP [3, 5, 9].
ITP is a heterogeneous disorder, therefore it has a diagnosis of exclusion [9, 10]. The ITP Bleeding Assessment Tool (ITP-BAT) is used as the standard diagnostic criteria. It is developed by the International Working Group and American Society of Hematology [11]. ITP World Impact Survey (I-WISH) and the Life Quality Index of ITP patients (ILQI) are used alongside to determine the health related quality of life of these patients [12, 13]. ITP has an unknown cause of pathogenesis encompassing both genetic and immunologic perturbations. This review summarizes the current knowledge about the immunologic, genetic, and epigenetic changes in ITP patients and the treatment options available.
Previously, it was assumed that onset of ITP was primarily due to platelet destruction [14, 15, 16]. Increasing investigations on different aspects of ITP have unveiled a number of pathogenic factors. It has been represented by a “Double-hit model” involving complex mechanisms that cause the impaired production and accelerated destruction of platelets. Thus, ITP is characterized as a highly complex and heterogeneous disorder [9, 17, 18]. Among the immunologic factors, cellular and humoral immunity are mainly responsible for increased destruction of platelets [15]. Molecular analysis has revealed the role of mutations in the immunity-related genes to be involved in ITP pathogenesis [19, 20].
Megakaryocytopoiesis is a complex process that occur within the bone marrow [15]. It involves complex cellular and molecular processes to produce platelets [21]. Impairment in these processes cause production and release of immature platelets (Fig. 1A) [7]. Investigations on ITP mice models indicated the presence of young agranular megakaryocytes containing degenerated nucleus in the blood. These megakaryocytes were produced and released due to apoptosis that inflated membranes, cytoplasm, mitochondria, and distended peripheral areas [19, 22]. Subsequently, megakaryocytic platelet production may be inhibited by autoantibodies which cause a significant qualitative and quantitative disruption in number and morphology of platelets [20]. This induces an emergency in the body. Vigorous signals to increase the production of platelets are sent to the bone marrow which ultimately defaults the homeostatic conditions causing ITP [21, 22].
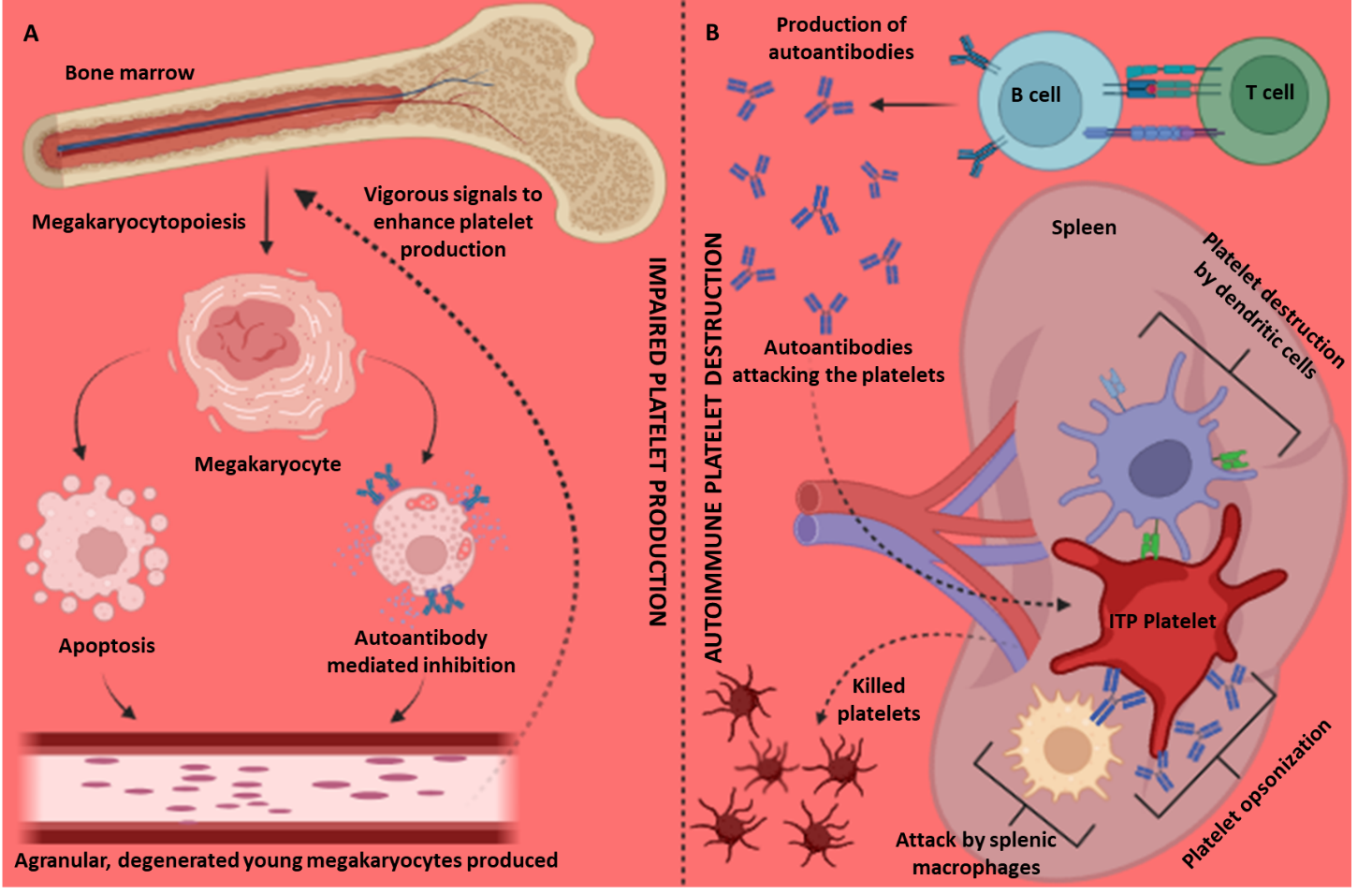 Fig. 1.
Fig. 1.
Immunologic cascade in Immune Thrombocytopenia (ITP). (A) Impaired megakaryocytopoiesis leading to the production of agranular, degenerated young megakaryocytes. (B) Autoimmune platelet destruction in the spleen caused by the anti-platelet autoantibodies produced by the defected B and T cells. Created with BioRender.com.
Impairment in the T and B lymphocytes induce the production of pathogenic anti-platelet autoantibodies. These antibodies drive the apoptotic mechanisms against platelets primarily in the spleen and liver (Fig. 1B) [19, 20]. Massive destruction may also occur by other mechanisms such as opsonization, complement activation, and phagocytosis directed by autoantibodies, splenic macrophages, dendritic cells, regulatory T and B cells, helper and cytotoxic T cells, B cells activating factor (BAFF), antigen presenting cells (APCs), natural killer cells (NK), imbalanced cytokines and myeloid derived suppressor cells (MDSC), and dysregulated helper and cytotoxic T cells (Th and Tc): Th1/Th2 and Tc1/Tc2 ratios (Table 1, Ref. [7, 17, 19, 20, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44]) [7, 8, 15, 19, 20, 23, 24, 25, 26, 45].
| Cell types | Subset | Dysregulation | Reference |
| T cells | |||
| Helper T cells | Th1 | Upregulated | [19, 20, 35, 36, 37, 38] |
| Th2 | |||
| Th17 | |||
| Th22 | |||
| Th1/Th2 | |||
| T follicular helper cells (TFH) | Splenic | Upregulated | [17, 33] |
| Circulating | |||
| Regulatory T cells | CD25+ | Downregulated | [7, 19, 20, 24, 31, 33, 39] |
| CD8+ | |||
| CD4+ | |||
| FoxP3+ | |||
| CD28– | |||
| Cytotoxic T cells | Splenic CD8+ T cells | Upregulated | [17, 20, 26, 33, 37] |
| Tryptophanyl-tRNA synthetase (TTS) | |||
| Tc1/Tc2 | |||
| B cells | |||
| B cell activating factor (BAFF) | - | Upregulated | [17, 19, 24] |
| Regulatory B cells | - | Downregulated | [7, 19, 23, 24, 33, 40] |
| Splenic B cells | - | Upregulated | [19, 41] |
| Antigen Presenting Cells (APC) | |||
| Macrophages | - | Upregulated | [32] |
| Dendritic Cells (DCs) | CD80, CD39, CD86 | Upregulated | [19, 23, 24, 42] |
| Plasmacytoid DCs (pDCs) | Downregulated | ||
| Indoleamine 2,3-dioxygenase 1 (IDO1) | |||
| Myeloid DCs | |||
| Natural Killer Cells | CD56+ CD3– NK cells | Downregulated in acute phase, Normal in chronic phase | [26, 27] |
| Monocytes | - | Upregulated | [19, 32] |
| Others | |||
| Toll-Like Receptors (TLRs) | TLR4 | - | [24] |
| TLR7 | |||
| C reactive protein (CRP) | - | Upregulated | [19, 20] |
| Myeloid-derived suppressor cells (MDSCs) | - | Downregulated | [23, 24] |
| Chemokine receptors (CXCR) | CXCR3 | Upregulated | [19, 34, 43] |
| CCR5 | |||
| CCR3 | Downregulated | ||
| CCL5 | |||
| CXCL5 | |||
| Cytokines | IL-1 | Upregulated | [19, 29] |
| IL-2 | Upregulated | [19, 20, 35] | |
| IL-4 | Downregulated | [35, 44] | |
| IL-6 | Upregulated | [20, 36, 37] | |
| IL-7 | Downregulated | [28] | |
| IL-10 | Downregulated | [44] | |
| IL-11 | Upregulated | [19] | |
| IL-12 | Upregulated | [44] | |
| IL-16 | Upregulated | [25] | |
| IL-17 | Upregulated | [19, 20, 31, 36, 37] | |
| IL-18 | Upregulated | [28] | |
| IL-21 | Upregulated | [20, 31, 33, 37] | |
| IL-22 | Upregulated | [19, 20] | |
| IL-37 | Upregulated | [30] | |
| IFN-G | Upregulated | [19, 20, 28, 35, 44] | |
| TNF- |
Upregulated | [19, 28, 44] | |
| TGF- |
Upregulated | [20, 36, 37] | |
| GATA3 | Downregulated | [44] | |
Th, helper T cell; CD, cluster of differentiation; NK, natural killer; IL,
interleukin; IFN-G, interferon gamma; TNF-
ITP is an organ specific disorder that fundamentally affects the reticuloendothelial system. The platelet lysis primarily occurs in the liver and spleen [16, 45, 46, 47]. Investigations on ITP (CD61 knockout) mice models have demonstrated the role of spleen in ITP pathogenesis. It is responsible for antibody response and migration patterns of lymphocyte subsets against platelets [48]. It involves mechanisms such as opsonization, complement activation, apoptosis, phagocytosis, desialylation, glycan modification, and repertoire cloning of platelets [49, 50, 51, 52, 53].
The lymphocytic repertoire caused by autoreactive autoantibodies occur at different stages of development. These may include immune tolerance defects categorized as central tolerance, differentiation blocks, and peripheral tolerance defects [4, 8, 54]. Antibody mediated platelet destruction mainly involves IgG and less frequently IgM and IgA antibodies [22, 52]. The defects in helper T cells cause the irregular proliferation of follicular T cells. These in turn cause the autoreactive splenic B cells to differentiate into antiplatelet autoantibodies [29, 52]. The autoantibodies phagocytize mature platelets and megakaryocytes to inhibit the production of new platelets [30, 50, 54].
The platelets and megakaryocytes undergo antibody dependent cellular cytotoxicity (ADCC) or complement dependent cytotoxicity (CDC). First, they are detected and opsonized by the antigen presenting cells (APCs: dendritic cells, natural killer cells, macrophages, monocytes) and then presented to T cells via major histocompatibility complex (MHC) II [31, 32, 52, 55]. However, these autoreactive cells could be detected in only 50–70% of the active ITP patients. This may be due to limitations in the detection methods, or rapid clearance of the platelet bound auto-antibodies for which advanced detection methods must be developed [4, 22, 31, 46].
Platelets are also rapidly cleared by T cell mediated reactions [56, 57]. Upregulated helper T cell subsets, follicular T cells, and cytotoxic T cells and downregulated regulatory T cells have been observed in several ITP patients (Table 1) [33, 58]. CD8+ cytotoxic T cells may be responsible for the direct lysis of platelets. The mTOR signaling pathway cause differentiation of CD8+ T cells to cytotoxic T lymphocytes (CTLs). The CTLs cause platelet destruction through desialylation and apoptosis by increasing the cytotoxicity effector proteins such as Granzyme A, Granzyme B, and Perforin [57, 59, 60].
In Fc-mediated platelet destruction, the GPIIbIIIa antibodies in peripheral
blood attack the platelet surface glycoproteins containing Fc gamma receptors
(Fc
Desialylation and apoptosis are the Fc-independent platelet clearance pathways.
The anti-GPIb
The complexity and heterogeneity of ITP is implicated by genetic predispositions and epigenetic changes [63, 64, 65]. Researchers have postulated the role of these genetic factors based on the identified gene polymorphisms in interleukin genes (IL-4, IL-6, and IL-10) in other autoimmune diseases (systemic lupus erythematosus, rheumatoid arthritis) [66]. Genetic analysis and functional assays have been employed to investigate predisposed factors involved in development of ITP [67, 68]. Single nucleotide polymorphisms (SNPs), copy number variations (CNVs), dysregulated expression of RNAs, and epigenetic changes have been identified (Table 2, (Ref. [42, 44, 63, 64, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110])) [63, 69, 70, 111, 112, 113, 114]. SNPs in the immunoregulatory genes encoding T cells, B cells, Fc receptors, cytokines, and chemokines have been found in ITP pathogenesis (Fig. 2) [63, 64, 72, 73, 74, 75, 115, 116, 117]. These variations may impose morphological, structural, and functional changes in the encoded proteins [71].
| Types | Genes | Functions | Variations | References |
| Single Nucleotide Polymorphisms | IFNA17 | Treg, TGF- |
9:21227622 A |
[91] |
| IFN-G | Encodes a soluble cytokine that is a member of the type II interferon class | (+874) TT genotype | [81] | |
| SOCS1 | Negative feedback loop to attenuate cytokine signaling | del 16p13.2p13.11 | [98] | |
| STAT1 | Mediates the expression of various immunoregulatory genes | rs1467199; 2:191015776 C |
[98, 99] | |
| TNF- |
Regulates cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation | −308G/A, −238G/−857C | [75, 79, 81, 83, 90] | |
| TNF- |
Mediates inflammatory, immunostimulatory, and antiviral responses and apoptosis | +252 A |
[82] | |
| TNFSF13B | Encodes a TNF related cytokine, acts as a potent B cell activator, play an important role in the proliferation and differentiation of B cells | –871C/T | [74] | |
| TNFAIP3 | Inhibit nuclear factor kappa-light-chain-enhancer of activated B (NF- |
rs2230926; 6:137874929 T |
[63, 94, 95] | |
| rs5029939; 6:137874586 C | ||||
| rs10499194; 6:137681500C | ||||
| TGFB1 | Encodes a secreted ligand of the TGF-beta superfamily of proteins | Codon 10, T |
[81] | |
| GBE1 | Increases the solubility of the glycogen molecule and reduces osmotic pressure within cells | rs117503120; 3:81303406 G |
[67] | |
| TENM4 | Establishes proper neuronal connectivity during development | rs4483616; 11:79113278 G |
[67] | |
| SYN3 | Encode neuronal phosphoproteins, Modulates release of neurotransmitters | rs5998634; 22:32773129T |
[67] | |
| RBM45 | Encodes a member of the RNA recognition motif | rs16866133; 2:178157883T |
[67] | |
| IL-1B-31 | Mediator of the inflammatory response, involved in various cellular activities, including cell proliferation, differentiation, and apoptosis | variant allele (T) high | [80] | |
| IL-1Ra | Modulates a variety of interleukin 1 related immune and inflammatory responses | het/hom variant high | [64, 75, 80, 81, 88, 89] | |
| IL-4 | Mediates allergic, anti-parasitic, wound healing, and acute inflammation, helps in the production of allergen specific Immunoglubulin E (IgE) | intron 3: RP1/RP2 genotype proportion | [66] | |
| IL-10 | Down-regulates the expression of Th1 cytokines, major histocompatibility complex (MHC) class II Ags, and costimulatory molecules on macrophages, enhances B cell survival, proliferation, and antibody production, blocks NF- |
rs1800872; 1:206773062 T |
[66, 75, 83, 86] | |
| IL-17F | Encodes a cytokine and induce the production of IL2, transforming growth factor-beta (TGFB)1/TGFB, and monocyte chemoattractant protein 1 | rs763780; 6:52236941 T |
[84, 85] | |
| IL-23R | Mediates JAK2/STAT3 signaling | rs1884444; 1:67168129 G |
[87] | |
| CTLA-4 | Encodes a protein which transmits an inhibitory signal to T cells | rs11571315; 2:203866178 T |
[72] | |
| rs3087243; 2:203874196 G | ||||
| rs5742909; 2:203867624 C | ||||
| FCRL3 | Encodes a member of the immunoglobulin receptor superfamily and is one of several Fc receptor-like glycoproteins | rs7528684; 1:157701026 A |
[63] | |
| rs11264799; 1:157700967 C | ||||
| PTPN22 | Encodes of member of the non-receptor class 4 subfamily of the protein-tyrosine phosphatase family that is a lymphoid-specific intracellular phosphatase | 1858 C |
[63, 73, 92, 96] | |
| 1123 G | ||||
| CARD9 | Mediates the production of pro-inflammatory cytokines | rs4077515; 9:136372044 C |
[97] | |
| HDAC3 | Histone deacetylase activity and regulates transcription that can down-regulate p53 function and modulate cell growth and apoptosis | rs2530223; 5:141634927 T |
[105] | |
| DNMT3A | Encodes a DNA methyltransferase that function in de novo methylation | −448 G/A | [106] | |
| DNMT3B | Encodes a DNA methyltransferase that function in de novo methylation | −149C/T; rs2424913; 20:32786453 C |
[106, 107] | |
| −579 G |
[70] | |||
| FCGR2A | A cell surface receptor on phagocytic cells, involved in the process of phagocytosis and clearing of immune complexes | 131H/R | [68, 76] | |
| FCGR2C | A transmembrane glycoprotein, involved in phagocytosis and clearing of immune complexes | −386 G/C | [78] | |
| FCGR2C −386C/−120T | ||||
| FCGR3A | Encodes a receptor for the Fc portion of IgG, removes antigen-antibody complexes from the circulation | 158 V/F | [77] | |
| VDR | Encodes vitamin D3 receptor | (Cdx2) G |
[103] | |
| CD28 | Provides co-stimulatory signals required for T cell activation and stimulation | rs1980422 | [71] | |
| CD40 | Encodes a receptor on antigen presenting cells (APCs) of the immune system and mediates various immune and inflammatory responses | rs1883832; 20:46118343 T |
[93] | |
| PRF1 | Encodes perforin that allow the release of granzymes and subsequent cytolysis of target cells | R28C rs141660796, N252S rs28933375 | [42] | |
| PD-1 | Programmed cell death protein | rs36084323 C |
[71] | |
| FAS | Induces apoptosis triggered by binding to FAS | rs56006128 | [101] | |
| SDF1/CXCL12 | Activates STAT3 and Akt signaling pathways | rs2839693 | [100] | |
| rs266085 | ||||
| TBX21 | Encode transcription factors involved in the regulation of developmental processes | c.595T |
[101] | |
| Epigenetics | Foxp3 | Regulates transcription | Hypermethylation of CpG sites in the promoter region | [108] |
| IL-1Ra | Modulates a variety of interleukin 1 related immune and inflammatory responses | VNTR polymorphism | [70] | |
| CD4+ T cells | Recognize antigens displayed by an APCs | Global H3K9 hypomethylation | [70] | |
| PD-1 | Immune regulator | Promoter hypermethylation | [102] | |
| NOTCH1 | Regulates interactions between physically adjacent cells | Hypermethylation of CpG sites | [44] | |
| MBD2 | Mediator of methylation signal, acts as a demethylase to activate transcription | Hypomethylation | [108] | |
| MBD4 | Bind specifically to methylated DNA, repress transcription from methylated gene promoters | Hypomethylation | [108] | |
| CD70 | Induces proliferation of co-stimulated T cells, enhances the generation of cytolytic T cells, and contributes to T cell activation | Hypomethylated promoter region | [109, 110] | |
| Copy Number Variations | FCGR2C-ORF | [69] | ||
| TNFSF15 | ||||
| TNFSF8 | ||||
| AKNA | ||||
| ITCH | ||||
| SAMHD1 | ||||
| Mosaic Duplications | 17q21.31 (0.2 Mb) | [69, 104] | ||
| 2p23.2 (0.34 Mb) | ||||
| 9q21.2 (10.89 Mb) | ||||
| 9q31.2 (17.77 Mb) | ||||
| 14q13.2-14q13.3 (0.37 Mb) | ||||
| 17p12 (1.46 Mb) | ||||
| 20q11.21-20q13.13 (14.24 Mb) | ||||
| 22q11.21 (0.16Mb) | ||||
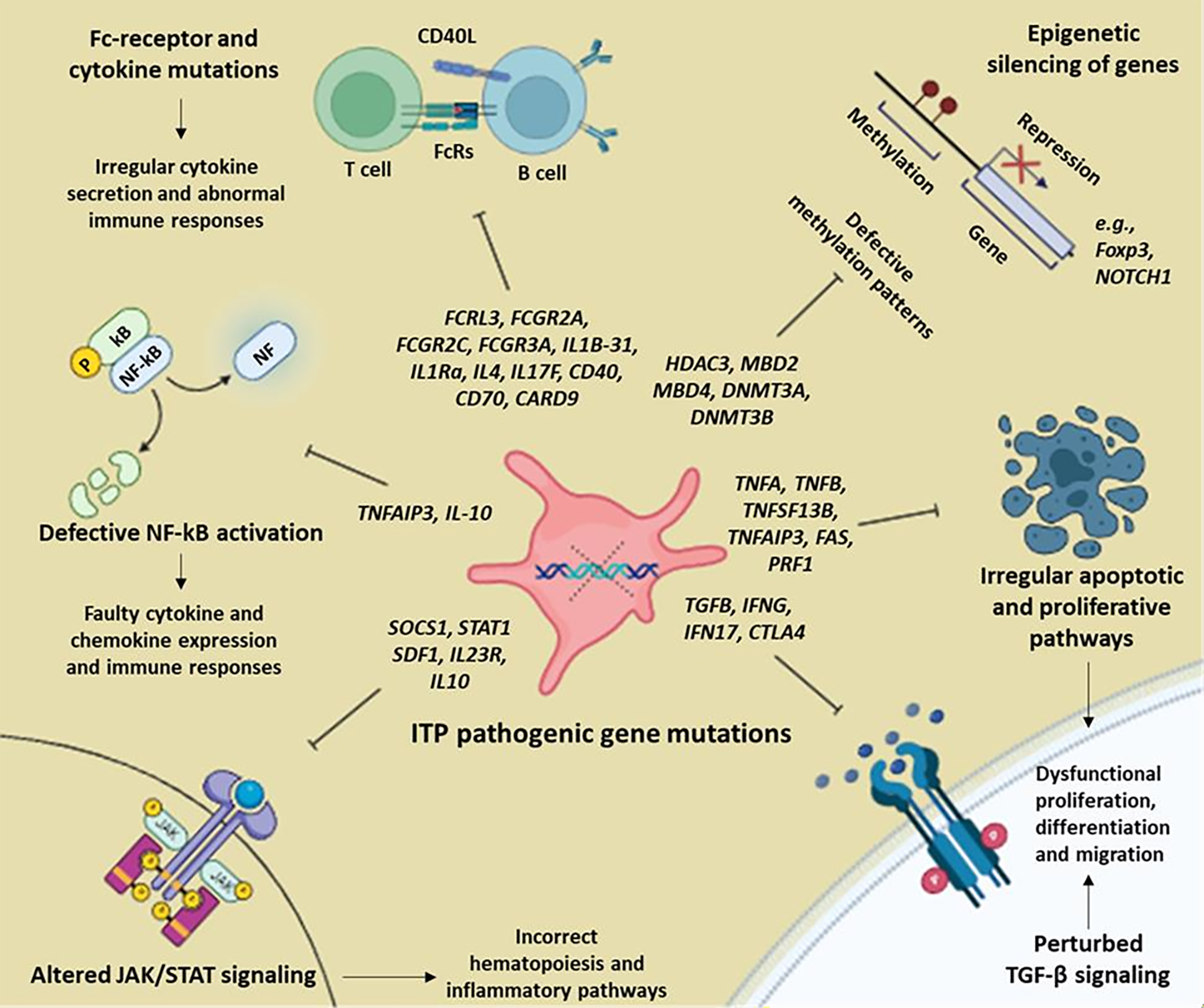 Fig. 2.
Fig. 2.
Polymorphisms and epigenetic mechanism involved in the
pathophysiology of Immune Thrombocytopenia (ITP) and alteration of various
signaling pathways. NF-
T cells function with MHC and other co-stimulatory and co-inhibitory molecules to recognize the antigens presented by the APCs [118]. Mutations have been found in T cells related genes which induce signaling cascades against platelets [119]. T-1993C mutation of T cell specific TBX21 gene has been found in the pathophysiology of ITP [120]. Chen et al. [72] investigated the genotype frequencies of CTLA4 rs11571315 and rs5742909. Significant differences between ITP cases and healthy controls were found, yet rs5742909 was only statistical significance in the group of patients with secondary ITP [72]. Subsequently, the pathogenic complexity of ITP has been indicated by comparison of recent findings with previous studies [121]. These studies conducted on different populations encompass a number of socio-demographic and genetic differences. Thereby, heterogeneity exists in the pathogenesis of ITP in different cohorts.
SNP -871 C
Mutations in Fc receptor encoding genes have been associated with ITP
pathogenesis. This is due to the involvement of Fc
The association of cytokine and chemokine encoding genes has been demonstrated
in various studies on ITP patients. These genes play important roles such as
stimulation of megakaryocytopoiesis, regulation of platelet production, and
generation of autoantibodies. Polymorphisms in IL-1B-31,
IL-1Ra, IL-4, IL-10, IL-17F, IL-23R,
TNF-
Despotovic et al. [91] revealed the role of inflammation related genes in the development of ITP by whole exome sequencing. IFNA17, IFNLR1, DGCR14, SMAD2, REL and CD83 genes playing roles in T cell differentiation, proliferation, and regulation are involved in the onset of the disease [91]. Polymorphisms of TNIP1, TNFAIP3, CD24, CD40, IRF5, CARD8, PTPN22 genes are also involved in the disease pathogenicity [92, 93, 94, 95, 96, 124, 127]. The presence of polymorphisms in CARD9 gene revealed decreased susceptibility to develop ITP along with reduced severity of the disease [97]. These results from different populations represent the significant role of various immune related gene mutations in the pathogenesis and susceptibility ITP among different individuals.
In a study, 3998 missense mutations involving 2269 genes in more than 10 individuals having ITP have been identified. These mutations mainly affected the Phosphatidylinositol 3 kinase/serine/threonine kinase B (PI3K/Akt) signaling pathways and platelet activation. They were reported as the important causative agents of ITP [65]. This is an important signaling pathway that regulates the cell cycle, proliferation and differentiation of platelets. Further investigations must be conducted to replicate the findings and find the definitive pathogenic agents.
Genome wide association studies (GWAS) in 200 Chinese ITP patients have revealed
862,620 SNPs across the autosomal region. Association of four novel loci have
been found with neuroactive ligand-receptor interactions, cancer, and the
Janus kinases/signal transducer and activator of transcription protein (JAK/STAT) pathway [67]. Haploinsufficiency of SOCS1 gene which is a
negative regulator of JAK/STAT pathway have been reported in an ITP cohort [98].
STAT1 gene polymorphisms have been significantly associated with
Interferon gamma (IFN-
Dysregulated apoptotic and proliferative pathways have led the researchers to investigate mutations present in FAS and perforin (PRF1) genes. These mutations have been associated with increased destruction and abnormal production of platelets leading to the onset of ITP [42, 101]. PD1 gene mutations have been associated with programmed cell death. These mutations alter the apoptotic and proliferative pathways causing pathophysiological changes in the individuals carrying the mutated genotypes [102].
Polymorphisms in genes encoding human platelet antigens (HPA), human leukocyte antigens (HLA), major histocompatibility complex (MHC), and mucins (e.g., MUC5B and MUC6) have been identified [129, 130, 131]. Mutations in the SDF1 gene have been found that significantly impact the process of megakaryopoiesis. This cause abnormal production of platelets leading to ITP [100]. An uncommon polymorphism in the vitamin D receptor (VDR) gene have been identified in Iranian ITP patients [103]. Copy number variations and mosaic duplications have also been identified in various chromosomal regions to be involved in ITP pathogenesis [69, 104]. Contrastingly, -448 SNP in DNMT3A and T allele of LAG3 rs870849 have been found to have protective effects against ITP [71, 106]. These studies indicate that the pathogenic mutations can be used as biomarkers to diagnose ITP while the protective polymorphism can also be used to develop important therapeutic strategies.
Autoimmune diseases are often associated with complex extrinsic factors due to their non-evident pathogenic causes. Scientists have explored the epigenetic changes and methylation patterns responsible for the expression and repression of different genes [70]. These epigenetic mechanisms are controlled by genes encoding DNA methyl transferases and histone acetylases. Investigations on ITP subjects have uncovered polymorphisms in DNMT3A, DNMT3B, HDAC3, MBD2, and MBD4 genes [64, 105, 106, 107, 132]. Dysregulated methylation patterns of FOXP3, CD70, NOTCH1, IL1Ra, MBD2, MBD4 and promoter region of PD1 genes have also been revealed that cause repression of these genes (Table 2) [44, 102, 108, 109, 110, 114]. All these genes have been found to have a significant role in the pathogenesis of ITP.
Immunoregulatory genes modulate the immune responses and signaling pathways. Presence of SNPs, CNVs, and epigenetic alterations influence the proper functioning of these genes. Studies conducted in different populations indicate the presence of variable pathogenic factors. These findings indicate that ITP is a highly complex and heterogeneous disorder. More studies are required to investigate the intrinsic and extrinsic immunoregulatory factors and replicate the findings. Genetic variations can serve as important diagnostic and therapeutic tools. The involvement of genetic factors can serve as important biomarkers in ITP diagnosis. Similarly, a number of therapeutic strategies can be devised to control the altered mechanisms and modulate the severity of the symptoms.
ITP treatment is fundamentally required to increase the platelet count to
maintain homeostasis. This is done to prevent frequent bleeding episodes or halt
any active bleeding [133]. ITP management guidelines suggest that a platelet
count of
The foremost management guidelines recommend that ITP treatments must have minimal toxicity so that patients’ health-related quality of life (HRQoL) could not be impaired [136, 137]. ITP patients are more acceptable of the treatments that manage symptoms long-term with minimal impact on daily lives [138]. The recommended first line treatment approach for ITP patients is the administration of corticosteroids, intravenous immunoglobulin (IVIg), anti-D immunoglobulins, and platelets transfusion [137, 138]. The non-responding patients requiring second line treatments must be treated with immunosuppressive agents such as rituximab (off-label), thrombopoietin receptor agonists (TPO-RAs), the spleen tyrosine kinase (SYK) inhibitor fostamatinib, or splenectomy [136, 137, 138].
The chronic or refractory ITP patients have indicated the need to develop new
therapeutic strategies. These must target different pathways involved in ITP
pathogenesis. Some of the novel treatment approaches to inhibit the destruction
of peripheral blood platelets are (i) neonatal Fc receptor inhibition
(Rozanolixizumab, Nipocalimab, and Efgartigimob); (ii) phagocytosis inhibition
targeting FC
ITP is a complex autoimmune disorder having unknown etiology. Both immunologic and genetic perturbations have been found to be involved in its pathophysiology. Among the immunologic factors, the autoantibodies produced by the defects in B and T cells cause destruction of platelets and defective megakaryocytopoiesis. The genetic analysis has revealed the presence of various single nucleotide polymorphisms, copy number variations, and epigenetic changes to be involved in the development of the disorder. The unknown pathophysiological cause indicates the need of individualized genetic testing to proceed with the development of personalized medicines, biomarker prediction, and therapeutic strategies.
ZT, MIQ and STO designed and conceptualized the article. ZT and KZ searched the literature and draw figures. MIQ and EVC provided help, streamlined the literature search and draw the tables. ZT and KZ wrote the original manuscript draft. MIQ and EVC reviewed and edited. EVC and STO revised the final version and provided funding. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We acknowledge Mr. Ghayyas Ud Din for providing support and guidance in preparation and publication of the manuscript.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.