



 , Elisa Pellencin 1, Aurora Romeo 1, Giorgio Giaccone 1, Giacomina Rossi 1, Sara Prioni 2,†, Paola Caroppo 1,†
, Elisa Pellencin 1, Aurora Romeo 1, Giorgio Giaccone 1, Giacomina Rossi 1, Sara Prioni 2,†, Paola Caroppo 1,†1 Neurology V and Neuropathology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milano, Italy
2 Clinical Neuropsychology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20133 Milano, Italy
†These authors contributed equally.
Abstract
Background: Microtubule-associated protein tau (MAPT) mutations are one of the main causes of genetic Frontotemporal dementia (FTD) and are characterised by high clinical heterogeneity. A behavioural variant of FTD is the principal phenotype, but other rarer phenotypes are described, mostly reported as single cases. In this review, we provide an overview of the clinical phenotypes associated with MAPT mutations in order to define their characteristics and explore genotype-phenotype correlations. Methods: We performed systematic bibliographic research on the Pubmed database, focusing on articles published between 1998 and 2022. We analysed the clinical phenotype of 177 patients carrying MAPT mutations, focusing on the rarest ones. We performed a narrative synthesis of the results. Results: Regarding language phenotypes, the most frequent were the non-fluent variant and the semantic variant of Primary Progressive Aphasia (nfvPPA, svPPA), approximately in the same proportion. Almost 20% of the whole group of patients present a clinical phenotype belonging to the corticobasal syndrome-progressive supranuclear palsy (CBS-PSP) spectrum. While no clear genotype-phenotype correlation could be identified, some mutations were associated with a specific phenotype, while others gave origin to multiple clinical pictures and mixed phenotypes. Conclusions: A high clinical heterogeneity exists in FTD associated with MAPT mutations without a clear phenotype-genotype correlation in most cases. However, some characteristics can be helpful to drive genetic testing. Deep phenotyping of patients, together with functional studies of single mutations, particularly those associated with atypical phenotypes, are necessary to better understand the biological mechanisms underlying this clinical variability.
Keywords
- MAPT
- FTD
- primary progressive aphasia
- AD
- CBS
- PSP
- phenotypes
Frontotemporal dementia (FTD) refers to a group of disorders characterised by striking clinical and pathological heterogeneity. Up to 40% of FTD are inherited forms caused by mutations in the progranulin (GRN), in the chromosome 9 open reading frame 72 (C9orf72) or in the microtubule-associated protein tau (MAPT) gene in most cases. The latter is responsible for approximately 5–10% of all familial FTD cases [1]. Almost 90 pathogenic MAPT mutations have been discovered so far (Human Gene Mutation Database, https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/; [2]), causing different effects on tau protein, with alteration of: (i) tau affinity for microtubules, leading to microtubule destabilisation; (ii) tau aggregation properties; (iii) the physiological proportion of 4R and 3R tau isoforms [3]. The clinical phenotypes associated with FTD are highly heterogeneous, with some differences between the three main genetic forms [4]. In MAPT mutation, the age at onset is usually earlier than in GRN and C9orf72 mutations and wide phenotypic variability, in terms of age of onset, clinical presentation and features of pathological tau deposits in the brain [1], is evident. The most common phenotype is the behavioural variant (bvFTD), while primary progressive aphasia (PPA) is less frequently described. bvFTD is characterised by progressive atrophy in the frontal and temporal lobes, causing behavioural, cognitive and personality alterations. PPAs include a heterogeneous group of diseases mainly affecting language. Depending on the brain area involved, symptoms vary from progressive loss of meaning and single-word comprehension deficits (semantic variant of PPA) to slow, effortful, hesitant and distorted speech (non-fluent variant of PPA). In addition, other phenotypes such as Corticobasal Syndrome (CBS), Progressive Supranuclear Palsy (PSP) and amnestic syndrome consistent with Alzheimer’s disease (AD-like) are associated with MAPT. CBS is characterised by asymmetric motor signs (rigidity, tremor, dystonia, and myoclonus) and is often associated with apraxia, cortical sensory deficits and alien limb phenomena. PSP affects body movements, causing early loss of balance, difficulty in walking or swallowing, slurred speech and eye movement impairment. AD-like phenotypes caused by MAPT mutations are usually characterised by predominantly episodic memory deficits at onset. In this review, we provide an overview of the clinical phenotypes associated with MAPT mutations, focusing on the rarest ones and on those belonging to the Frontotemporal Lobar Degeneration (FTLD) spectrum, trying to better define their characteristics and to explore genotype-phenotype correlations.
We conducted systematic bibliographic research on the Pubmed database. The search focused on articles published between 1998 (the year of the discovery of the MAPT gene) and 2022. We applied the following terms of search: (MAPT) AND (FTD), (MAPT) AND (frontotemporal dementia), (MAPT) AND (FTDP-17), (MAPT) AND (bvFTD), (MAPT) AND (behavioural variant), (MAPT) AND (PPA), (MAPT) AND (primary progressive aphasia), (MAPT) AND (svPPA), (MAPT) AND (semantic variant), (MAPT) AND (semantic dementia), (MAPT) AND (nfvPPA), (MAPT) AND (non-fluent variant), (MAPT) AND (lvPPA), (MAPT) AND (logopenic variant), (MAPT) AND (phenotype), (MAPT) AND (PSP), (MAPT) AND (progressive supranuclear palsy), (MAPT) AND (CBD), (MAPT) AND (CBS), (MAPT) AND (corticobasal syndrome) and (MAPT) AND (corticobasal degeneration). The Title/Abstract filter was used (see PRISMA flow-chart, Fig. 1)
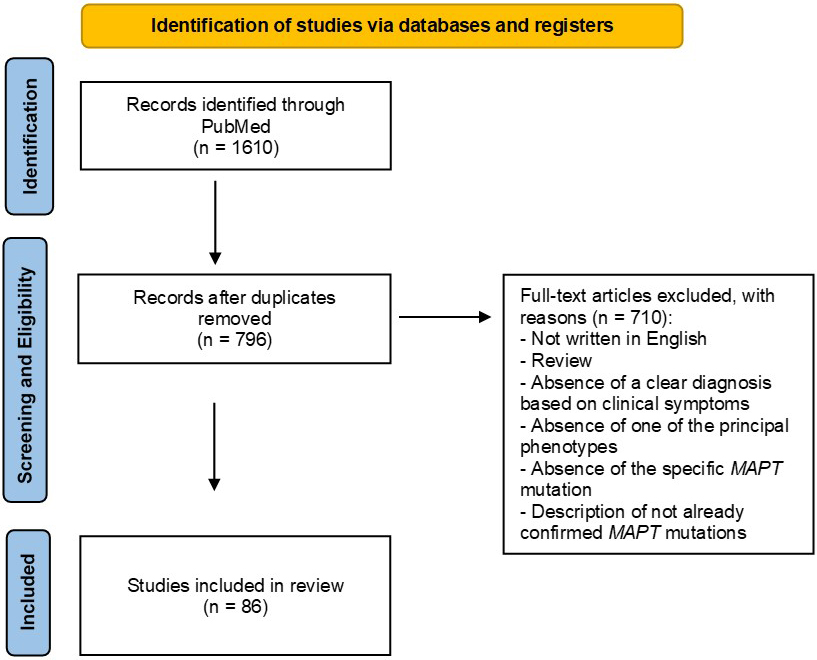 Fig. 1.
Fig. 1.PRISMA flow-chart of the Pubmed search. MAPT, microtubule-associated protein tau.
Since we primarily focused on the FTLD spectrum, we did not include the terms “AD” or “Alzheimer Disease” in our research. The few articles that included these terms that we found were described here because they were somehow included in an FTLD cohort or accidentally found while studying FTLD patients.
We found a total of 1610 articles. All the articles not written in English, the reviews and the studies without a clear description of the clinical phenotypes and/or without a formal diagnosis were excluded. We also excluded articles that did not specify the MAPT mutation or that described just haplotypes or risk factors or variants with uncertain significance (Fig. 1).
Where possible, the current bvFTD, PPAs, PSP and corticobasal degeneration (CBD) criteria [5, 6, 7, 8] were applied retrospectively.
We selected 86 articles, including 177 patients. Among them, 28 patients showed one of the PPAs diagnosis (including semantic variant and non-fluent variant), 23 had PSP phenotype, 12 CBS phenotype, 30 had AD-like phenotype, and 84 were diagnosed with bvFTD. Some of these papers are listed in different groups, as the same article often comprises patients with different disease phenotypes. We analysed the clinical data of the patients, including age at onset, familial history, neuroimaging data and MAPT mutations. We compared the ages at onset between groups by using ANOVA with Bonferroni correction. A narrative synthesis of the results has been performed. All MAPT mutations were reported referring to the NCBI Reference Sequence Database (RefSeq) transcript NM_005910.6.
We identified 21 articles describing 28 patients with different linguistic
phenotypes (Table 1, Ref. [9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29]), including two patients recently reported by
our group [19, 27]. Ten patients had the clinical diagnosis of nfvPPA (35.71%)
and ten subjects of svPPA (35.71%). Six patients exhibited clinical symptoms
consistent with the right temporal variant of FTD (rtvFTD) (21.43%). In three of
them, we revised the diagnosis on the basis of the recent rtvFTD criteria [30].
Two patients had a clinical diagnosis characterised by linguistic symptoms plus
other non-verbal symptoms (7.14%): one patient with Primary Progressive Apraxia
of Speech and one subject with a Primary Progressive Anarthria or Apraxia of
Speech. In the whole PPA group, familial history was reported as positive in 23
cases (82.14%). Age at onset varied between 24 and 69, with a mean age of onset
of 51.68 (
| Phenotype | Age at onset | Symptoms/deficit at onset | Other symptoms/deficits | MRI atrophy | Family history | MAPT mutation | Reference |
| svPPA | 46 | word-finding, semantic | N.A. | bilateral frontotemporal | y | P301L | [9] |
| svPPA | 69 | word-finding, semantic | N.A. | bilateral frontotemporal | y | P301L | [9] |
| svPPA | 43 | word-finding, semantic | N.A. | bilateral frontotemporal | y | P301L | [9] |
| rtvFTD | 46 | prosopoagnosia | memory, behavioural disorders | right temporopolar | n | V363I | [10] |
| Primary Progressive Anarthria or AOS | 60 | dysarthria, orofacial apraxia | N.A. | N.A. | y | P332S | [11] |
| rtvFTD | 50 | memory, attention, anomia | N.A. | bilateral frontotemporal | y | P332S | [11] |
| rtvFTD | 49 | memory, prosopoagnosia, depression | semantic | bilateral anterior temporal pole | y | P332S | [11] |
| svPPA | 38 | memory, anomia | semantic, behavioural disorders | bilateral frontotemporal | y | G389R | [12] |
| svPPA | 46 | word-finding, naming | behavioural disorders | anterior temporal pole |
y | P301L | [13] |
| svPPA | 53 | semantic | N.A. | N.A. | y | P301L | [13] |
| svPPA | 48 | semantic | behavioural disorders | N.A. | y | P301L | [13] |
| nfvPPA | 65 | non-fluent aphasia and parkinsonism | anxiety, cognitive decline | bilateral cortical | y | K298E | [14] |
| nfvPPA + CBD | 60 | word-finding, naming, stuttering | parkinsonism, right-hand apraxia, stimulus-sensitive myoclonus | frontotemporal |
y | R5H | [15] |
| nfvPPA | 31 | N.A. | N.A. | frontotemporal |
y | L266V | [16] |
| nfvPPA | 69 | stuttering | N.A. | NA | y | V363I | [17] |
| nfvPPA + AOS (later CBS) | 55 | AOS | bradykinesia | left frontal | n | V363I | [18] |
| svPPA | 47 | loss of semantic knowledge | N.A. | N.A. | y | P301L | [19] |
| nfvPPA | 54 | naming, prosopoagnosia | N.A. | left temporal | y | P301L | [20] |
| PPA + AOS | 69 | naming, disprosodia | parkinsonism, lower motor neuron disease | N.A. | y | K317N | [21] |
| svPPA | 56 | memory, anomia, comprehension, surface dyslexia | seizures | frontotemporal |
y | P301L | [22] |
| nfvPPA | 24 | memory, agrammatism, AOS | behavioural disorders | frontotemporal |
n | G389R | [23] |
| nvfPPA | 57 | naming | N.A. | left temporal | n | D177V | [24] |
| nfvPPA | 37 | non-fluent aphasia | behavioural disorders | N.A. | N.A. | S305I | [25] |
| nfvPPA | 65 | word-finding, dysartria | N.A. | N.A. | y | G304S | [26] |
| svPPA | 37 | semantic, judgment | N.A. | left temporopolar | y | Q336H | [27] |
| rtvFTD | 59 | memory deficits, behavioural changes | prosopagnosia and atypical parkinsonism | frontotemporal | y | S352L | [28] |
| rtvFTD | 52 | episodic memory deficits, depression, apathy | anomia and several behavioural problems | bilateral temporal; right |
y | R406W | [29] |
| rtvFTD | 62 | behavioural changes (inappropriate, neglectful of other feelings, gluttonous) | prosopagnosia, single-word comprehension deficit and object naming, then familiar face recognition | marked anterior temporal pole; right |
y | P301L | [29] |
MRI, Magnetic resonance imaging; y, yes; n, no; N.A., not available.
Ten patients of our series had the diagnosis of nfvPPA. The average age at onset
was 51.7 years (
Ten patients had the diagnosis of svPPA; the main symptoms at onset were
language deficits (anomia and word finding) and memory disturbances. Average age
at onset was 48.3 (
Interestingly, the diagnosis of rtvFTD was found in six patients. The mean age
at onset was 53 years (
Two patients showed a PPA-plus phenotype, in which language disturbances were present but not predominant. These two subjects presented Primary Progressive Apraxia of Speech or Anarthria [11, 21] linked to P332S and K317N MAPT mutations. The first report [11] was the mother of the two subjects carrying the same mutation and who developed right temporal variant FTD. Her first symptom was a modification in her voice followed by orofacial apraxia, brisk face reflexes and hypernasal dysarthria; across the 25 years of illness, the patient had never manifested cognitive or behavioural modifications. The subject of the second paper [21] presented with difficulty in expression and production of sounds. Over time, the patient developed PSP-like symptoms, such as vertical gaze palsy, axial rigidity, backward falls and apraxia of eyelid opening. We also found one CBS case with nfvPPA signs at onset carrying the V363I MAPT mutation; even though the main diagnosis was actually CBD, the patient showed unspecified primary progressive aphasia, left-sided parkinsonism and CBS [31].
We identified 12 patients with CBS phenotype (Table 2, Ref. [31, 32, 33, 34, 35, 36, 37, 38]). Mean age
at onset was 48.82 years (
| Phenotype | Age at onset | Symptoms/deficit at onset | Other symptoms/deficits | MRI atrophy | Family history | MAPT mutation | Reference |
| CBS | 70 | right-sided dexterity impairment, slowed gait, imbalance | dysarthria, asymmetric parkinsonism, apraxia, right-sided neglect | biparietal | y | V363I | [31] |
| CBS-PPA | late 50s | left-sided parkinsonism and aphasia | N.A. | N.A. | n | V363I | [31] |
| CBS | 27 | left arm dystonia and rigidity | N.A. | right parieto-frontal | y | P301S | [32] |
| CBS/PSP | 38 | slowness, writing disability | N.A. | bilateral frontal | y | P301S | [33] |
| CBS/PSP | 38 | psychomotor slowness, gait and posture disorder | N.A. | bilateral frontal | y | P301S | [33] |
| CBS | 68 | Right-hand clumsiness | language, depression, mood changes | left parieto-temporal | y | P301T | [34] |
| CBS | 49 | gait instability | lack of motor coordination in the right-hand | mild frontotemporal | y | P301T | [34] |
| CBS | 43 | speech difficulties and cognitive decline | motor difficulties |
left posterior parietal | y | P301T | [34] |
| CBS | 54 | asymmetric parkinsonism at right, micrographia | memory deficits | bilateral symmetric putaminal hyperintense signals | y | P301L | [35] |
| CBS/PSP | 63 | memory and mood | N.A. | N.A. | y | N410H | [36] |
| CBS-AOS | 47 | speech difficulty | AOS, orofacial apraxia and dysphagia | left posterior frontal lobe | y | C291R | [37] |
| CBS | 40 | clumsiness and tremor of the left-hand | stiffness, movement slowness, speech impairment, cognitive disturbances | right fronto-parietal | n | G389R | [38] |
y, yes; n, no; N.A., not available; CBS, corticobasal syndrome; PPA, primary progressive aphasia; PSP, progressive supranuclear palsy; AOS, apraxia of speech.
Casseron and colleagues [33] reported two cases of P301S mutation found in a large family with a diagnosis of CBD. Both patients had an age of onset in their late 30s. The patient described by Bugiani and colleagues [32] had, in turn, a very early age of onset (27 years-old) and presented asymmetric symptoms from the beginning. Interestingly, his father was diagnosed with schizophrenia and neuropathologically showed frontotemporal atrophy with widespread neuronal tau inclusions. Erro and colleagues [34] reported three cases carrying another mutation at the same codon, the P301T, with an older age of onset and, in one case, with speech difficulties at the beginning. Most patients in this group had a positive family history of neurodegenerative/psychiatric diseases (83.33%).
We found 23 patients with PSP phenotype, whose mean age at onset was 43.09
(
| Phenotype | Age at onset | Symptoms/deficit at onset | Other symptoms/deficits | MRI atrophy | Family history | MAPT mutation | Reference |
| PSP | 40 | behavioural, memory and attention disturbances | N.A. | diffuse cortico-subcortical in late-stage | n | N279K | [39] |
| PSP | 41 | behavioural, attention disturbances | N.A. | normal | y | N279K | [39] |
| PSP + PLS | 45 | gait disturbance | N.A. | asymmetric frontotemporal, left hippocampus. mesencephalic | y | P301T | [34] |
| PSP | 40 | dystonia and supranuclear gaze palsy | N.A. | normal | y | S285R | [40] |
| PSP | 41 | gait unsteadiness | N.A. | N.A. | y | S285R | [40] |
| PSP | 40 | fatigue and micrographia | N.A. | N.A. | n | IVS 10+16C |
[41] |
| PSP | 48 | cognitive decline, changes in speech | gait and postural disturbances, vertical supranuclear gaze palsy | left frontal and temporal | y | IVS10+3G |
[42] |
| PSP | 40 | unstable gait, behavioural change, memory problems | N.A. | unremarkable | y | N279K | [43] |
| PSP | 41 | shuffling gait, bradykinesia | N.A. | N.A. | y | N279K | [43] |
| PSP | 43 | difficulty in walking | N.A. | N.A. | y | N279K | [43] |
| PSP | 46 | difficulty speaking and breathing | gait disturbances, limb bradykinesia | N.A. | n | S285R | [44] |
| PSP | 42 | parkinsonism, personality changes, postural tremor | micrographia, shuffling gait | N.A. | y | N279K | [44] |
| PSP | 44 | Right-hand clumsiness and oscillopsia | horizontal pendular nystagmus | N.A. | y | N279K | [44] |
| PSP-RS | 40 | bradykinesia, resting tremor at right-hand | smaller voice and handwriting | frontal and temporal | y | N279K | [45] |
| Atypical PSP | 38 | forgetfulness, word-finding problems, and slowness | N.A. | mild diffuse cerebral atrophy | y | DelN296 | [46] |
| Atypical PSP | 39 | altered behaviour and parkinsonism, and progressive clumsiness of his left limbs | N.A. | mild diffuse cerebral atrophy | y | DelN296 | [46] |
| PSP | 62 | gait disorder, postural instability, dysarthria, micrographia | N.A. | unspecified cerebral atrophy | N.A. | R5L | [47] |
| PSP | 43 | personality changes | memory, backward falls | N.A. | y | L284R | [48] |
| PSP | 37 | akinetic-rigid syndrome, gait disturbance, falls | micrographia, dysarthria, no upgaze, apraxia of eyelid | N.A. | y | G303V | [49] |
| PSP | 36 | antecollis, dysarthria, postural instability, slowing of ocular movements, increased deep tendon reflex | N.A. | N.A. | y | DelN296 | [50] |
| PSP | 56 | diplopia due to oculomotor dysfunction with supranuclear ophthalmoparesis | N.A. | midbrain and mesencephalon | y | V363A | [18] |
| PSP | 41 | leg stiffness and en-bloc turning | bradykinesia, instability, hypomimia, vertical supranuclear gaze palsy | mild temporal | y | N279K | [51] |
| PSP | 48 | dystonia of the left arm, slurring of speech | writing with right-hand difficulties, walking up and downstairs | mild over the cerebrum vertex | y | S305S | [52] |
y, yes; n, no; N.A., not available; PSP, progressive supranuclear palsy; CBS, corticobasal syndrome; PLS, primary lateral sclerosis; PSP-RS, progressive supranuclear palsy Richardson syndrome.
The most frequent MAPT mutation associated with the PSP phenotype was N279K (39.13%), followed by the N296del and the S285R mutations (both 13.04%). The other eight cases were characterised by different MAPT mutations. MAPT N279K mutation was described in Japanese families [43, 44]. Comparing their phenotypic characteristics with sporadic PSP patients, Ogaki and colleagues [43] found that the visual grasping symptom could be a sign specifically related to the presence of a MAPT mutation.
Pastor and colleagues [46] described two patients with atypical PSP linked to the N296del MAPT mutation. This mutation could be a risk factor for Parkinson’s disease [46] in the homozygous state and, interestingly, even in the heterozygous state [50].
Two MAPT mutations at the same residue, V363I and V363A, are associated respectively with CBS [31] and PSP phenotypes [18]. In the latter study, the PSP patient had a mixed diagnosis of CBS, PPA and left-sided parkinsonism, with complete anarthria and severe dysphagia in the advanced stage.
We identified 15 articles describing 30 patients with an AD-like phenotype at
onset linked to a MAPT mutation (Table 4, Ref. [9, 18, 27, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64]). One
patient has been previously reported by our group [27]. The mean age at onset was
55.73 (
| Phenotype | Age at onset | Symptoms/deficit at onset | Other symptoms/deficits | MRI atrophy | Family history | MAPT mutation | Reference |
| AD | 59 | memory | parkinsonism | bilateral temporo-frontal | n | P301L | [9] |
| AD | 46 | memory | parkinsonism | N.A. | y | P301L | [9] |
| AD | 56 | memory, loss and calculation difficulties | parkinsonism | N.A. | y | R406W | [53] |
| AD | 59 | withdrawal and difficulty adapting to new environments | parkinsonism | N.A. | y | R406W | [53] |
| AD | 62 | memory | parkinsonism | N.A. | y | R406W | [53] |
| AD | 48 | topographical disorientation, word-finding difficulties | N.A. | N.A. | y | R406W | [53] |
| AD | 58 | mild cognitive impairment progressing to amnesia for faces and names | parkinsonism | N.A. | y | R406W | [53] |
| AD | 55 | memory | N.A. | hippocampal | y | R406W | [54] |
| AD | 56 | memory | N.A. | bilateral medial temporal lobes | y | R406W | [55] |
| AD | 45 | memory, disorientation, loss of interest | N.A. | N.A. | y | R406W | [55] |
| AD | 51 | memory | N.A. | temporal medial lobes, hippocampus and parahippocampal | y | R406W | [55] |
| AD | 68 | memory | behavioural | medial temporal lobe | y | R406W | [56] |
| AD | 59 | memory | behavioural | frontotemporal | y | R406W | [57] |
| AD | 56 | memory, disorientation | depression, verbal perseveration | bilateral temporal | y | IVS10+16C |
[58] |
| AD | 49 | memory, disorientation | falls, stiff neck, occasional choking | N.A. | y | IVS10+16C |
[59] |
| AD | 40 | memory, irritability | depression | N.A. | y | Q351R | [60] |
| AD | 65 | memory | N.A. | bilateral temporal | y | R406W | [61] |
| AD | 65 | memory | N.A. | cortical and central | y | R406W | [61] |
| AD | 52 | prosopoagnosia | N.A. | bilateral medial temporal | y | R406W | [61] |
| AD | 55 | memory, personality change, depression | behavioural | N.A. | y | R406W | [62] |
| AD | 62 | memory, personality change, anxiety | behavioural | hippocampal and medial temporal lobe atrophy | y | R406W | [62] |
| AD | 60 | memory, restlessness | behavioural | hippocampal and medial temporal lobe atrophy | y | R406W | [62] |
| AD | 54 | memory, personality change, childish behaviour, irritability | behavioural | hippocampal and medial temporal lobe atrophy | y | R406W | [62] |
| AD | 56 | memory, childish behaviour | behavioural | hippocampal and medial temporal lobe atrophy | y | R406W | [62] |
| PCA | 51 | prosopoagnosia | visuospatial deficits | right parietal and temporal regions | y | V363I | [18] |
| AD | 59 | memory | N.A. | bilateral, medial temporal lobe and hippocampi | y | R406W | [63] |
| AD | 65 | memory | N.A. | medial temporal | y | Q336H | [27] |
| AD | 53 | prosopoagnosia | behavioural, language, parkinsonism | medial temporal lobe and parahippocampal | y | R406W | [64] |
| AD | 58 | memory | behavioural, AOS | N.A. | y | R406W | [64] |
| AD | 50 | dyscalculia, social withdrawal, lack of initiative | behavioural, language, parkinsonism | medial temporal hippocampus |
y | R406W | [64] |
y, yes; n, no; N.A., not available.
The R406W MAPT was the most frequent mutation with AD-like phenotype (76.67%). Two patients carried the P301L MAPT mutation. All the remaining patients (23.33%) carried rarer mutations. The almost totality of the AD-like group was characterised by a positive family history of neurodegenerative diseases (96.67%). It’s worth mentioning the only article describing a phenotype consistent with posterior cortical atrophy (PCA) [18]: a 54-year-old woman who developed difficulties in recognising faces and visuospatial impairment at the age of 51 and carried the V363I mutation.
We identified 84 patients with bvFTD phenotype at onset (Supplementary
Table 1, Ref. [16, 20, 43, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95]). More than half of patients had a family
history of dementia (71.43%), considering that several papers did not specify
this information (25%). The mean age at onset was 47.62 (
A general overview of the whole cohort is reported in Table 5. Using ANOVA, we
compared ages at onset between groups, and we only found a significant difference
between AD and all other phenotypes (p
| Phenotype | N° of patients | Positive family history | Mean AAO | |
| PPA | 28 | 82.14% | 51.68 ( | |
| nfvPPA | 10 | 60%* | 51.7 ( | |
| svPPA | 10 | 100% | 48.3 ( | |
| rtvFTD | 6 | 83.33% | 53 ( | |
| PPAOS/Primary Progressive Anarthria | 2 | 100% | 64.5 ( | |
| bvFTD | 84 | 71.43%* | 47.62 ( | |
| PSP | 23 | 82.61% | 43.09 ( | |
| CBS/CBD | 12 | 83.33% | 48.82 ( | |
| AD-like | 30 | 96.67% | 55.73 ( | |
AAO, age at onset; PPAOS, Primary Progressive Apraxia of Speech.
*some articles did not specify a family history of disease.
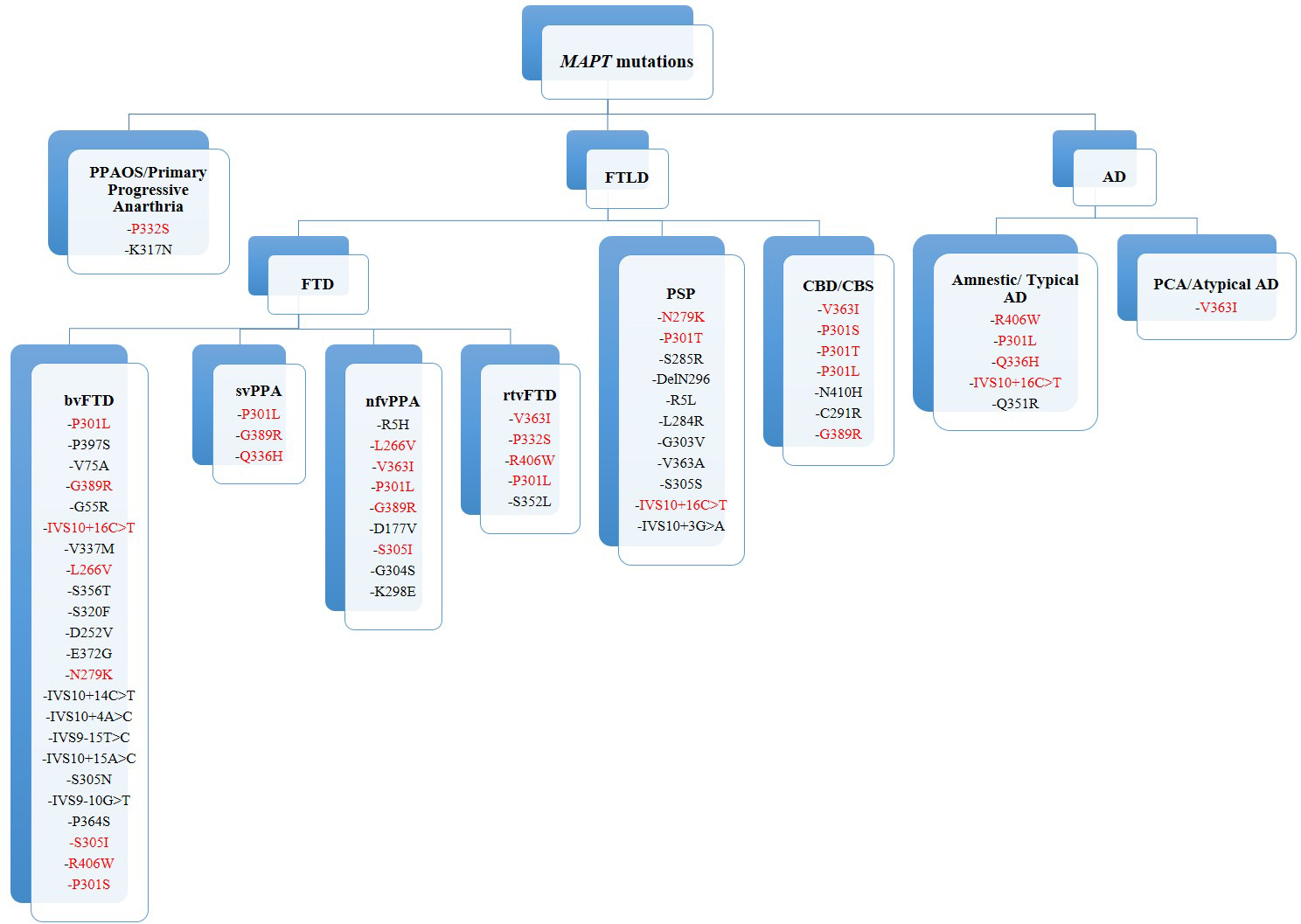 Fig. 2.
Fig. 2.MAPT mutations and phenotypes. In red: MAPT mutations associated with different phenotypes. FTLD, frontotemporal lobar degeneration; FTD, frontotemporal dementia; bvFTD, behavioural variant frontotemporal dementia; svPPA, semantic variant primary progressive aphasia; nfvPPA, non-fluent variant primary progressive aphasia; rtvFTD, right temporal variant frontotemporal dementia; PPAOS, primary progressive apraxia of speech; PSP, progressive supranuclear palsy; CBD/CBS, corticobasal degeneration or syndrome; AD, Alzheimer’s disease; PCA, posterior cortical atrophy.
Notably, some of the mutations are associated with multiple phenotypes, in
particular CBS, PPA and bvFTD. The P301L appears to be a cross-group mutation, as
it was found at least once in every phenotype group except for the PSP group.
IVS10+16C
In this review, we analysed the clinical phenotypes of patients carrying the MAPT mutations reported in the literature between 1998 and 2022 to delineate possible clinical-genetic correlations, paying particular attention to the rarest clinical presentations such as PPAs, atypical parkinsonism and AD-like phenotypes.
Among the entire group of patients included in this review, 52.54% carried a phenotype rarer than bvFTD: AD-like phenotype (16.95%), followed by the PPAs group (15.82%), which may reflect the fact that atypical phenotypes are more fully characterised in the literature than the classical ones.
Interestingly, considering all patients, seven had an onset before their 30s, most of them with a diagnosis of bvFTD, in agreement with previous findings on very early age at onset FTD, in which most of the subjects present the bvFTD phenotype [96]. In this review, we extend the findings of very early onset FTD to other phenotypes, with 18 MAPT patients with PPAs and PSP/CBS having age at onset at or before their 40s. Early age at onset does not have a clear link with specific mutations.
Regarding the language phenotypes, we found almost the same number of patients with svPPA and nfvPPA, while logopenic variant was absent. An almost equal ratio of svPPA and nfvPPA was found in MAPT mutations in one of the largest worldwide cohorts of genetic FTD [4]. This result differed from genetic TDP-43 proteinopathies, where the predominant language phenotype was the logopenic variant in a large cohort of GRN carriers [97] and the nfvFTD in C9orf72 expansion [4, 98]. Unlike MAPT patients, svPPA is exceptional in these other two genetic forms of FTD [97]. This result suggests a relative homogeneity of pathology for the semantic phenotype with a major vulnerability of the temporal anterior lobe for tau accumulation [99].
Another difference with TDP43 genotypes is that the mean age at onset in PPA patients by our review is earlier than PPA linked to C9orf72 expansion and GRN mutations reported in the literature [97].
Interestingly, although the criteria for rtvFTD have only recently been defined, we identified at least six patients whose clinical presentation was consistent with this phenotype. RtvFTD is rarer than the left variant and represents a “unique”, usually considered sporadic and associated with TDP-43 pathology [100]. The recognition of this clinical entity has become even more important as the involvement of the right temporal lobe seems to be associated with a more widespread neurodegeneration than in patients with main involvement of the left temporal lobe and a shorter survival, possibly linked to a diagnostic delay [101]. RtvFTD has also been associated, although rarely, with GRN mutations, suggesting that these two genes should be tested first.
Among the PPA group, we classified five patients as “PPA-plus” because of other neurological symptoms associated, mainly parkinsonism, myoclonus and dystonia. Conversely, we could not classify patients as mixed PPAs, a diagnostic category largely represented in PPA, because of the lack of a detailed neuropsychological assessment in several cases.
Considering the overall group of PPA, the most frequent MAPT mutation was the P301L. This is the most frequent mutation worldwide, and it is found to be linked to bvFTD phenotypes [4]. In our review, P301L was also found associated with svPPA, with one case of nfvPPA, with one rtvFTD and one CBS. The rarer V363I mutation has been found in four PPA cases, half of them also characterised by CBS signs. This mutation has also been described in bvFTD [102], showing high heterogeneity in its clinical presentation. Some authors hypothesise that the rare presence of this mutation in the general population can be due to an incomplete penetrance and late disease onset [31].
In this paper, we included a large number of patients with PSP and CBS phenotypes, usually defined as sporadic; genetic PSP has also been associated with mutations in parkinsonism-related genes [103, 104] and very rarely with C9orf72 and GRN mutations. PSP patients from our review had a mean age at onset earlier than that observed in sporadic forms or in PSP associated with other FTD genes [105], and almost 40% of PSP linked to MAPT had a very early age at onset, before their 40s, an important feature that can lead to suspect a MAPT mutation in PSP phenotypes. Among patients with CBS phenotype, relatively “pure” CBS was found in more than 50% of patients, while the others showed mixed clinical symptoms, classifiable as PSP-CBS according to MDS criteria [106]. Therefore, we could suggest that co-occurrence of PSP and CBS features and early age at onset may orient the diagnosis toward a genetic tauopathy.
We identified 30 patients with an AD-like phenotype presenting with memory
deficits but later developing behavioural modifications with or without
parkinsonism, thus orienting the diagnosis towards FTD. The MAPT
mutation more frequently associated with this phenotype was the R406W, but we
also found two patients carrying P301L, two IVS10+16C
Several mutations are associated with multiple phenotypes, showing no clear genotype-phenotype correlation. However, some mutations are predominantly associated with atypical forms such as AD-like presentations or parkinsonisms (Fig. 2). However, a positive family history, the presence of other associated symptoms, and an early age of onset may be suggestive of a MAPT mutation.
MAPT mutations can have different impacts on tau protein, altering its ability to promote microtubule assembly, affecting its aggregation properties or determining the ratio between 4R and 3R isoforms [107], such as mutations associated with PSP and CBS phenotypes that often lead to 4R pathology, similarly to sporadic cases (see Supplementary Fig. 1). However, the mechanisms by which the mutations can give origin to different clinical forms are not known yet. A recent interesting study on iPS cells showed that one of the more frequent mutations associated with parkinsonism (N279K) has an impact on mitochondrial functions at variance with other mutations [108]. This suggests that the clinical variability could be explained by mutation-related specific biological mechanisms that need to be further explored.
Our review has several limitations. First, the research terms included the name of the diseases and of the gene but not every single symptom or tau pathology. In addition, some papers lacked detailed clinical descriptions and neuropsychological assessment. However, to include the majority of articles, we searched in depth in the reference list of every paper and, in some articles written before the appearance of current clinical criteria, we deduced the diagnoses based on clinical description. In addition, due to the heterogeneity of the clinical phenotypes, except for age at onset, we did not perform a metanalysis of the results but a narrative descriptions, and we discussed the results in light of previous findings on other genetic forms. Despite these limitations, we could provide an overview and an update of clinical-genetic associations of the cases described in the literature, analysing the phenotypes of a large number of MAPT patients. We underlined the high clinical heterogeneity of FTD associated with MAPT mutations without clear phenotype-genotype correlations in most cases. Deep phenotyping of patients, together with the study of the pathogenetic mechanisms of single mutations, particularly those associated with atypical phenotypes, are necessary to better understand the biological basis of the clinical and neuropathological phenotypic variability of neurodegenerative diseases associated with MAPT mutations.
All data underlying the results are available as part of the article.
CV and EP acquisition of data, analysis and interpretation of data, drafting the manuscript. PC conception and design, analysis and interpretation of data, drafting the manuscript. GR and GG, analysis and interpretation of data and reviewing it critically for important intellectual content. SP conception and design, analysis and interpretation of data drafting the manuscript. AR acquisition of data, analysis and interpretation of data, reviewing it critically for important intellectual content. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors give final approval of the version to be published.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Given the role as Guest Editor, Giorgio Giaccone had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Marek Kieliszek.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.