

1 Department of Biology, School of Life Sciences, BK21 FOUR KNU Creative BioResearch Group, College of Natural Sciences, Kyungpook National University, 41566 Daegu, Republic of Korea
2 Research Institute for Dok-do and Ulleung-do Island, Department of Biology, School of Life Sciences, Kyungpook National University, 41566 Daegu, Republic of Korea
3 Brain Science & Engineering Institute, Kyungpook National University, 41944 Daegu, Republic of Korea
Abstract
Background: Neurodegenerative diseases, such as diabetic retinopathy
(DR) and glaucoma, induce retinal neuron loss. Acetylcholine-containing
cholinergic neurons, known as starburst amacrine cells (SACs), play critical
roles in the generation of precise neuronal activity in the retina and are
located in the inner nuclear layer (INL, conventional) and ganglion cell layer
(GCL, displaced). Methods: This study investigated the loss of and
morphological changes in SACs in the retinas of streptozotocin (STZ)-induced
diabetic and insulin-deficient C57BL/6-Tg(pH1-siRNA
Keywords
- starburst amacrine cells
- diabetic retinopathy
- streptozotocin
- choline acetyltransferase
- calcium-binding proteins
- immunocytochemistry
Diabetic retinopathy (DR) is a major complication of diabetes that can lead to progressive vision loss and blindness [1, 2, 3, 4]. Approximately one-third of individuals with diabetes develop DR, the treatment of which requires multiple options due to its complex and multifactorial pathogenesis [3, 5]. Hyperglycemia, which induces superoxide production and oxidative stress, plays a central role in the development of vascular alterations [6, 7]. Diabetic microvascular complications typically include neuropathy, nephropathy, and retinopathy, whereas macrovascular complications include cerebrovascular, cardiovascular, and peripheral complications [8, 9]. DR is also a strong predictor of microvascular and macrovascular complications [8, 10, 11].
DR has been classified into two types: non-proliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR). NPDR occurs in the first stage of DR and is characterized by altered vascular permeability, basement membrane thickening, pericytes loss, and acellular capillary formation. NPDR can be further divided into three types based on its progression: mild NPDR, moderate NPDR, and severe NPDR. PDR is an advanced stage of DR and is characterized by neovascular complications and may lead to retinal detachment. In any stage of DR, diabetic macular oedema may develop due to the accumulation of exudative fluid in the macula [1, 3, 12, 13].
A growing body of research have reported that neurodegeneration plays a significant role in the development of DR [6, 14]. DR is a highly specific neurovascular complication accompanied by progressive damage to retinal neurons, including such as photoreceptors, horizontal, bipolar, amacrine, and retinal ganglion cells (RGCs), and glial cells, such as Müller cells, astrocytes, and microglia [2, 15]. Previous studies have shown that the loss of retinal cells is associated with increased apoptosis, decreased thickness, and cell density reduction [16, 17, 18, 19, 20, 21]. Neurodegeneration in DR occurs in early stages even before vascular lesions can be detected [3, 22, 23]. However, there is still a debate over whether microvasculopathy causes neurodegeneration or vice versa or if they are mutually independent [3, 22, 24, 25].
Streptozotocin (STZ), initially isolated from Streptomyces achromogenes, is a naturally occurring chemical agent that is particularly toxic to the insulin-producing beta cells in the pancreatic islet and is the most prominent diabetogenic chemical compound in diabetes research [26]. Many studies have used STZ-induced diabetic animal models, especially mouse and rat models of DR [16, 17, 18, 20, 27, 28, 29, 30]. For example, periodic progression of cellular and vascular lesions in STZ-induced DR has been reported [27]. In mice with STZ-induced diabetes, the number of cells in the ganglion cell layer (GCL) was reduced [16, 18]. In addition, in rats with STZ-induced diabetes, retinal cell apoptosis was increased, whereas the total retinal thickness was decreased [17, 20, 28]. The relationship between vascular damage and neuronal changes, such as apoptosis of retinal neurons, has also been investigated using mice with STZ-induced diabetes [29]. Basement membrane thickening of arterial and venous capillaries has been observed in the retina of rats with STZ-induced diabetes [30].
Starburst amacrine cells (SACs) release two neurotransmitters, the excitatory neurotransmitter acetylcholine and the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). SACs are also called cholinergic amacrine cells because they are the only retinal cells that release acetylcholine [31, 32, 33, 34]. SACs are distributed regularly in the retina [34, 35, 36, 37]. SACs in the GCL are called displaced SACs or OFF-SACs, and those in the inner nuclear layer (INL) are called conventional SACs or ON-SACs. SACs play several important roles, including a major role in the detection of motion images in the retina [38, 39].
Calcium ions participate in several important neuronal actions, and calcium-binding proteins (CBPs) are known to mediate the functions of calcium [40, 41]. Calbindin-D28K (CB), calretinin (CR), and parvalbumin (PV) are CBPs distributed in a large subpopulation of retinal neurons [42, 43]. The CBP expression of choline acetyltransferase (ChAT)-immunoreactive (IR) neurons differs among animals. For example, ChAT-IR neurons express CB in the lungfish retina [44], bat retina [35], ground squirrel retina [45], and human and marmoset retina [46], whereas CR is expressed in the lungfish retina [44], rat retina [47], and ground squirrel retina [45]. However, ChAT-IR neurons only express PV in the ground squirrel retina [45] and rabbit retina [48].
The death of several retinal cell types with the progression of DR and the loss
of ChAT-IR neurons in the retinas of 24-week-old Ins2
All experiments involving animals were approved by the Animal Care and Use Committee of Kyungpook National University (permission no. 2020-0158). Animals were group housed under a 12-h light:12-h dark cycle until used for studies. Temperature and humidity levels in animal housing facilities ranged from 23 °C to 26 °C and from 45% to 65%, respectively.
Adult mice (C57BL/6J, 8–10 weeks old, weighing 20–30 g) were examined. After 4
h of fasting, we measured their weight and blood glucose levels and randomly
categorized the animals into experimental and control groups. Experimental mice
were intraperitoneally administered 6 mg/ml STZ dissolved in 50 mM sodium citrate
buffer (pH 4.5), and control animals were intraperitoneally administered 50 mM
sodium citrate buffer (pH 4.5) in reference to standard protocol of previous
study [27]. We calculated the amount of injection solution at ratio of 40 mg/kg
of weight of each mouse and injected via intraperitoneal injection using 1 mL
syringe. The aforementioned process was repeated for 5 days. After injection, we
checked the weight and fasting blood glucose level of each mouse every 5 days.
Blood was obtained from the tail vein. Two weeks after the injection, mice with
blood glucose levels
| Animals | Duration (week) | Group | Number | BGL (mg/dL) |
|---|---|---|---|---|
| 4–6 | Diabetic | 3 | 450.00 | |
| STZ injected mice | Control | 3 | 178.33 | |
| (C57BL/6J mice) | 42 | Diabetic | 3 | 404.67 |
| Control | 3 | 160.33 | ||
| IDCK mice | 20 | Diabetic | 3 | 265.00 |
| (C57BL/6N mice) | Control | 3 | 166.67 | |
| BGL, blood glucose level; STZ, streptozotocin; IDCK, insulin-deficient
C57BL/6-Tg(pH1-siRNA | ||||
IDCK mice were obtained from the National Institute of Food and Drug Safety
Evaluation (Cheongju, Korea). IDCK mice with blood glucose levels
C57BL/6J mice at 4–6 and 42 weeks after STZ injection and IDCK mice at 20 weeks
after birth were euthanized with isoflurane inhalation (5% in O
Retinal whole mounts were processed free-floating in small vials at room temperature with gentle agitation. With three rinses in ice-cold 0.1 M PB between each step, the retinas were processed as follows: (1) freezing and thawing three times for better antibody penetration; (2) preincubation in 1% sodium borohydride for 20–30 min; (3) incubation in 0.1 M PB with 4% normal serum from the host of secondary antibody with 0.5% Triton X-100 for 1 day; (4) incubation in the primary antiserum in blocking solution for 3 days; (5) incubation in the secondary antiserum in blocking solution for 1 day; and (6) staining of the nuclei of the retinas with DAPI (1:1000). The primary antibodies used in this study were rabbit anti-CB (1:250, Sigma-Aldrich, St. Louis, MO, USA), rabbit anti-CR (1:200, Sigma-Aldrich), rabbit anti-PV (1:250, Swant, Burgdorf, Bern, Switzerland), and goat anti-ChAT (1:200, Millipore, Burlington, MA, USA). For CB, CR, and PV, fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG (Jackson ImmunoResearch Inc., Baltimore, PA, USA) secondary antibody was used. To identify ChAT, FITC-conjugated goat anti-rabbit IgG secondary antibody (Millipore) and Cy3- conjugated donkey anti-goat IgG (Jackson ImmunoResearch Inc.) secondary antibodies were used.
The procedures for single-cell injection following immunocytochemistry have been
described in our previous reports [51, 52, 53]. Immunocytochemistry before the
single-cell injection procedure was performed without Triton X-100 because this
detergent can influence the diffusion of the dye in the neuronal membrane. The
fixed tissues were incubated in a 1:200 dilution of goat anti-ChAT (Millipore)
primary antibody in 0.1 M PB for 2 h at room temperature and then incubated in a
1:50 dilution of FITC-conjugated donkey anti-goat IgG secondary antibody (Jackson
ImmunoResearch Inc.) in 0.1 M PB for 2 h at room temperature after three rinses
with 0.1 M PB. The dish containing the immunolabeled retinal tissue was placed on
a microscope stage, and FITC-labeled cholinergic neurons were viewed under a
Zeiss 40
We counted ChAT-IR neurons manually in whole-mounted retinas from the center to
the periphery. As the density of retinal neurons changes with eccentricity, we
carefully chose counting areas by positioning at the same retinal regions between
control and experimental mice. We counted cells in fourteen sample areas at 300
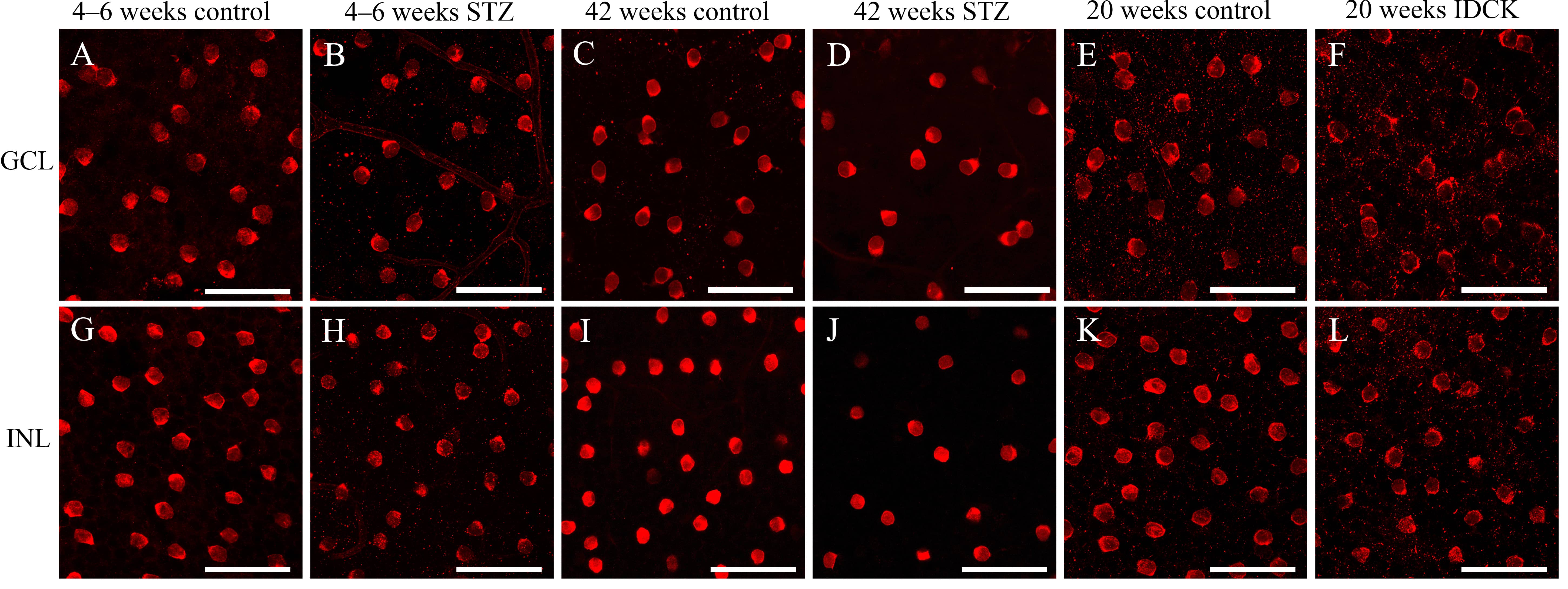
The typical distribution of ChAT-IR neurons in the mouse retina has been described in our previous study [19]. The density of ChAT-IR neurons was reduced in the GCL and INL in STZ-induced diabetic mice at 4–6 and 42 weeks and in IDCK mice at 20 weeks compared with that in control mice (Figs. 1,2, Table 2, Supplementary Tables 1–3). Compared with the findings in control mice, the number of cells in the experimental group decreased by 8.34% at 4–6 weeks (8.15% in GCL, 8.48% in INL) and by 14.89% at 42 weeks (15.73% in GCL, 14.14% in INL). Meanwhile, the cell count was 16.80% lower in IDCK mice at 20 weeks (21.12% in GCL, 13.14% in INL) than in control mice. Fig. 1 shows the representative areas in GCL and INL in control and experimental mice at approximately same retinal regions. The densities of ChAT-IR neurons in GCL and INL were lower in experimental mice than in control mice. Decrease of ChAT-IR neurons is more significant in GCL and INL in STZ-induced diabetic mice at 42 weeks than at 4–6 weeks (Figs. 1,2).
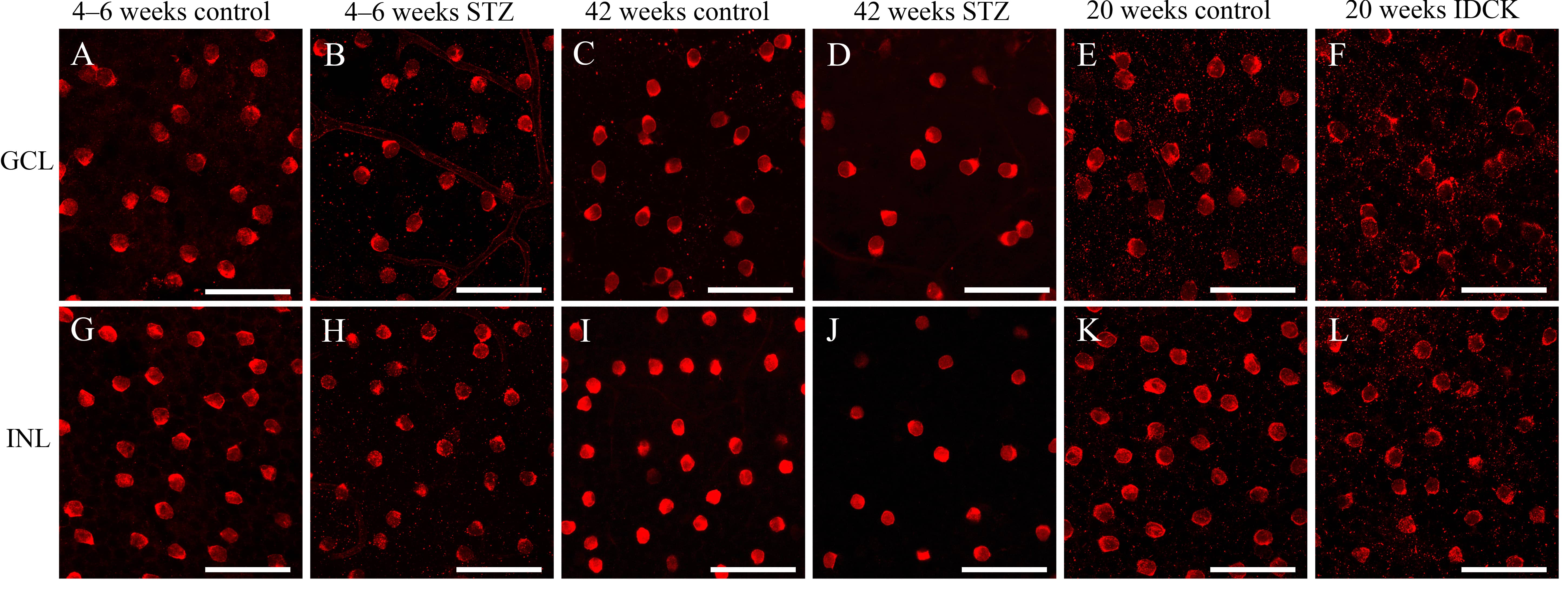 Fig. 1.
Fig. 1.High-power photomicrographs of the ChAT-IR neuron distribution
in the mouse retina. (A–F) ChAT-IR neurons in the GCL of the mouse retina.
(G–L) ChAT-IR neurons in the INL of the mouse retina. (A,G) ChAT-IR neurons in
mice in the control group at 4–6 weeks after STZ injection. (B,H) ChAT-IR
neurons in mice in the experimental group at 4–6 weeks after STZ injection.
(C,I) ChAT-IR neurons in mice in the control group at 42 weeks after STZ
injection. (D,J) ChAT-IR neurons in mice in the experimental group at 42 weeks
after STZ injection. (E,K) ChAT-IR neurons in 20-week-old C57BL/6N mice in the
control group. (F,L) ChAT-IR neurons in 20-week-old IDCK mice in the experimental
group. The density of ChAT-IR neurons was reduced in all experimental groups
compared with the control groups. STZ, streptozotocin; IDCK, insulin-deficient
C57BL/6-Tg(pH1-siRNA
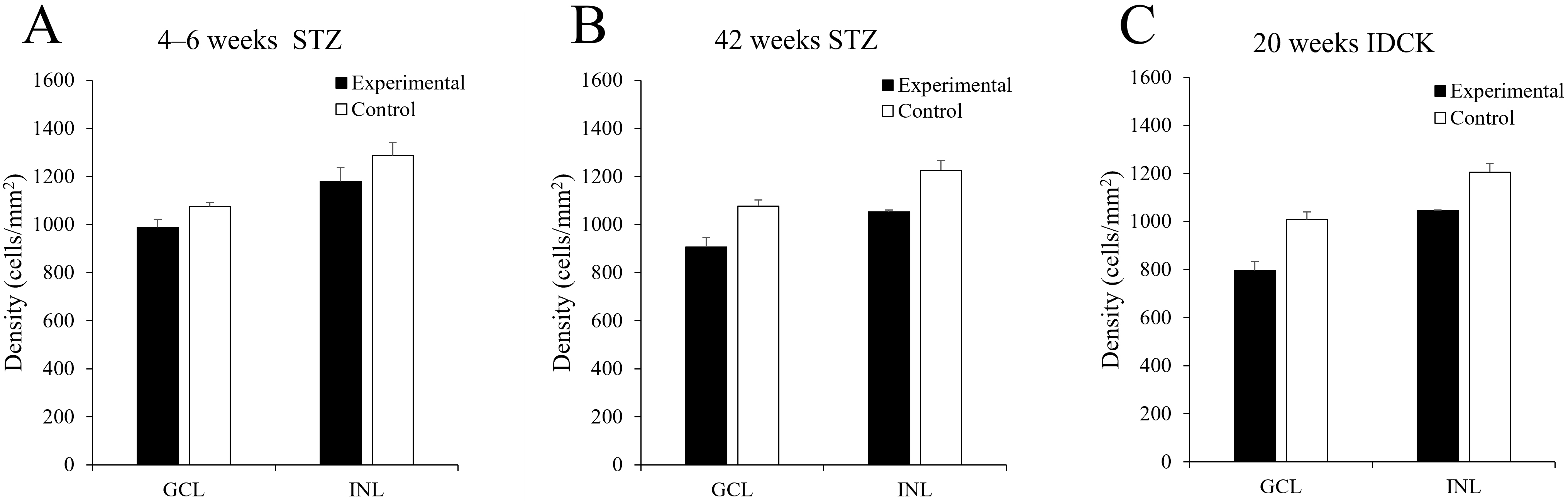
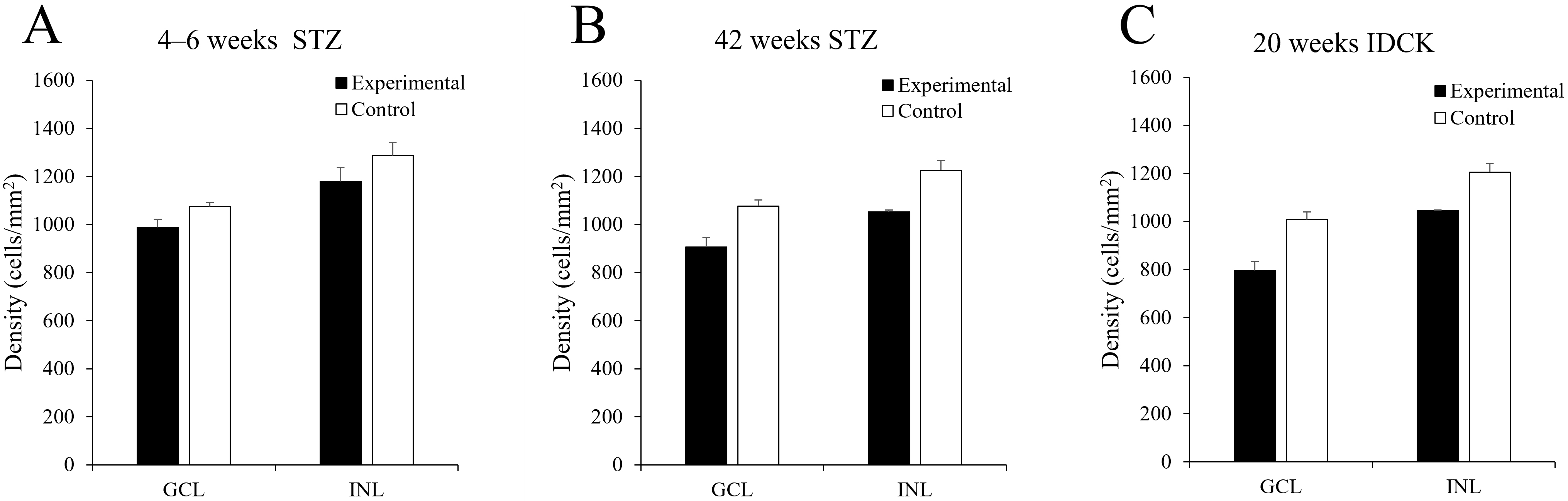 Fig. 2.
Fig. 2.Histogram showing mean density of ChAT-IR neurons in the mouse
retina. (A) At 4–6 weeks after STZ injection, ChAT-IR neuron counts decreased
by 8.15% (GCL) and 8.48% (INL) in the experimental group compared with the
control group. (B) At 42 weeks after STZ injection, ChAT-IR neuron counts
decreased by 15.73% (GCL) and 14.14% (INL) in the experimental group compared
with the control group. (C) In 20-week-old IDCK mouse retinas, ChAT-IR neuron
counts decreased by 21.12% (GCL) and 13.14% (INL) compared with the control
group. Data are presented as the mean
| Group | Retina | No. of ChAT-IR neurons counted (GCL) | No. of ChAT-IR neurons counted (INL) | No. of ChAT-IR neurons counted (total) | ||||||
| Experimental | Control | % Reduction | Experimental | Control | % Reduction | Experimental | Control | % Reduction | ||
| 4–6 weeks STZ | Retina #1 | 575 | 611 | 5.89 | 692 | 750 | 7.73 | 1267 | 1361 | 6.91 |
| Retina #2 | 545 | 594 | 8.25 | 662 | 723 | 8.44 | 1207 | 1317 | 8.35 | |
| Retina #3 | 540 | 602 | 10.30 | 626 | 690 | 9.28 | 1166 | 1292 | 9.75 | |
| Mean |
553 |
644 |
8.15 |
660 |
721 |
8.48 |
1213 |
1323 |
8.34 | |
| 42 weeks STZ | Retina #1 | 486 | 586 | 17.06 | 584 | 669 | 12.71 | 1070 | 1255 | 14.74 |
| Retina #2 | 531 | 615 | 13.66 | 594 | 712 | 16.57 | 1125 | 1327 | 15.22 | |
| Retina #3 | 507 | 607 | 16.47 | 589 | 678 | 13.13 | 1096 | 1285 | 14.71 | |
| Mean |
508 |
603 |
15.73 |
589 |
686 |
14.14 |
1097 |
1289 |
14.89 | |
| 20 weeks IDCK | Retina #1 | 432 | 544 | 20.59 | 585 | 664 | 11.90 | 1017 | 1208 | 15.81 |
| Retina #2 | 470 | 575 | 18.26 | 586 | 698 | 16.05 | 1056 | 1273 | 17.05 | |
| Retina #3 | 434 | 575 | 24.52 | 587 | 663 | 11.46 | 1021 | 1238 | 17.53 | |
| Mean |
445 |
565 |
21.12 |
586 |
675 |
13.14 |
1031 |
1240 |
16.80 | |
| ChAT, choline acetyltransferase; IR, immunoreactive; GCL, ganglion cell layer;
INL, inner nuclear layer; STZ, streptozotocin; SD, standard deviation; IDCK,
insulin-deficient C57BL/6-Tg(pH1-siRNA | ||||||||||
Fig. 2 shows the result of the paired student t-test in the graph.
Statistically significant differences in mean density per retina between
experimental and control groups in STZ-induced diabetic mice at 4–6 weeks and 42
weeks and in 20-week-old IDCK mice were identified (p
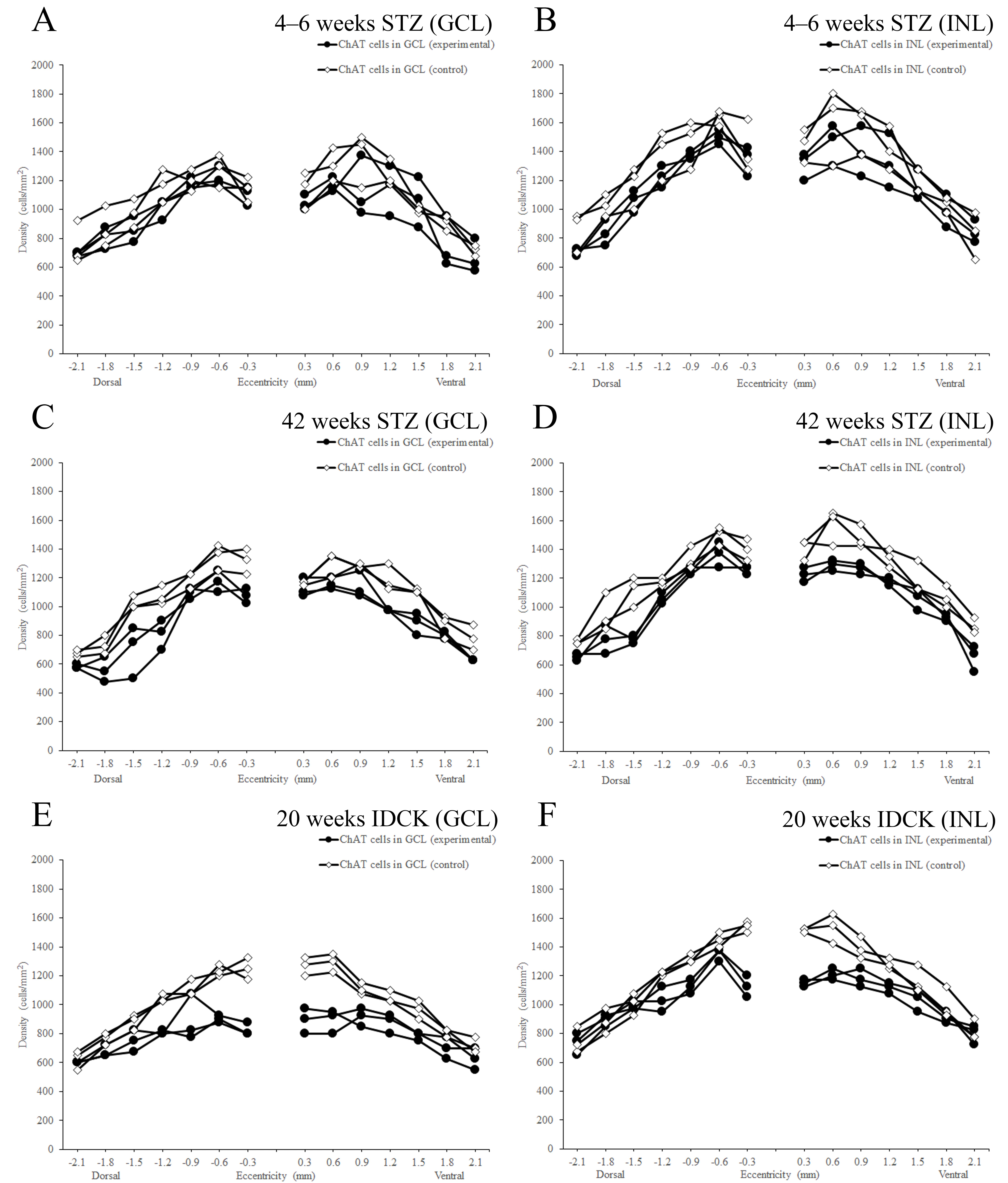
ChAT-IR neuron distributions along the dorsoventral axis of the retina in
experimental and control mice at 300
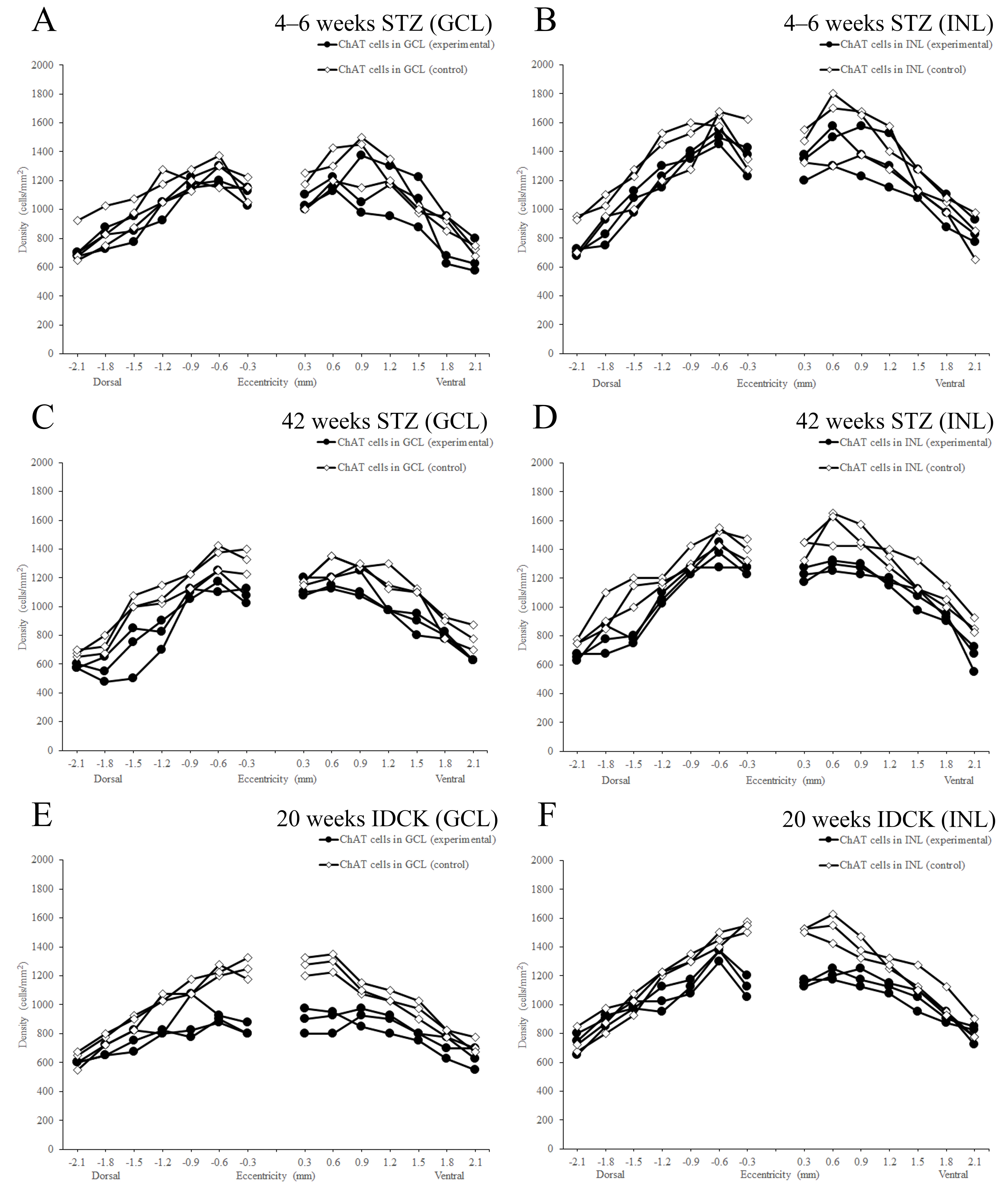 Fig. 3.
Fig. 3.Histogram of the ChAT-IR neuron distribution along the
dorsoventral axis in the mouse retina. (A) ChAT-IR neurons in the GCL in control
and experimental mice at 4–6 weeks after STZ injection. (B) ChAT-IR neurons in
the INL in control and experimental mice at 4–6 weeks after STZ injection. (C)
ChAT-IR neurons in the GCL in control and experimental mice at 42 weeks after STZ
injection. (D) ChAT-IR neurons in the INL in control and experimental mice at 42
weeks after STZ injection. (E) ChAT-IR neurons in the GCL in 20-week-old IDCK
control and experimental mice. (F) ChAT-IR neurons in the INL in 20-week-old IDCK
control and experimental mice. The line of DR mice is located below that of
normal mice on average. STZ, streptozotocin; GCL, ganglion cell layer; ChAT,
choline acetyltransferase; INL, inner nuclear layer; IDCK, insulin-deficient
C57BL/6-Tg(pH1-siRNA
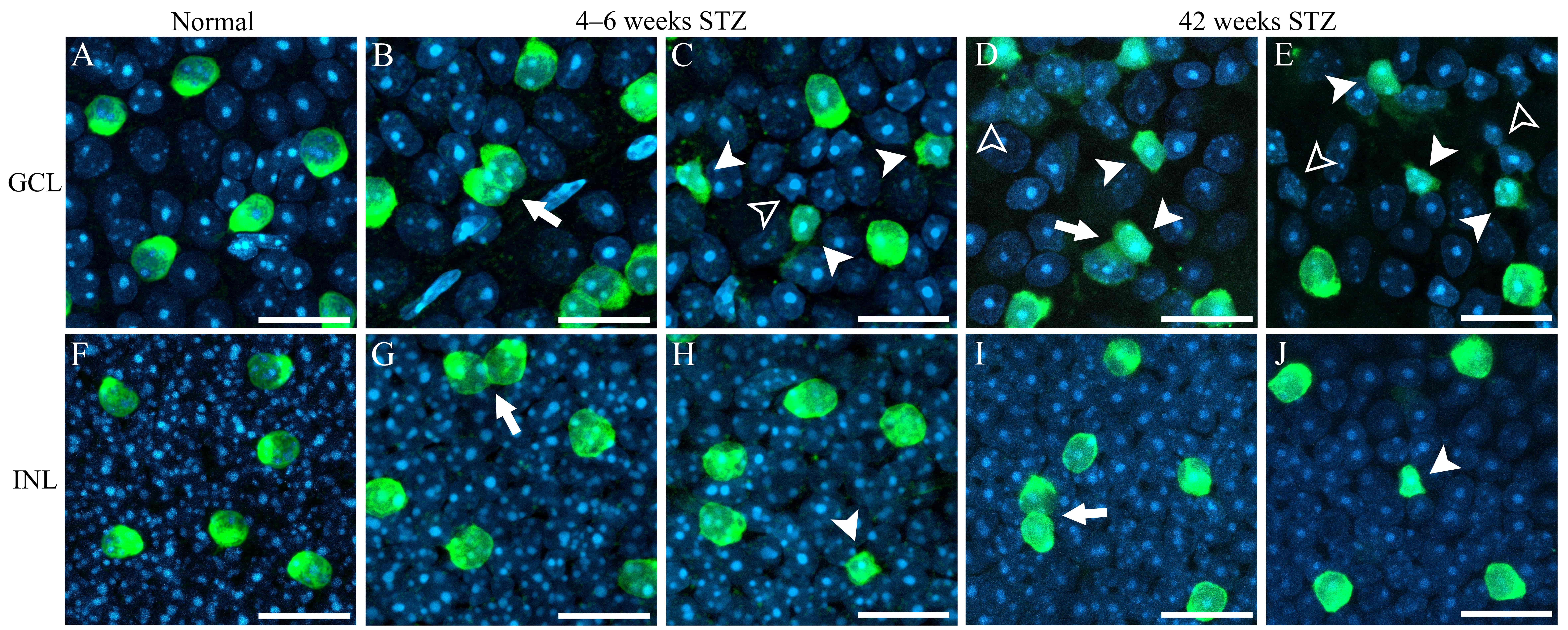
Abnormal cell aggregation and cell body deformation in the retina were observed in mice with STZ-induced diabetes (Fig. 4). Fig. 4A,F show the normal cells in GCL and INL, respectively, in C57BL/6J mouse retina. Cells with round/oval cell bodies were appeared to be arranged with adequate distances. They appeared to form independently spaced retinal mosaics. In early-stage (4–6 weeks) and late-stage DR (42 weeks), some ChAT-IR neurons in the GCL (filled arrowheads in Fig. 4C–E) and INL (filled arrowhead in Fig. 4H,J) became distorted. Some other retinal cells (empty arrowheads in Fig. 4C–E) stained with DAPI were also distorted. Abnormal ChAT-IR neuron aggregation was induced by DR in the GCL (arrows in Fig. 4B,D) and INL (arrows in Fig. 4G,I), but this phenomenon was rarely observed in normal retinas.
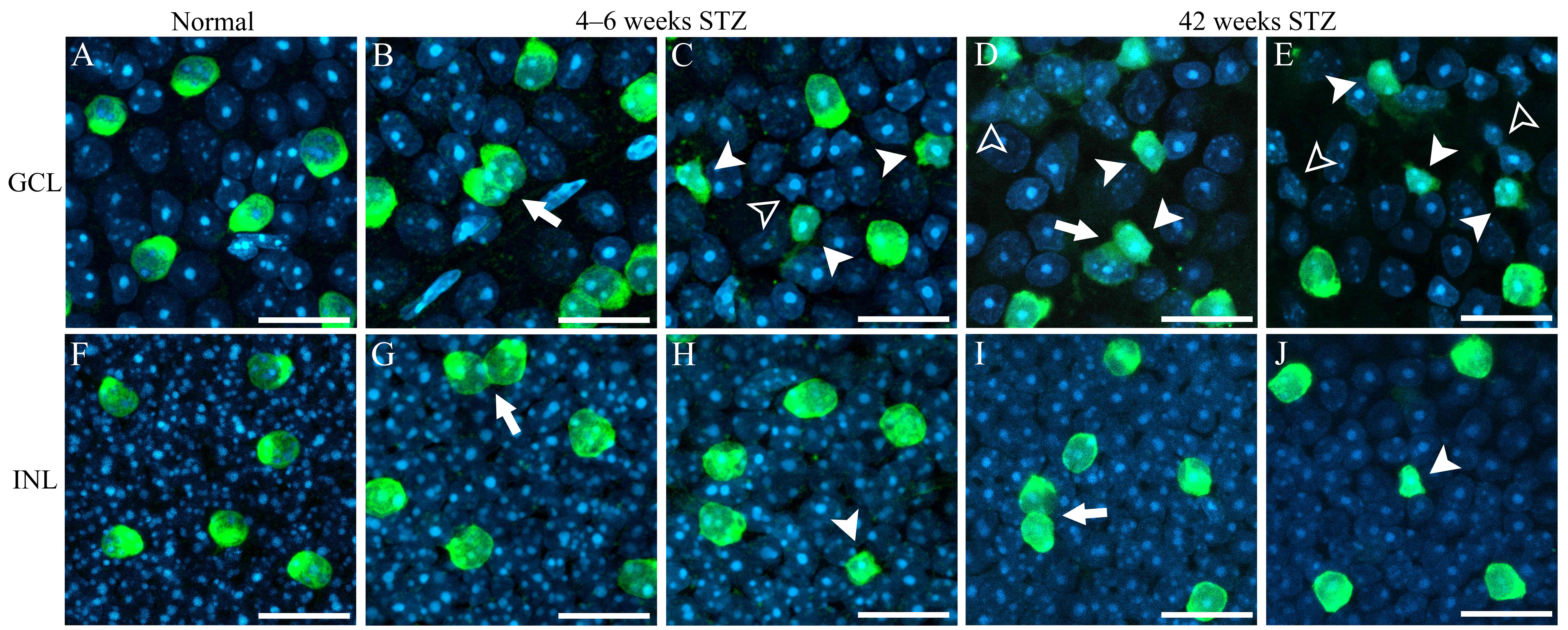 Fig. 4.
Fig. 4.High-power photomicrographs of ChAT-IR neurons. (A) Normal GCL.
(B,C) GCL in retina of mice with STZ-induced diabetes at 4–6 weeks. (D,E) GCL in
retina of mice with STZ-induced diabetes at 42 weeks. (F) Normal INL. (G,H) INL
in retina of mice with STZ-induced diabetes at 4–6 weeks. (I,J) INL in retina of
mice with STZ-induced diabetes at 42 weeks. Aggregation of ChAT-IR neurons caused
by DR (arrows in panels B, D, G, and I). Distorted cell bodies of ChAT-IR neurons
(filled arrowheads in panels C, D, E, H, and J). Some other retinal neurons
became distorted (empty arrowheads in panels C, D, and E). STZ, streptozotocin;
GCL, ganglion cell layer; INL, inner nuclear layer; ChAT, choline
acetyltransferase; IR, immunoreactive; DR, diabetic retinopathy. Scale bar = 20
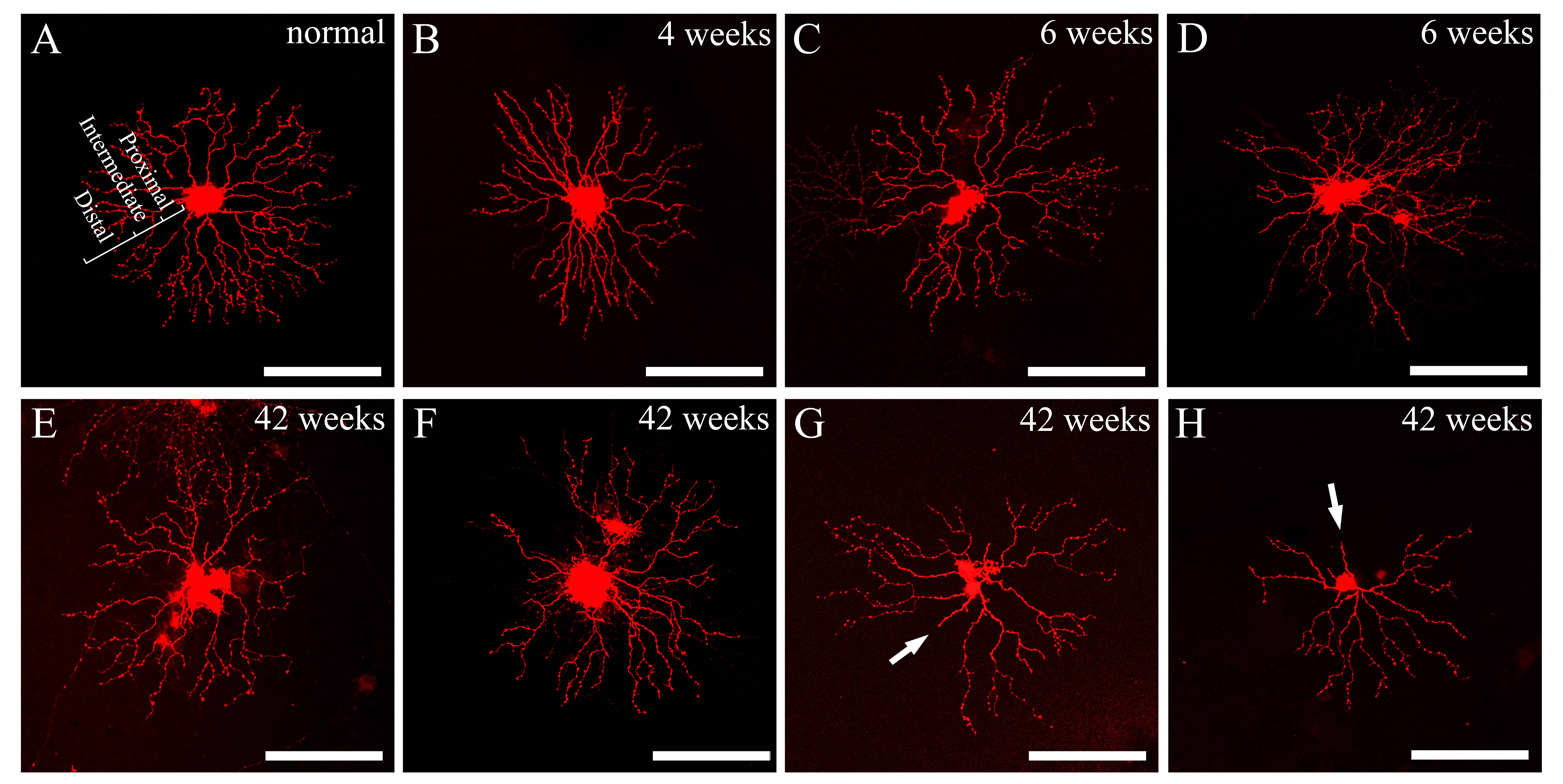
Fig. 5 presents the dendritic morphological differences between normal (Fig. 5A) and damaged (Fig. 5B–H) ChAT-IR neurons from the retinas of control and experimental mice. The distinctive dendritic morphology of SACs has been described in many previous studies [54, 55, 56, 57]. SACs are known to possess four or five primary dendrites that extend radially from the cell body, and dendritic arbors with many branches generally form circular dendritic fields. The dendritic arbors are concentrically and regularly distributed around the soma, and there is almost no overlap of dendritic arbors. The dendritic field has been categorized into three distinct annular zones, namely proximal, intermediate, and distal, which progress radially outward from the soma. The dendrites of the proximal and intermediate areas are relatively thin, and they taper with progression radially outward. The distal area is characterized by terminal varicosities and boutons [55]. Compared with the dendrites of normal ChAT-IR neurons, those of damaged ChAT-IR neurons in DR start to lose their regular pattern. Fig. 5B,C show the dendritic branches that lost their dendrites at 4 and 6 weeks. Fig. 5D reveals that some of the dendritic fields were clearly altered, and the dendritic branches were entangled and overlapped in space at 6 weeks. Fig. 5E,F reveal significant reductions in dendritic branches in ChAT-IR neurons at 42 weeks. The dendritic arbors varied considerably, and the deprivation of dendritic branches in the distal and intermediate areas was more obvious. Fig. 5G,H show extensively reduced dendritic branches at 42 weeks. Because of the prominent loss of terminal varicosities and boutons and the intermediate area, only the proximal zone of dendrites was observed in some cells (arrows in Fig. 5G,H).
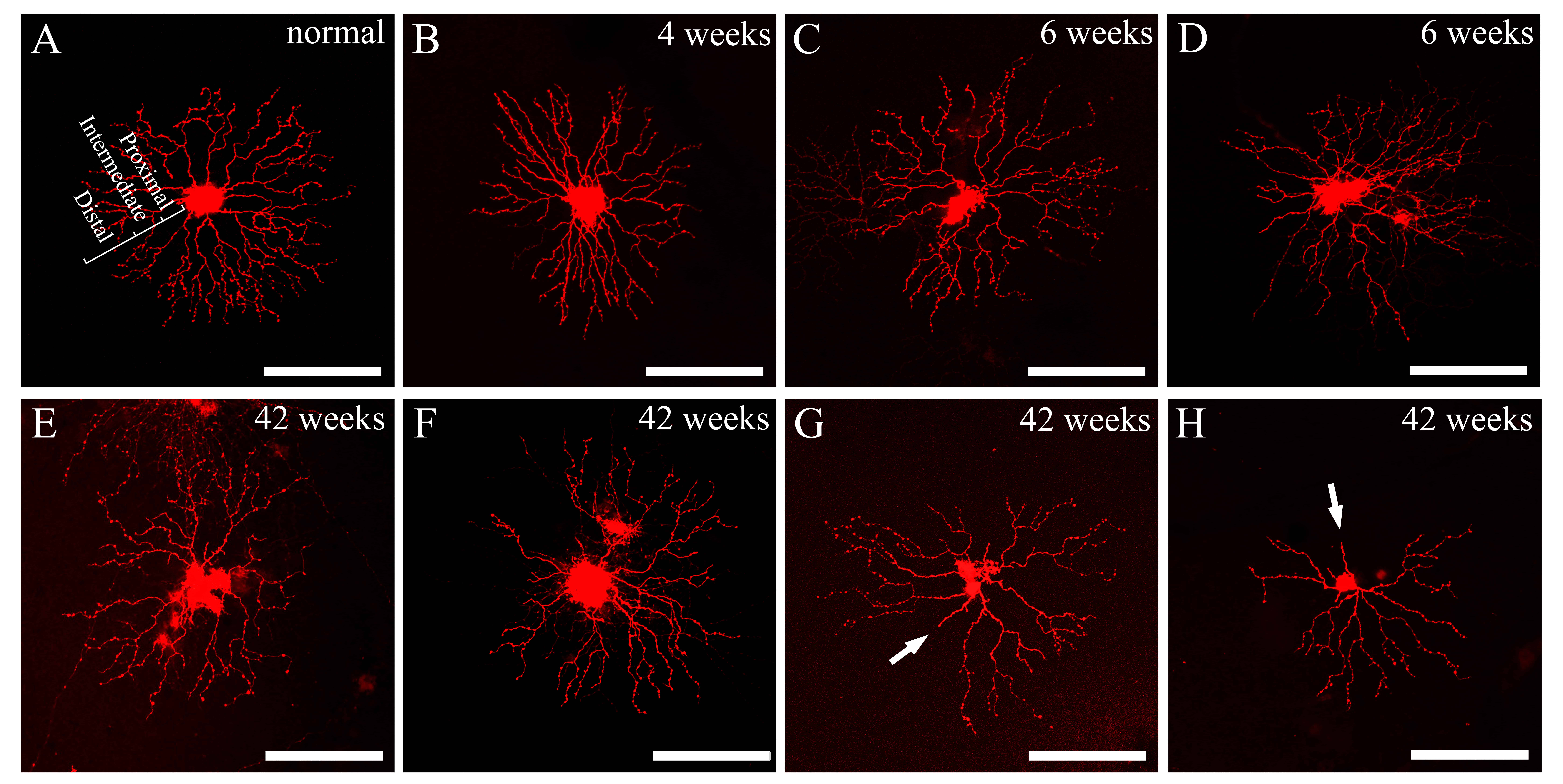 Fig. 5.
Fig. 5.Fluorescence confocal photomicrographs of DiI-injected ChAT-IR
neurons. (A) Normal ChAT-IR neurons in the mouse retina. The dendritic field has
been categorized three distinct annular zones: proximal, intermediate, and
distal. (B) Damaged ChAT-IR neurons in STZ-induced diabetic mice at 4 weeks.
(C,D) Damaged ChAT-IR neurons in STZ-induced diabetic mice at 6 weeks. (E–H)
Damaged ChAT-IR neurons in STZ-induced diabetic mice at 42 weeks. Arrows indicate
proximal area after the dendritic loss. ChAT, choline acetyltransferase; IR,
immunoreactive; STZ, streptozotocin. Scale bar = 50
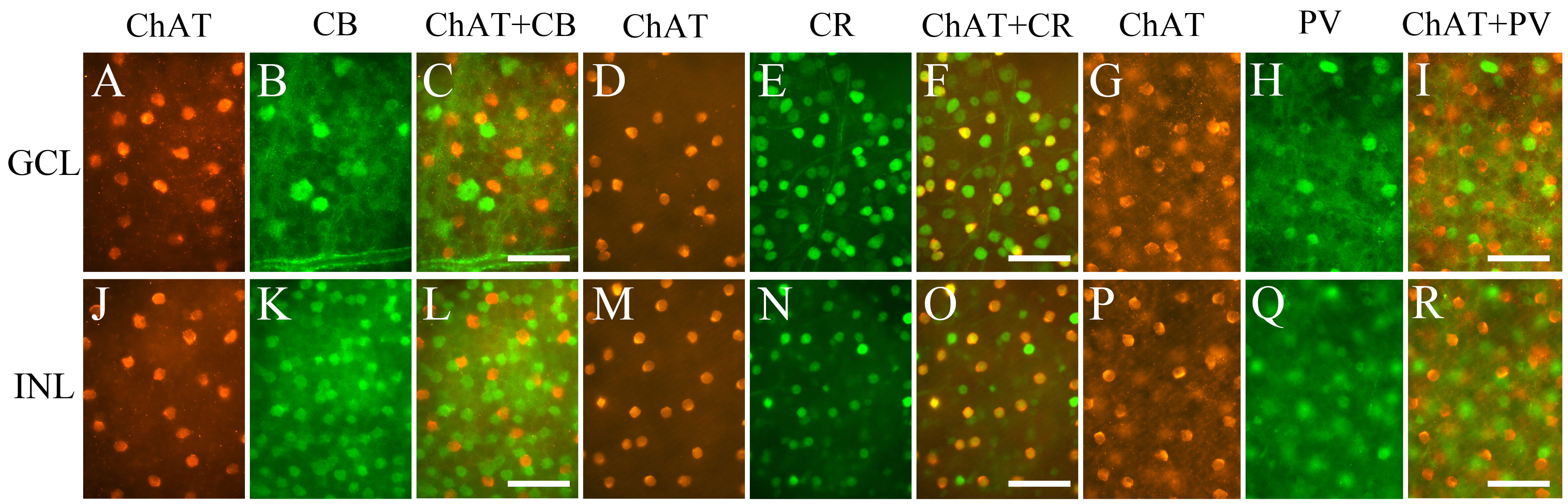
We double-labeled ChAT with CB, CR, or PV in the mouse retina (Fig. 6). Fig. 6 shows cells expressing ChAT (Cy3), CB (FITC), CR (FITC), and PV (FITC) in both in GCL (Fig. 6A–I) and in INL (Fig. 6J–R). To confirm colocalization, we superimposed the images which were photographed at same location and same focal plane. All ChAT-IR neurons both in GCL (Fig. 6F) and INL (Fig. 6O) contained CR. No ChAT-IR neurons were colocalized with CB (Fig. 6C,L) or with PV (Fig. 6I,R).
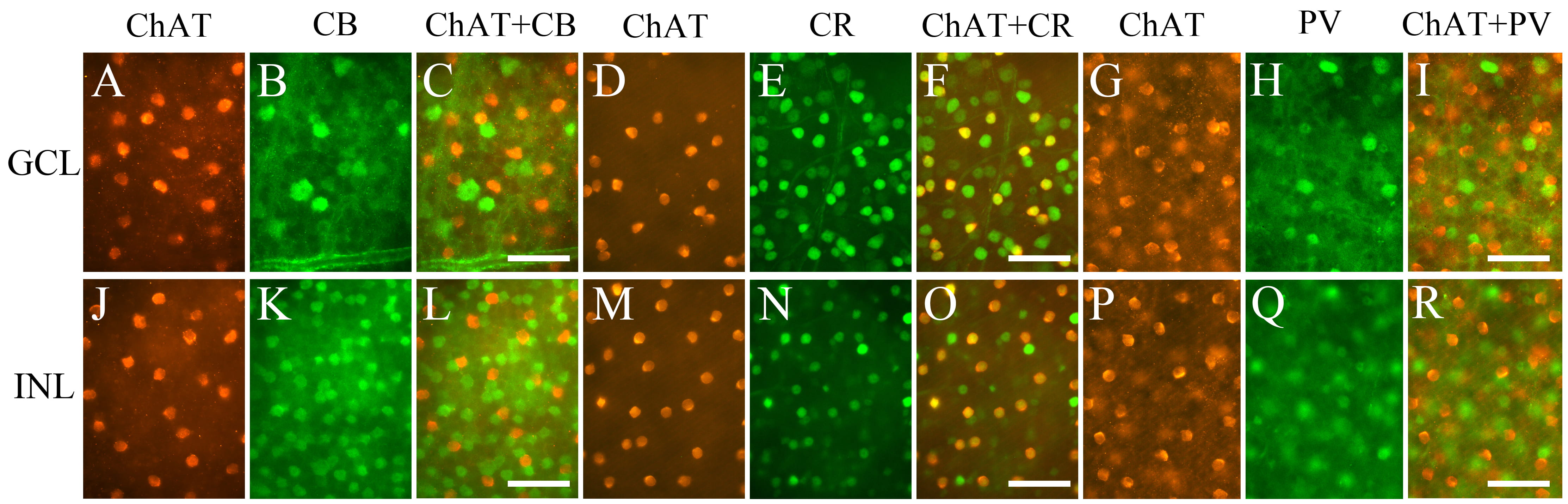 Fig. 6.
Fig. 6.Fluorescence photomicrographs of retinal whole mounts
immunolabeled for ChAT and CBPs. Images of ChAT (A,D,G,J,M,P), CB
(B,K), CR (E,N), and PV (H,Q). Superimposition of the images of ChAT with those
of CB, CR, and PV (C,F,I,L,O,R). All ChAT-IR neurons expressed CR but not CB and
PV. Panels A and B, D and E, G and H, J and K, M and N, and P and Q were
photographed on the same focal plane. ChAT, choline acetyltransferase; CB,
calbindin-D28K; CBP, calcium-binding protein; CR, calretinin; PV, parvalbumin;
GCL, ganglion cell layer; INL, inner nuclear layer. Scale bar = 50
In this study, we observed changes in the density of ChAT-IR neurons in the retinas of mice with STZ-induced diabetes and IDCK mice. We also detected abnormal cell aggregation, cell body deformation, and morphological changes in dendrites caused by DR. All ChAT-IR neurons expressed CR but not CB and PV.
Amacrine cells represent the most diverse neuronal type in the retina, and
approximately 30–60 different types of amacrine cells have been reported in the
retina [32, 58]. Of these cell types, SACs are one of the most intensively
studied interneurons in the retina [59, 60]. SACs can be categorized into two
subpopulations based on their location (INL or GCL). In the present study,
ChAT-IR neurons in retina of mouse with STZ-induced diabetes decreased total
8.34% at 4–6 weeks, and total 14.89% at 42 weeks, respectively. ChAT-IR
neurons decreased total 16.80% in retina of 20-week-old IDCK mouse compared to
age-matched control mouse retina. Decreased ChAT-IR neuron counts were detected
in the early and late phases of DR in STZ-induced diabetic mice and in IDCK mice.
In addition, the loss of these cells was greater in the late phase than in the
early phase. Furthermore, the loss of ChAT-IR neurons in STZ-induced diabetic
mice was slightly higher in the GCL than in the INL at 42 weeks, whereas almost
no difference was detected at 4–6 weeks. The loss of ChAT-IR neurons in IDCK
mice was also greater in the GCL in the present study. However, the loss of
ChAT-IR neurons in 24-week-old Ins2
Previous studies have noted retinal cell loss, decreased retinal thickness, and
increased apoptosis in mice with STZ-induced diabetes [18]. Increased apoptosis
has been demonstrated with TUNEL and caspase-3 staining in the retinas of
STZ-induced diabetic rats, Ins2
Abnormal aggregation and distorted cell bodies were found in ChAT-IR neurons in DR in the present study. These changes have been reported in CB-, CR-, and PV-IR neurons in the retinas of ischemic rabbits [67]. Abnormal aggregation has also been detected in some CR-IR neurons of the entorhinal cortex in Alzheimer’s disease [68]. Hence, abnormal cell aggregation might represent a symptom of neurodegenerative diseases. In the present study, the length of dendrites and the number of branch points in the dendrites of ChAT-IR neurons were decreased in DR. In accordance with the present findings in the retina, the dendritic branching and spine density of neurons have been reported to be reduced in the parietal cortex of rats with diabetes [69]. The number of branch points and the total dendritic length of hippocampal CA3 pyramidal neurons were reduced in diabetic rats [70]. The total dendritic length and spine density of the pyramidal neurons of the prefrontal cortex, occipital cortex, and hippocampus were also lower in rats with diabetes [71]. Therefore, the pathological condition of DR might affect the alterations in somatic and dendritic structural elements. The reduced number of branches indicates lowered connectivity, suggesting reduced activity in the neurons.
In the present study, although abnormal aggregation, distorted cell bodies, dendritic branch loss, and SAC death were apparent in DR progression, the mechanism underlying these changes is yet to be determined. Previous studies have shown that amacrine cells die via apoptosis in DR [49]. Cell shrinkage and pyknosis are characteristic features of apoptosis and change of cytoskeleton occurs during apoptosis [72]. Thus, the morphological changes and death of SACs in the present study may be due to apoptosis. There is also a possibility that the morphological changes of SACs are due to losses of structural support of other retinal cell types. In DR, various other amacrine cell types, including dopaminergic and AII amacrine cells, and supporting glial cells were lost [2]. However, more studies are necessary to fully understand the mechanisms underlying changes and death of SACs in DR in the present study.
SACs in the retina secrete both GABA and acetylcholine. In many brain areas of
mammalian species, GABAergic interneurons can be classified into subpopulations
based on their expression of certain CBPs, such as CB, CR, and PV [73, 74, 75]. In the
present study, all ChAT-IR neurons were expressed only in CR. Recently,
transcriptomic analysis using high-throughput single-cell RNA sequencing has
profiled 63 types of amacrine cells in mouse retina including SAC. They assessed
the expression of ChAT including some other molecular markers in SAC but have not
assessed the expression of CR to characterize the SAC [32]. It will be necessary
to observe whether the CR expression in SAC agrees with single-cell genomics
transcription analysis in the future. CBP expression in ChAT-IR neurons greatly
differed among species [35, 44, 45, 46, 47, 48, 76, 77]. In some animals, ChAT-IR neurons
expressed only one of the CB [35], CR [47, 77], or PV [48], whereas ChAT-IR
neurons expressed two or three of CB, CR, or PV in the other animals [45, 76].
The reason for various expression profiles among animals is unclear. Members of
EF-hand family has different structure within their EF-hand motifs. And this
structural variety provides different Ca
Although its function is unknown, CR is a major CBP in the central visual system and has been used to label discrete neuron populations with distinctive morphology and electrophysiology in mice [52, 82, 83]. It has been suggested that CR is important in calcium buffering and transport similar to other CBPs, e.g., CB and PV [40, 41, 84, 85]. CR is involved in sharpening the timing of action potentials and is associated with many biological processes, such as cell proliferation, differentiation, and cell death [85, 86]. A recent study reported that CR may play a significant role in promoting synaptic efficacy during high-rate activity [87]. As SACs are important bridging cells along with direction-selective RGCs [88, 89], it will be interesting to examine if CR is related to sharpening action potential timings and promoting synaptic efficacy in motion pathways.
The functional aspects of SACs in the DR have not been thoroughly investigated. Previous studies have observed that ChAT-IR neurons constituted approximately 3% of all amacrine cells in the INL and approximately 20% of all amacrine cells in the GCL, comprising one of the largest populations of amacrine cells in the retina [36, 37, 59, 90]. This finding implies that SACs have key roles in retinal function. In DR, the loss of SACs may cause the decrease in the optokinetic response [91]. In particular, SACs have been extensively studied as the key elements of the mechanism underlying direction selectivity, an essential neural computation that occurs in the retina for detecting a moving object [34, 92, 93]. Direction selectivity involves anatomically symmetric cholinergic and asymmetric GABAergic synaptic connectivity from SACs [34, 87]. Thus, the loss of SACs and deprivation of dendritic branches caused by DR can interfere with direction-selective circuits in the retina. In the central visual system, diabetes disrupts functional connectivity between the primary visual cortex and higher visual regions [94]. Furthermore, diabetes causes brain atrophy, which results in a lower gray matter volume in the occipital lobe [95, 96, 97]. Therefore, these data indicate that diabetes disrupts the accuracy of visual function. However, more detailed studies are needed for understanding the impact of diabetes on vision in the retina and other visual areas.
Retinal cell death seems to be an evident clue in the progression of DR. Our results showed that the number of SACs, both in the GCL and INL, in DR mouse decreased in a time-dependent manner. Abnormal aggregation and distorted cell bodies were also found in SACs in the present study. Moreover, some of the dendritic arbors varied considerably revealing reduced and entangled dendritic branches. SACs in mouse retina expressed CR but not CB and PV. The findings of the present study are expected to contribute to a better understanding of changes of SACs in DR and may guide the development of diagnostic and therapeutic strategies for DR in the future.
BGL, blood glucose level; CB, calbindin-D28K; CBP, calcium-binding protein;
ChAT, choline acetyltransferase; CR, calretinin; DR, diabetic retinopathy; FITC,
fluorescein isothiocyanate; GABA, gamma-aminobutyric acid; GCL, ganglion cell
layer; IDCK, insulin-deficient C57BL/6-Tg(pH1-siRNA
All data generated or analyzed during this study are included in this published article.
JRS and CJJ designed the research study. JRS and MJL performed the research. JRS analyzed the data. JRS, MJL, and CJJ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
All experiments involving animals were approved by the Animal Care and Use Committee of Kyungpook National University (permission no. 2020-0158).
We thank National Institute of Food and Drug Safety Evaluation for providing
C57BL/6-Tg(pH1-siRNA
The work has been supported by National Research Foundation of Korea (NRF), funded by Ministry of Education (NRF-2020R1F1A1069293).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.