




1 College of Osteopathic Medicine of the Pacific, Western University of Health Sciences, Pomona, CA 91766, USA
2 Department of Emergency Medicine, Corona Regional Medical Center, Corona, CA 92882, USA
3 Department of Emergency Medicine, St. Barnabas Hospital Health System, Bronx, NY 10457, USA
4 Your Energy System, Palo Alto, CA 94301, USA
Abstract
Immunothrombosis has emerged as a dominant pathological process exacerbating morbidity and mortality in acute- and long-COVID-19 infections. The hypercoagulable state is due in part to immune system dysregulation, inflammation and endothelial cell damage, as well as a reduction in defense systems. One defense mechanism in particular is glutathione (GSH), a ubiquitously found antioxidant. Evidence suggests that reduction in GSH increases viral replication, pro-inflammatory cytokine release, and thrombosis, as well as decreases macrophage-mediated fibrin removal. The collection of adverse effects as a result of GSH depletion in states like COVID-19 suggest that GSH depletion is a dominant mechanism of immunothrombosis cascade. We aim to review the current literature on the influence of GSH on COVID-19 immunothrombosis pathogenesis, as well as the beneficial effects of GSH as a novel therapeutic for acute- and long-COVID-19.
Keywords
- glutathione
- GSH
- COVID-19
- SARS-CoV-2
- immunothrombosis
- HIV
- diabetes
- microclot
- thrombosis
The COVID-19 pandemic has claimed roughly 6.7 million lives as of January 2023 per the World Health Organization. SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus that spreads through direct, indirect, or close contact with infected hosts by respiratory droplets. The initial entry of the virus into human cells was found to be via the SARS-CoV-2 spike (S) protein binding to the cell surface receptor angiotensin-converting-enzyme-2 (ACE2) expressed by type II pneumocytes [1]. Binding and entry leads to a cascade of steps resulting in inflammation and ultimately systemic disease [2].
While the disease severity can range from mild to severe, commonly reported symptoms include loss of smell, fever, fatigue, and dizziness. Severely affected patients exhibit acute respiratory distress syndrome (ARDS) leading to oxygen deficiency [3]. Researchers have identified one key pathologic component associated with infection severity and course, termed immunothrombrosis. Immunothrombosis describes the interconnectivity between the body’s natural coagulation processes and the innate immune system. The overall effect is the formation of thrombi within small and large blood vessels. In states of hyperinflammation, like COVID-19, patients are marked by a dysregulated and hyperactive innate immune system as well as a hypercoagulable state. The immunothrombosis cascade normally depends on a number of cytokines and inflammatory mediators which is intended to provide a protective role. When dysregulated, the process can lead to the development of excessive cytokine release, termed cytokine storm [4]. After observing the cardiopulmonary tissue of a subset of patients with severe COVID-19 who also developed ARDS, immunothrombosis was identified as a significant contributing factor to these patients critical conditions [5, 6]. Specifically, the presence of amyloid fibrin microclots which are resistant to fibrinolysis and found microvessels in both acute and Long-COVID-19 [6]. Immunothrombosis is dependent on platelet aggregation and mediators of platelet aggregation, one of which is the ubiquitous antioxidant glutathione in its reduced form (GSH). Studies have shown that GSH may act as an inhibitor of platelet aggregation and in many cases of COVID-19, a deficiency in GSH has been observed [7, 8].
GSH is an important antioxidant that naturally works to limit the levels of
dangerous reactive oxygen species (ROS). Decreased levels of GSH and elevated
levels of ROS have been associated with COVID-19 pathogenesis [9]. COVID-19 is
also associated with elevated levels of D-dimer, a breakdown product of fibrin
clots, and has been used as a specific biomarker to indicate severity of COVID-19
[10]. With exacerbated oxidative stress and cytokine storm, increased levels of
interleukin-6 (IL-6) and decreased levels of interferon-
The significance of these findings identify an additional pathologic component of COVID-19 and also serve to identify novel adjunctive therapies. Confirming what role GSH plays in the pathophysiology of COVID-19 and immunothrombosis can serve as a scaffold for future therapeutic research. We aim to review the role of GSH in COVID-19 pathogenesis, immunothrombosis, and immune system dysregulation.
This article is a comprehensive review investigating the role of glutathione in the COVID-19 immunothrombosis cascade. Information was obtained using PubMed and NCBI databases from December 2022 to February 2023. Search results included terms such as: “Glutathione”, “COVID-19”, “SARS-CoV-2”, “Immunothrombosis”. Search terms specific to respective subsections included “glutathione peroxidase”, “oxidized glutathione”, “interleukin 6”, “transforming growth factor beta”, “macrophage”, “D-dimer”, “cytokines”, “reactive oxygen species” and “thrombosus”. Research included in this article were selected based on quality and significance of results. Review articles included were chosen based on comprehensiveness of the topic of interest. Exclusion criteria included non-relevance, poor sample size, and inconclusive data and significance. A total of 226 articles were identified, 122 were excluded based on exclusion criteria, and 104 articles were included in qualitative synthesis.
Thrombosis is the formation of a clot inside blood vessels which leads to partial or complete vessel occlusion, preventing bleeding after vessel injury. Clots are formed when vascular damage activates tissue factor (TF), which leads to recruitment of platelets and initiation of the coagulation cascade. Finally, the clot is stabilized using fibrin and thrombin. TF is continuously delivered to the clot to participate in its growth [13]. Interestingly, immune cells such as neutrophils and monocytes can also interact with platelets and the coagulation cascade to induce clot formation in blood vessels [1]. Neutrophils release neutrophil extracellular traps (NETs) which bind to and activate multiple coagulation factors, attract platelets, and bind TF to amplify the clotting cascade [1]. Monocytes release microparticles that express intravascular TF to further activate coagulation and create microthrombi matrices which immune cells use to assist them in recognition, containment, and destruction of pathogens in a process termed immunothrombosis [14]. Research shows that intravascular TF may also be found in neutrophils, eosinophils, and platelets [14].
Hyperinflammatory states such as those seen in COVID-19 can trigger
immunothrombosis [15]. Severe COVID-19 induces extensive pulmonary inflammation,
triggering a macrophage activation syndrome (MAS)-like event with proinflammatory
cytokines, macrophage and lymphocyte recruitment, and subsequent endothelial
damage [15]. Endothelial dysfunction is associated with decreased bioavailability
of nitric oxide (NO) due to an increase in ROS, with a similar physiological
change seen in cases of decreased GSH pools [16]. The decrease in NO increases
the expression of transcription factor nuclear factor kappa B (NF-
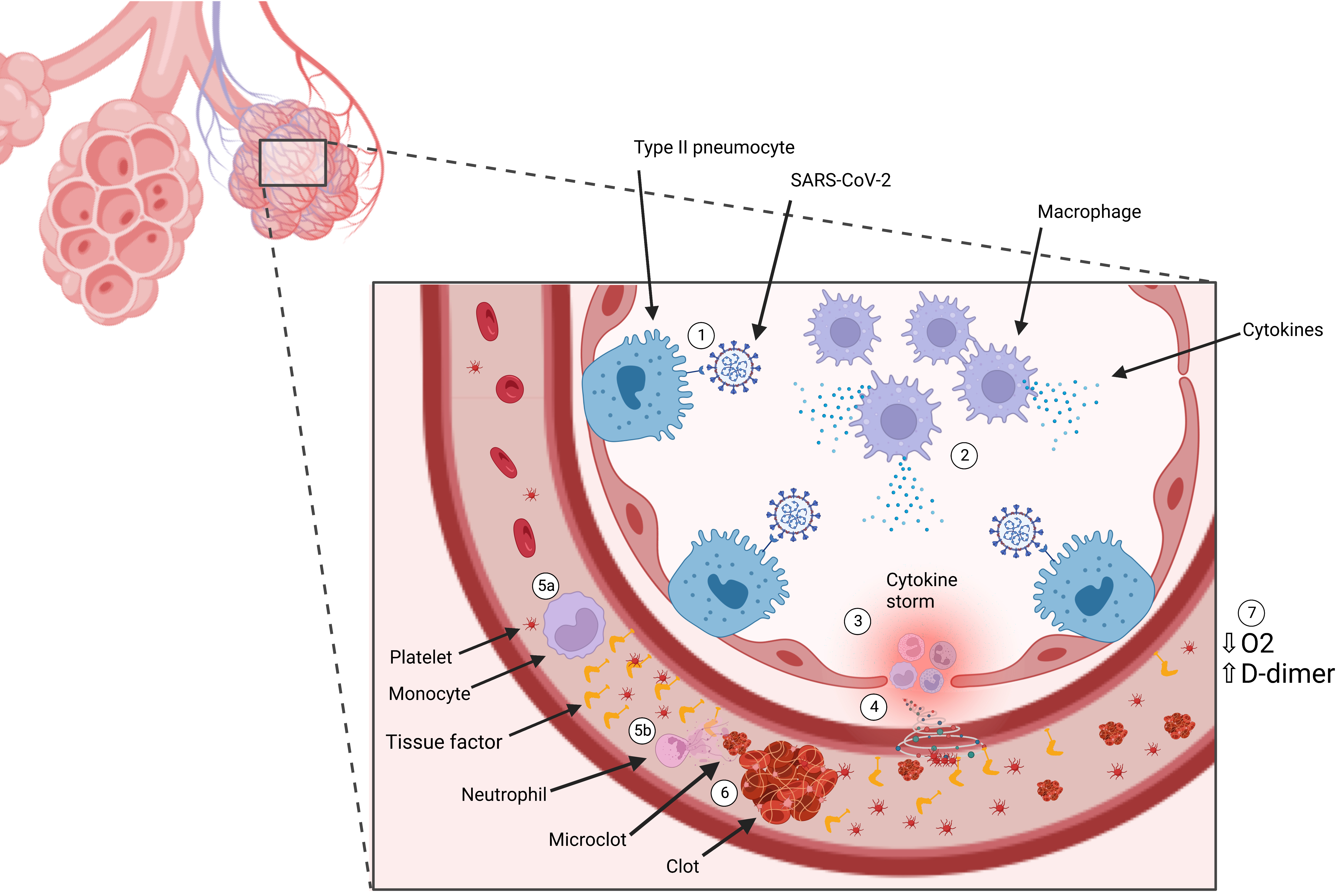 Fig. 1.
Fig. 1.Immunothrombosis induction. (1) SARS-CoV-2 binds to ACE-2 receptor on type-2 pneumocyte cells. (2) Infection with SARS-CoV-2 results in recruitment of immune cells, including macrophages, which release cytokines. (3) Cytokines recruit more immune cells which produce excess cytokines, otherwise known as a cytokine storm. (4) The cytokine storm results in endothelial inflammation and damage, releasing tissue factor (TF) into circulation. (5) Cytokines recruit (a) monocytes, which release microparticles expressing intravascular TF, and (b) neutrophils, which release neutrophil extracytoplasmic traps (NETs). (6) The combination of TF and NETs activate immunothrombosis, generating clots and microclots. (7) Clotting and impaired blood flow results in impaired oxygen delivery and elevated D-dimer levels.
Severe COVID-19 is marked by high levels of D-dimer, a fibrin byproduct that can indicate prevalence of thrombotic formation. Severe COVID-19 patients experience a 9-fold increase in the prevalence of alveolar-capillary microthrombi when compared to patients with influenza [19]. It is important to identify therapies to prevent the thromboembolic complications of COVID-19. Early reports on the use of low molecular weight heparin (LMWH) for severe COVID-19 cases with elevated D-dimer have demonstrated a reduction in mortality [20]. However, according to the American College of Chest Physicians (CHEST) guidelines, thromboprophylaxis therapy is only recommended for hospitalized patients. Unfortunately, patients remain at an increased risk for thromboembolic complications even after resolution of acute infection. Patients with new or persistent symptoms following the acute phase of COVID-19 should be followed to rule out thromboinflammatory disease [21]. A systematic literature review conducted by Overton et al. [21] revealed that there is little global standardization of thromboembolism reporting and a low rate of acute COVID-19 vasculopathy detection, suggesting that current diagnostic methods for identifying pulmonary vascular disease may not be sufficient. Further development of tools to detect abnormalities in pulmonary vasculature and differentiate between thromboinflammatory disease from thromboembolic disease is needed [21]. Development of therapeutics to prevent the hyper-thromboinflammatory state seen in COVID-19 is also of high value.
Glutathione (GSH) is a low molecular weight antioxidant present in nearly all cells that functions to limit the impact of oxidative stress on vital cellular components such as lipids, proteins, and DNA through redox reactions [22]. GSH can be found in the mitochondria, nucleus, and cytosol of cells, as well as plasma and extracellular spaces like the fluid which lines pulmonary alveoli [22]. GSH is increased when the body is exposed to oxidants and electrophiles and, when decreased, yields a vulnerable state for some diseases [22]. There is an association between chronic pulmonary inflammation and low GSH levels, exemplified in cystic fibrosis patients who produce less GSH and smokers who have been found to have lower levels of GSH [22].
Glutathione has been found to be directly related to thrombosis and thrombotic events through many mechanisms, generally related to platelet attenuation and activity on the coagulation cascade [23, 24]. GSH is utilized by glutathione peroxidase (GPX) to reduce free hydrogen peroxide and lipid hyperoxides. Jin et al. [25] found that knockout of plasma GPX, GPX-3, resulted in increased platelet-dependent thrombosis in murine models. A deficiency in GPX-3 reduces metabolism of ROS, partly due to a reduction in NO which results in increased platelet adhesion and aggregation, as mentioned earlier [25]. The antioxidant properties of GPX-3 are also thought to protect against post-translational modifications of fibrinogen by ROS and NO-derived oxidants resulting in increased thrombogenicity [26]. Furthermore, Dayal et al. [27] found that overexpression of the most abundant GPX, GPX-1, served a protective role from platelet hyperactivity and age-dependent increased susceptibility to venous thrombosis after inferior vena cava ligation in murine models.
Thomas et al. [28] evaluated the relationship between GSH in
unstimulated platelets from diabetic versus control subjects, finding that
diabetic subjects have lower GSH levels in their platelets than control subjects.
They also measured the amount of thromboxane A2 (TXA2) produced by activated
platelets in diabetic versus control subjects, finding that diabetic subjects
produce more TXA2 than control subjects. TXA2 stimulates the activation of new
platelets, increases platelet aggregation, and is a vasoconstrictor. In states of
increased oxidative stress, there is increased oxidized GSH (GSSG) [29]. Essex
et al. [30] have found that decreased GSH or a mixture of GSSG and GSH
can increase platelet aggregation. Furthermore, they found that GSSG alone is
able to increase platelet aggregation [30]. They suggested an agonist-induced
mechanism whereby the addition of GSSG to platelets generated sulfhydryls in the
Recently, Wang et al. [31] discovered another connection between GSH
and platelet aggregation. Protein disulfide isomerase (PDI) is an endoplasmic
reticulum (ER)-resident oxioreductase found in platelets that is critical for
platelet aggregation. PDI is oxidized by ER oxidoreductin-1
Pacchiarini et al. [7] evaluated the effect of GSH on platelet functions. They found GSH concentrations of 3 mM or 10 mM were able to modify platelet aggregation, TxB2 production, and PDGF release by platelets. Of these three parameters, TxB2 production was significantly reduced at both doses, PDGF release was significantly reduced at 10 mM, and collagen-induced platelet aggregation was not significantly reduced [7]. Thomas et al. [32] found that GSH is able to inhibit human platelet aggregation induced by adenosine diphosphate (ADP), collagen, and arachidonic acid. Together, these findings suggest that GSH may serve to inhibit platelet activation when administered exogenously. Furthermore, activated platelets produce ROS through NADPH oxidase (NOX) signaling and induction of mitochondrial dysfunction. This sets up a cycle of repeated ROS production, platelet activation, adhesion, and recruitment which contributes to the prothrombotic risk seen in inflammatory conditions like COVID-19 [33].
Studies on patients that suffered both atherosclerotic and cardioembolic stroke found that low plasma levels of GSH could be an independent risk factor for stroke severity. During acute ischemia following a stroke, tissue recovery is based on antithrombotic activity of the body, as well as resistance to oxidative stress caused by the ischemia. GSH is a low molecular weight aminothiol, protecting other cellular thiols from oxidative damage, and helping reduce reactive oxygen species the cell is exposed to. In addition to this it is heavily involved in platelet attenuation [23].
In experimental stroke models, platelet aggregometry and thromboelastography (ROTEM) demonstrated activity of vWF that cross links platelets in arterial thrombi, the end product of the coagulation cascade that promotes clot formation [34]. Diabetic patients are known to have a multifactorial hypercoagulable state related to increased inflammatory markers, and increased platelet aggregation (and indeed, an increased risk for downstream clinical effects such as stroke, heart attack, and other thrombotic events). GSH levels have not only been found to be low in these patients, but replacement with N-acetylcysteine (NAC), a synthetic precursor for GSH, have been found to reduce thrombin activation and platelet activation [24]. Wang et al. [35] found that the percentage of circulating blood-platelet leukocyte aggregates (PLAs) were significantly elevated in diabetes, with platelets having lower GSH and GPX-1 levels. These findings were also associated with increased methylglyoxal (MG), which is normally cleared by GSH. They found that administration of NAC enhanced platelet GSH and GSH-dependent MG elimination, as well as corrected levels of GPX-1 [35].
The depletion of GSH may also allow viruses to replicate, and therefore is a
major factor in the pathology of COVID-19 [36]. When SARS-CoV-2 invades the body
it elevates proinflammatory markers such as IL-6 and TGF-
Viruses have been shown to induce oxidative stress by depleting GSH [42]. A major characteristic of SARS-CoV-2 infection is oxidative stress, which leads to inflammation and vascular dysfunction. SARS-CoV-2 potentiates all three components of Virchow’s triad—hypercoagulability, blood flow stasis, and endothelial cell damage—to increase risk of thrombosis [43]. Wichmann et al. [44] found high incidence of thromboembolic events in complete autopsies from COVID-19 patients at an academic medical center in Germany. Of note, venous thromboembolism, arterial thrombosis, and microvascular thrombosis have all been described as complications of COVID-19 [45]. In addition, D-dimer levels were significantly elevated with increasing severity of COVID-19 [38].
Research has shown that GSH, the most abundant antioxidant, is deficient in
COVID-19 [8, 37, 38, 46]. Oxidative stress leads to activation of
pro-inflammatory cytokines [47]. Increased amounts of cytokines like IL-6 and
TGF-
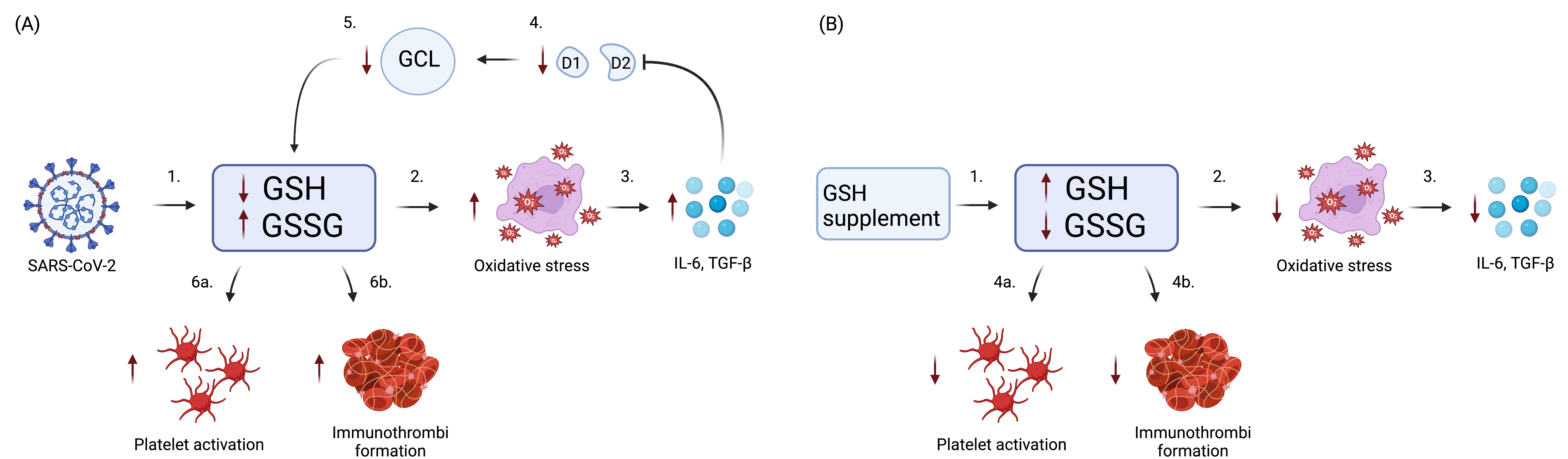 Fig. 2.
Fig. 2.Role of GSH in COVID-19 Immunothrombosis. (A) SARS-CoV-2
infection causes GSH deficiency, eventually leading to platelet activation and
immunothrombi formation. (1) Initially, SARS-CoV-2 reduces GSH and increases
GSSG. (2) reduction in GSH and increase in GSSG causes an increase in oxidative
stress. (3) In turn, this elicits the release of pro-inflammatory cytokines IL-6
and TGF-
The main structural components of SARS-CoV-2 are the S protein, membrane, envelope, and nucleocapsid proteins. The S protein is responsible for viral entry into host cells via the ACE-2 receptor [50]. The S1 spike protein of SARS-CoV-2 binds to ACE-2 receptors, allowing the S2 spike protein to facilitate fusion of the viral membrane with the host cell membrane [51]. Higher rates of viral fusion into the host cells of those with comorbidities leads to significantly higher risk of morbidity and mortality [52]. Zhang et al. [53] found that SARS-CoV-2 induced platelet activation, aggregation, and dense granule release via the binding of the S protein to the ACE2 receptor. Ryu et al. [54] found that the addition of purified, recombinant SARS-CoV-2 S1 protein to a coagulation-competent normal plasma is sufficient to induce formation of anomalous clots that are resistant to fibrinolysis. These clots are identical to clots that have been found in Long COVID-19 patients and have been postulated to block capillaries, causing symptoms such as breathlessness, coagulopathies, and inflammation [6].
The other structural components of SARS-CoV-2, particularly the nucleocapsid protein, may play a role in inflammation. The nucleocapsid proteins of SARS-CoV-2 stimulate the release of IL-6 in a dose-dependent manner [55, 56]. As mentioned before, this cytokine can decrease GSH and result in a cycle of GSH depletion and pro-inflammatory cytokine production [36]. IL-6 is associated with the cytokine storm that occurs in severe infections [11].
Lage et al. [8] found that markers associated with inflammatory responses persisted in COVID-19 patients after recovery, which was depicted as 52 days after infection onset. Particularly, the intracellular GSH levels were still found to be reduced compared to healthy control individuals. This suggests that decreased GSH not only contributes to acute COVID-19 but may also play a role in Long COVID.
There are varying results on effective therapies for COVID-19-induced thrombosis. In a few studies, early anticoagulation therapy in acute COVID-19 has been shown to improve patient outcomes, when compared to patients without any anticoagulation therapy [57, 58, 59, 60, 61]. Between patients who have received usual-care pharmacologic thromboprophylaxis and those who received therapeutic dose prophylactic anticoagulation, the INSPIRE, ACTION, and REMAP-CAP trials showed no difference in clinical outcomes [62]. Major bleeding occurred in a higher percentage of patients assigned to therapeutic-dose anticoagulation compared to those assigned to usual-care pharmacologic thromboprophylaxis [62].
A study of COVID-19 hospitalized patients showed that compared with patients who did not receive antiplatelet therapy, patients receiving acetylsalicylic acid had a significantly lower cumulative incidence of in-hospital death [45]. Another study found that antiplatelet therapy was associated with lower mortality rates, when compared to patients with no antiplatelet therapy and no anticoagulation therapy [63]. However, the RECOVERY trial found that patients treated with acetylsalicylic acid were not associated with reductions in mortality but had a slightly shorter duration of hospitalization and a higher proportion of these patients were discharged from the hospital alive within 28 days. Furthermore, the allocation to acetylsalicylic acid was associated with an increased risk of major bleeding and a decreased risk of thromboembolic complications [64]. In all, it appears that an ideal treatment would target thrombosis or platelet aggregation, without increasing the risk for bleeding. Findings suggest an oxidative stress pathway as a potential target for host-directed therapy to mitigate COVID-19 hyperinflammation and associated sequelae. Horowitz et al. [65] describes the use of GSH therapy in relieving dyspnea associated with COVID-19 pneumonia in two patients. It is likely that GSH supplementation, via a macrophage-induced pathway, plays a critical role in the removal of fibrin clots, which will be discussed later.
Interleukins are cytokines released by leukocytes and other types of cells in
the body in response to biological threats [66]. Interleukins function as
modulators for growth, differentiation and activation of inflammatory and immune
responses due to their anti- and pro-inflammatory inherent capabilities [67].
While many interleukins exist, we will focus on IL-6, a proinflammatory cytokine
which induces oxidative stress and systemic inflammation [68], and its effects on
GSH. IL-6 expression is often strictly controlled at transcriptional and
post-transcriptional levels, yet the dysregulated continual synthesis of IL-6 can
contribute to pathological effects of tissue injury and hyperinflammatory states
[69]. As previously discussed, the formation of thrombosis in hyperinflammatory
state is influenced by the level of GSH, and recent studies have shown that GSH
level is negatively affected by increased levels of IL-6 and TGF-
Recently, the SARS-CoV-2 viral N protein, a nucleocapsid protein [70] was shown
to upregulate the production of IL-6 via increased activation of its promoter in
A549 human lung cells in a dose-dependent manner [36, 55, 56]. Other studies
emphasize this connection by showing positive correlation between elevated IL-6
level and increased severity of COVID-19, such as exacerbated respiratory
failure, hypercytokinemia and rapid progression to acute respiratory distress
syndrome (ARDS) [11, 71]. The increase in proinflammatory cytokines is
hypothesized to deplete GSH to facilitate replication of SARS-CoV-2 and other
viruses, leading to exacerbated symptoms [36]. The current proposed mechanism
through which viruses such as SARS-CoV-2 accelerate their own replication is via
the reduction of GSH level and increasing the production of reactive oxygen
species [36]. Evidence supports that IL-6 has an ability to suppress the enzymes
iodothyronine deiodinases type I (D1) and II (D2), reducing the conversion of
prohormone thyroxine T4 to its active form T3, which ensures the deregulation of
glutamate cysteine ligase (GCL) and the successive reduction of GSH synthesis
[72, 73]. The focal point drawn from all these studies combined suggests the
formation of a repeating cycle of GSH depletion, leading to increased levels of
IL-6 and TGF-
The relationship between IL-6 and formation of thrombosis was assessed in a study measuring levels of IL-6 in patients with deep vein thrombosis (DVT). Zhang et al. [74] found that IL-6 expression was increased while miR-338-5p, a small segment of non-coding RNA, was decreased in patients with DVT, suggesting a negative correlation. They were able to replicate the negative correlation in murine models where miR-338-5p knockdown increased IL-6 expression [74]. Although the exact mechanism is unclear, this finding is consistent with the suggestion that thrombosis is also inflammatory-mediated. Senchenkova et al. [75] also shows the relationship between IL-6 and abnormalities in platelet production, where thrombocytosis response, platelet hyperreactivity, and accelerated thrombus development were absent in IL-6-deficient mice.
TGF-
D-dimer is a protein fragment found in the plasma when a blood clot undergoes
degradation by fibrinolysis. Although D-dimer can exist at low levels in the
plasma of healthy individuals from the physiologic breakdown of fibrin, elevated
levels develop in a number of pathologic conditions [82]. Levels
A state of oxidative stress is associated with diabetes, aging, cancer, and COVID-19, among others, and result in GSH depletion. The increased ROS level impacts the integrity of the RBC membrane, which impacts the red blood cell (RBC) function, leading to impaired hemostasis and thrombosis. The RBC aggregation that results leads to a hypercoagulable state [86]. The endothelial cell lining also becomes dysfunctional in a state of increased ROS, triggering platelet adhesion and activation. This phenomenon can be seen in aging, which is characterized by an overproduction of ROS [86]. It also presents with a higher incidence of thromboembolism and venous thrombosis [87]. Erythrocyte oxidation stress leads to thrombotic events in these conditions, marked by an elevated D-dimer. This suggests an important relationship between GSH and D-dimer levels.
Nwose et al. [88] found a statistically significant lower level of GSH and statistically significant higher D-dimer in diabetic and pre-diabetic patients compared to controls. There was also a significantly negative correlation between GSH and D-dimer levels in diabetic and pre-diabetic patients [88]. This finding can be attributed to erythrocyte oxidative stress induced by hyperglycemia which depletes GSH.
Elevated D-dimer is a well-studied biomarker for disease severity and mortality in COVID-19 [89]. Yao et al. [38] found that D-dimer elevation was present in 74.6% of patients in Remnan Hospital of Wuhan University, Wuhan, China, and was the only variable associated with increased mortality odds. Shah et al. [89] also found an association between elevated D-dimer levels and COVID-19 severity, with 15% of recovered COVID-19 patients retaining persistently elevated D-dimer level after a median of 3 months following infection. Kryukov et al. [46] found a negative association between D-dimer levels and GSH levels, and identified an association between low total GSH and risk of severe COVID-19. This observation is secondary to the elevated incidence of thromboembolic complications in COVID-19 patients. There currently exists a lack of diagnostic method to accurately determine the presence of microemboli. As such, testing for D-dimer may serve as a method of guiding clinical suspicion and monitoring response to therapy.
Macrophages have a wide variety of functions, both in physiological processes and in disease pathogenesis. Macrophages have immunological function in bacterial, viral and parasitic infections, and they also function in inflammatory and hemostatic processes [90, 91]. The role of macrophages in immunothrombosis is thought to be related to their role in fibrinolysis. Degradation of fibrin clots is mostly associated with the conversion of plasminogen by tissue-plasminogen activator to a serine protease plasmin. The degradation of fibrin by plasmin and the deposition of fibrin products is a process that has been well described, however, there remains a less studied pathway in which extravascular fibrin deposits are ultimately removed and degraded [92]. The clearance of fibrin from the blood has been viewed to involve the phagocytosis of microclots, presumably by macrophage [93]. The clearance mechanism of which macrophage remove fibrin has been closely tied to the activity of plasminogen and plasmin. It has been shown that once activated, plasmin activates matrix metalloproteinases that allows for macrophage movement through the extracellular matrix [94]. In several instances, the deficiency of plasminogen has shown to cause a decrease in the recruitment of macrophages [94, 95]. Beyond macrophage migration, it has also been shown the lack of plasmin activity leads to the inability of macrophage to clear fibrin deposits [95]. This process has further been demonstrated by a study from Motley et al. [92] that describes a novel pathway of macrophage endocytosis of fibrin. They found that there is a specific macrophage population, CCR2 positive, that is responsible for endocytosis of fibrin and elimination of these cells results in decreased cellular fibrin uptake. Furthermore, these macrophages are morphologically distinct from collagen degrading macrophages. They also confirmed the findings of previous studies in implicating plasmin/plasminogen in the function of macrophagic endocytosis of fibrin, noting that plasminogen is needed for the fragmentation of fibrin to expose cellular binding sites for endocytic uptake but also that plasmin directly stimulates macrophage phagocytosis. In a model of plasminogen-deficient mice, they showed that there was a reduction in endocytosis of fibrin, suggesting that leukocytic fibrinolytic pathways are largely, although not entirely, dependent on plasminogen [92]. Similarly to the loss of plasminogen, the loss of enzymatic activity of plasmin had similar findings of reduced fibrin endocytosis by leukocytes, further demonstrating the need for plasmin cleavage of fibrin prior to cellular uptake. Following endocytosis, degradation of fibrin occurs within lysosomes of macrophage. This process involves the binding of the amino-terminus alpha chain of fibrin to cell surface receptors on macrophage [90]. Beyond the direct role of macrophage in fibrin clot removal, macrophage and other phagocytes have also been shown to modulate thrombosis through the removal of active coagulation factors and activated platelets thus decreasing the ability for thrombus formation [96].
The interaction between macrophages and GSH relates both to the function of GSH as the main antioxidant of ROS and its role of signaling within the innate immune system [36]. Failure of detoxification will result in ROS reacting with cellular components and ultimately leading to impaired cellular function [11]. Control of infections such as Mycobacterium tuberculosis (M. tb) are dependent upon macrophages and their ability to inhibit pathogen growth with ROS [11]. Individuals with HIV and type 2 diabetes mellitus (T2DM) have shown to have decreased levels of GSH due to the increased production of free radicals depleting GSH [97, 98]. This subsequently leads to impaired macrophage productivity and decreased control of M. tb infection in these individuals [12, 99]. Increased M. tb survival in these patients can also be related to the role of GSH in the signaling of the innate immune system. IFN-gamma is responsible for the activation of macrophages, enhanced antigen presentation, and the induction of nitric oxidated mediated killing mechanisms [99]. Individuals with T2DM or HIV have been shown to have decreased levels of IFN-gamma. Supplementation of these individuals with L-GSH has shown to significantly increase production of IFN-gamma and decrease levels of IL-10. The combined efforts of increased macrophage activation from IFN-gamma and reduced inhibition from IL-10 leads to increased control of M. tb infection in these patients [12, 99].
Therefore the role of macrophages in immunothrombosis appears to be both in disease control as well as resolution of immunothrombosis through fibrin clot degradation. The function of macrophages within these processes is tightly connected to the levels of GSH. It would be expected then that supplementation of GSH in patients would enhance the productivity of macrophage both in disease control to prevent immunothrombosis and in increased clot dissolution in immunothrombosis.
There is sufficient evidence that SARS-CoV-2 infection decreases GSH, but the exact mechanisms by which this occurs remains unclear. Bartolini et al. [100] demonstrated that SARS-CoV-2 lowered uptake of the GSH precursor Cys and increased efflux of thiols, effectively lowering GSH by impairing metabolism of cellular GSH. In addition, they found that blocking viral replication successfully prevented GSH depletion [100]. SARS-CoV-2 also inhibits nuclear factor-erythyroid 2 p45-related factor 2 (Nrf2), a primary transcription factor critical in increasing expression of glutamate cysteine ligase (GCL) [101, 102, 103]. As mentioned previously, COVID-19 also depletes GSH through intracellular radical generation and inhibition of BRCA1. Impaired metabolism coupled with increased consumption of GSH results in decreased levels of GSH and increased levels of GSSG. GSH may also be reduced prior to SARS-COV-2 infection as reduced levels are seen in old age, other inflammatory conditions, and vitamin D deficiency, representing significant risk factors for severe SARS-CoV-2 infection [101].
Decreased GSH potentiates increased oxidative stress and increased release of
cytokines IL-6 and TGF-
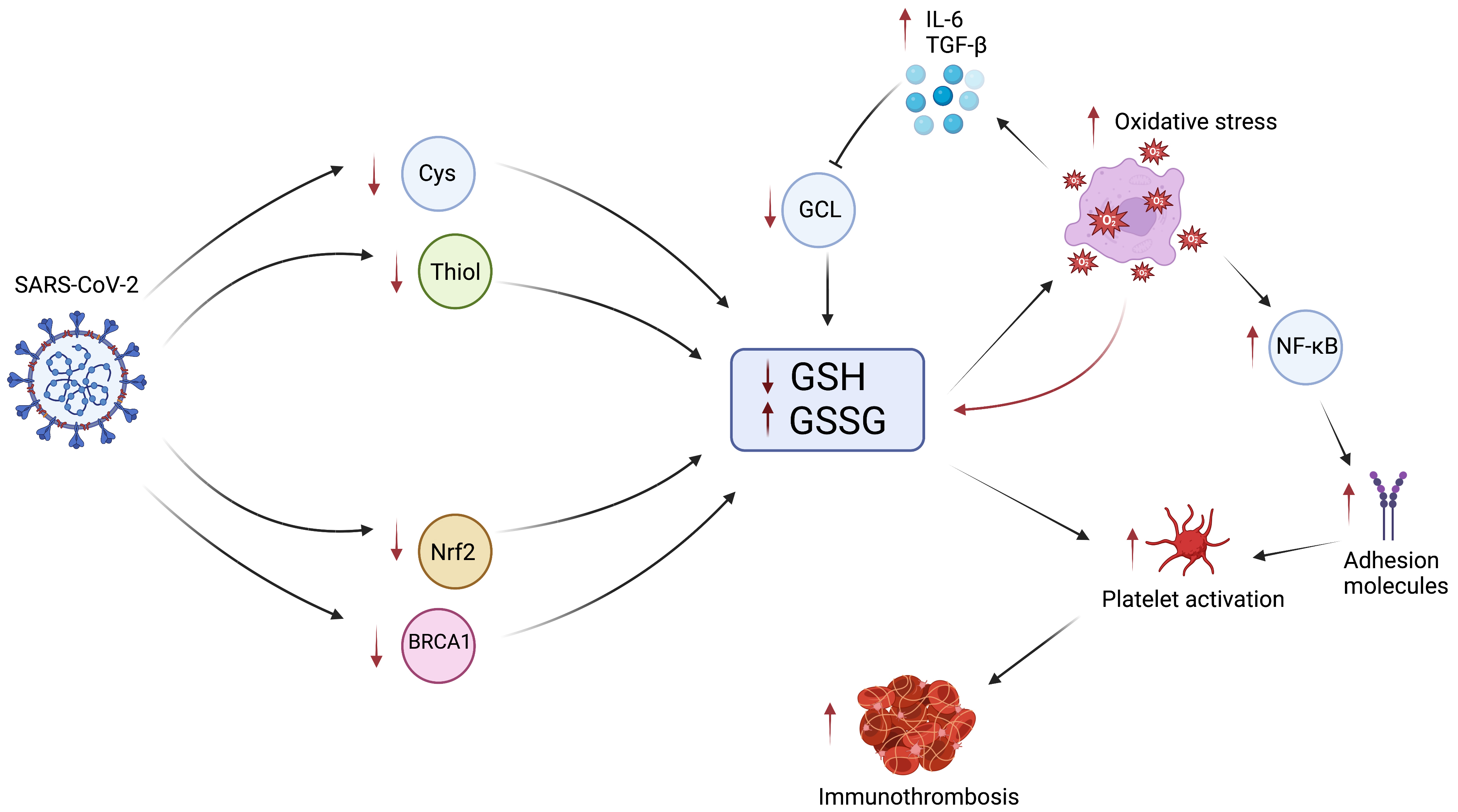 Fig. 3.
Fig. 3.The Role of GSH in COVID-19 Immunothrombosis. Infection with
SARS-CoV-2 results in decreased intracellular Cys and thiol due to decreased Cys
uptake and increased thiol efflux, reducing GSH synthesis. Infection also
inhibits Nrf2 and BRCA1, further reducing GSH synthesis. The reduction in GSH and
increase in GSSG results in impaired ability to control oxidative stress.
Increased oxidative stress further propagates the biochemical cascade by
consuming additional GSH, increasing the production of cytokines IL-6 and TGF-
Immunothrombosis is an important pathological feature of inflammatory conditions and infections like COVID-19. While immunothrombosis can serve as a natural defense mechanism by immune cells like neutrophils and monocytes, dysregulated immune responses can create a thrombotic cascade. Excessive cytokine release seen in COVID-19 can cause inflammation of endothelial cells, resulting in damage and release of TF in pulmonary microvessels, yielding microclots. These microclots can block capillaries, creating a hypoxic environment which furthers the coagulation cascade, or they can remain as emboli and circulate in the blood. Patients who experience severe acute- or long-COVID-19 infection are at an increased risk of adverse thrombotic events due to the hypercoagulable state. Diagnosing the presence of microclot formation from immunthrombosis in late-COVID-19 proves challenging, as current diagnostic modalities for evaluating the presence of a thrombus are not sufficient for identifying the circulating microclots and those embedded in capillary beds. The presence of D-dimers in blood may serve as an important biomarker in both monitoring infection course and estimating long-term adverse thrombosis risk.
Research has previously evaluated the role of GSH in the maintenance of ROS and
its association with cytokine levels in states such as HIV and diabetes, but had
not yet evaluated GSH depletion as a dominant cause of immunothrombosis in
COVID-19. SARS-CoV-2 has been shown to decrease GSH by reducing Cys uptake,
increasing thiol efflux, inhibiting Nrf2, and inhibiting BRCA1. There is
sufficient evidence that there is a negative correlation between the cytokines
IL-6 and TGF-
Conceptualization—VV, FG, MS, IG; methodology—VV, FG, MS, IG; resources—IG; writing - original draft preparation—IG, NL, MM, CA, AA, AG, SM, MS, FG, JR; writing - review and editing—VV, MS, IG; visualization—IG, CA; supervision—VV, MS, FG, IG; project administration—VV, MS, FG, IG. All authors have read and agreed to the published version of the manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
Figures created with BioRender.com (https://www.biorender.com).
We appreciate the funding support from the NIH (R15 HL143545/HL/NHLBI).
The authors declare no conflict of interest. VV is serving as one of the Guest editors of this journal. We declare that VV had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to ESH.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.