1. Introduction
There is a growing dissatisfaction with the lack of progress in the treatment
and prevention of Alzheimer’s disease [1]. This is widely accepted as arising
from a poor conceptualization of the relevant processes forming the biological
underpinnings of neuronal and cognitive loss in the course of dementia. For 40
years, research and targeted treatment in Alzheimer’s disease have focused on the
overproduction of amyloid- plaques and hyperphosphorylated tau tangles
[2]. A plethora of wider pathophysiological processes have data supporting their
role in Alzheimer’s disease, including alterations driven by immune/glia
inflammation, circadian disruption, obesity, diet, stress, sub-optimal
mitochondrial function and gut microbiome-derived products [3, 4, 5, 6, 7, 8]. The amyloid
hypothesis is also significantly challenged by amyloid- being an
endogenous antimicrobial, suggesting that its overproduction may be ‘too much of
a good thing’ in the course of heightened inflammation and toll-like receptor
(TLR)2/4 signaling [1, 9]. This is further supported by the high
amyloid- levels evident in other diverse medical conditions, including
glioblastoma [10], breast cancer [11], type 1 diabetes mellitus (T1DM) [12],
Parkinson’s disease [13] and amyotrophic lateral sclerosis [14]. The heightened
amyloid- levels in Parkinson’s disease and Lewy Body diseases can drive
the increased -synuclein aggregation classically defining these
diseases [13], indicating a role for excessive amyloid- production in
upregulating other pathophysiological processes. Such data, coupled to the role
of systemic processes in neurodegeneration has formed the underpinnings of a
growing consensus that a more holistic perspective of Alzheimer’s disease is
required, including the incorporation of the heightened levels of
amyloid- and hyperphosphorylated tau. This is laced with the hope that a
systemic conceptualization not only embraces the complexity of data highlighted
above but will also provide more feasible and achievable targets for treatment
and prevention.
This article highlights the wide array of systemic processes, including gut
microbiome and white adipocyte products as well as the
hypothalamus-pituitary-adrenal (HPA) axis and pineal/local melatonergic pathway
in the regulation of astrocyte modulation of neuronal activity and survival.
Classical central nervous system (CNS) areas associated with neuronal and
cognitive loss, such as the cortex and hippocampus, are highlighted as well as
non-classically associated brain areas, such as the hypothalamus. This provides a
pathoetiological model allowing systemic processes to alter CNS function, with an
important hub being the interface of astrocytes and neurons. Such a systemic
perspective also incorporates how other currently classified conditions, such as
polycystic ovary syndrome (PCOS) [15, 16], major depressive disorder (MDD) [17],
bipolar disorder [17], neuroticism [18], obesity [19, 20], stress/post-traumatic
stress disorder (PTSD) [21], discrimination stress [22], migraine [23] and type 2
diabetes mellitus (T2DM) [24] are associated with an increased risk of dementia.
Such wide arrays of data highlight the problems of a current classification
system that is based on endpoint ‘catastrophes’ and the importance of
investigating, assessing and seeking biomarkers for physiological processes
across current medical classifications.
The pineal and local mitochondrial melatonergic pathway is an important aspect
of Alzheimer’s disease pathoetiology and is briefly reviewed next.
2. Tryptophan-Melatonin Pathway
There is a gradual decrease in pineal melatonin over aging culminating in a
dramatic 10-fold lower level at night in people in their ninth decade of life,
compared to adolescence [25]. As night-time melatonin is credited with dampening
any residual low-level inflammatory activity and optimizing mitochondrial
function, the suppressed capacity of melatonin at night over aging has
consequences for the pathoetiology of most medical conditions, including cancer
[26] and cardiovascular disorders [27], as well as dementia [28]. The relevance
of melatonin is highlighted by its powerful efficacy in preventing dementia in
preclinical models [29, 30, 31].
Notably, melatonin seems produced in all body cells, primarily within
mitochondria [32], where its synthesis is intimately associated with the capacity
of mitochondria to upregulate the pyruvate dehydrogenase complex (PDC), which
increases the conversion of pyruvate to acetyl-CoA, thereby increasing adenosine
triphosphate (ATP) production by the tricarboxylic acid (TCA) cycle and oxidative
phosphorylation (OXPHOS). As acetyl-CoA is a necessary cosubstrate for the
conversion of serotonin to N-acetylserotonin (NAS) in the initiation of the
melatonergic pathway, the mitochondrial melatonergic pathway is intimately linked
to mitochondrial function. The capacity of cells to upregulate the melatonergic
pathway seems important to their capacity to resist challenge, either
environmental/systemic and/or from within the microenvironment in which they
reside [33]. Many medical conditions, including ‘autoimmune’/‘immune-mediated’
disorders may arise from the suppressed capacity of a given cell to upregulate
melatonin, thereby preventing melatonin from regulating PINK1/parkin mediated
mitophagy. Dysregulated mitophagy leads to major histocompatibility complex
(MHC)-1 induction and the MHC-1 driven chemoattraction of CD8 t cells that
drive the ‘autoimmune’ destruction of cells, including substantia nigra pars
compact dopamine neurons in Parkinson’s disease [33, 34]. The tryptophan-melatonin
pathway, and how it links to processes driving plaques and tangles, is shown in
Fig. 1.
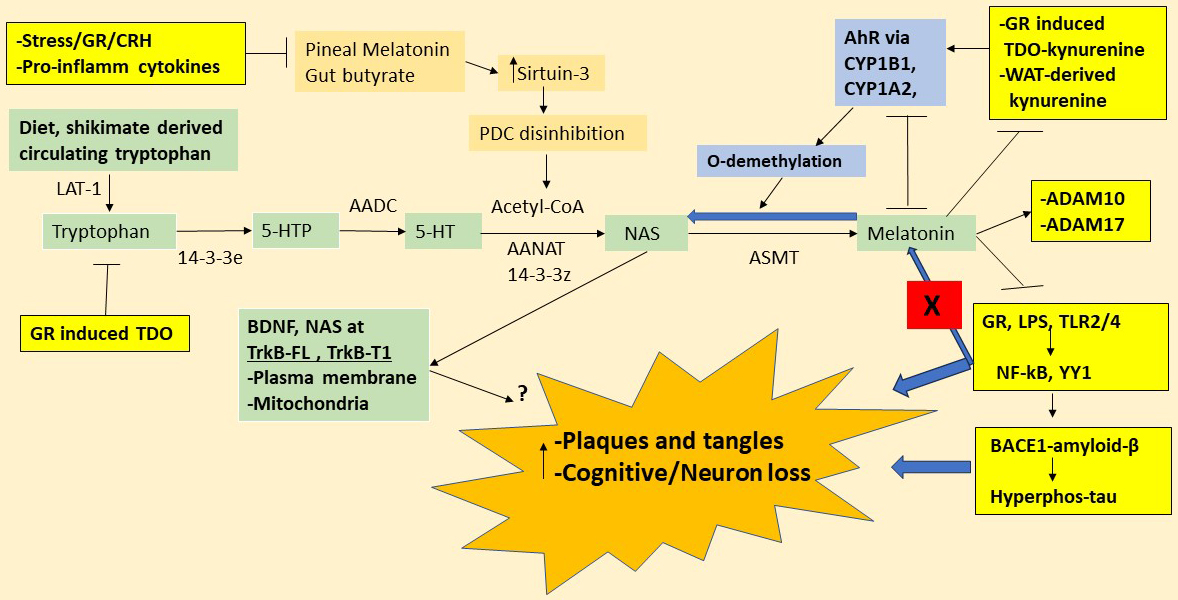 Fig. 1.
Fig. 1.
Tryptophan-melatonin pathway interactions. Shows the
tryptophan-melatonin pathway (green shade) and how it is intimately linked to key
systemic processes relevant to dementia pathoetiology, including: (1) Stress/HPA
axis activity drives GR activation and GR/TDO-kynurenine/AhR induction. Stress/GR
drives gut-derived LPS and other TLR2/4 activators that upregulate the
transcription factors, NF-kB and YY1, thereby inducing-BACE1-amyloid-
and hyperphosphorylated tau; (2) white adipocyte (WAT)-derived kynurenine
activates the AhR as well as inducing neurotoxic kynurenine pathway products; and
(3) gut microbiome-derived butyrate and circadian melatonin upregulates sirtuin-3
to disinhibit PDC, thereby optimizing mitochondrial function, whilst also
providing acetyl-CoA as a necessary cosubstrate for the conversion of serotonin
to NAS in the initiation of the melatonergic pathway. Tryptophan is mainly
diet-derived but is also produced by the shikimate pathway of the gut microbiome.
The large amino acid transporter (LAT)-1 takes circulating tryptophan into cells,
including astrocytes, with TPH1 and TPH2 needing to be stabilized by 14-3-3e to
allow tryptophan to be converted to 5-HTP. AADC then converts 5-HTP to 5-HT
(serotonin). 14-3-3z stabilized AANAT metabolizes 5-HT to N-acetylserotonin
(NAS), in the presence of acetyl-CoA as a necessary cosubstrate. NAS is converted
to melatonin by ASMT. Acetyl-CoA levels are highly dependent upon optimized
mitochondrial function arising from PDC disinhibition allowing the induction of
the mitochondrial melatonergic pathway to be intimately linked to mitochondrial
metabolic function. Pineal melatonin and the gut microbiome-derived short-chain
fatty acid, butyrate, induce sirtuin-3, which deacetylates and disinhibits PDC,
thereby enhancing the conversion of pyruvate to acetyl-CoA. Stress/CRH/GR
activation and pro-inflammatory cytokines suppress pineal melatonin and increase
gut permeability/dysbiosis, thereby lowering butyrate levels. The AhR, via CYP1A2
and CYP1B1, can O-demethylates melatonin to NAS, with AhR induced
CYP1B1/CYP1A2/CYP1A1 also able to hydroxylate melatonin to 6-hydroxymelatonin via
modifiable protein-protein interactions. The AhR can therefore suppress melatonin
by a couple of processes as well as raising the CYP1NAS/melatonin ratio. NAS and
melatonin have many common but important differential effects given that NAS
activates the BDNF receptor, TrkB, as well as inducing BDNF, being a couple of
means whereby NAS enhances TrkB activation. TrkB activation may be beneficial in
dementia, although this may be dependent upon the full-length (TrkB-FL) isoform
as the truncated isoform (TrkB-T1) can contribute to apoptosis. Notably, TrkB-FL
and TrkB-T1 effects will be dependent upon location on the mitochondrial and/or
plasma membranes. Melatonin affords powerful protection in dementia models,
partly mediated by its suppression of the gut-permeability associated LPS and GR
nuclear translocation, thereby preventing the GR from inducing TDO-kynurenine and
BACE1-amyloid-. Melatonin also suppresses hyperphosphorylated-tau both
directly and via decreased amyloid-. Melatonin also stimulates the
non-amyloidogenic -secretase activities of ADAM10 and ADAM17, whilst
inhibiting the expression of the amyloidogenic - and
-secretases. The melatonergic pathway is therefore in intimate
interactions with systemic processes linked to dementia, with the incapacity of
NF-kB and YY1 to induce melatonin from the melatonergic pathway (red shade X)
being crucial to dementia pathophysiology. Abbreviations: 5-HT, serotonin;
5-HTTP, 5-hydroxytryptophan; AADC, aromatic-L-amino acid decarboxylase;
acetyl-CoA, acetyl-coenzyme A; AANAT, aralkylamine N-acetyltransferase; AhR, aryl
hydrocarbon receptor; ASMT, N-acetylserotonin O-methyltransferase; BACE1,
-site amyloid precursor protein-cleaving enzyme 1; BDNF, brain-derived
neurotrophic factor; CRH, corticotrophin releasing hormone; CYP, cytochrome P450;
GR, glucocorticoid receptor; HPA, hypothalamus-pituitary-adrenal; LAT-1, large
amino acid transporter 1; LPS, lipopolysaccharide; NAS, N-acetylserotonin; NF-kB,
nuclear factor kappa-light-chain-enhancer of activated B cells; TLR, toll-like
receptor; TrkB-FL, tyrosine receptor kinase B-full length; TrkB-T1, tyrosine
receptor kinase B-truncated; TDO, tryptophan 2,3-dioxygenase; PDC, pyruvate
dehydrogenase complex; ADAM, A disintegrin and metalloproteinase domain-containing protein; YY, yin yang.
The melatonergic pathway is evident in all mitochondria-containing cells, where
it is induced by two transcription factors that drive amyloid-
production, namely Nuclear factor kappa-light-chain-enhancer of activated B cells
(NF-B) and yin yang 1 (YY1), via -site amyloid precursor
protein-cleaving enzyme (BACE)1 induction [35, 36]. The suppressed capacity of
NF-B and YY1 to synchronize melatonin production and release in
association with BACE1 and amyloid-, will prolong amyloid-
production whilst maintaining astrocyte and microglia reactivity, thereby driving
microbial/alarmin signalling and associated heightened local inflammation [1].
The loss of synchronized glia autocrine and paracrine melatonin will contribute
to ongoing CNS inflammation. The elimination of amyloid- by anti-amyloid
antibodies would not be expected to impact on such dysregulated inflammatory
processes, which data on the poor efficacy of anti-amyloid antibodies
pharmaceuticals would seem to indicate [37]. Melatonin also stimulates the
expression of the non-amyloidogenic alpha-secretase activities of A disintegrin
and metalloproteinase domain-containing protein (ADAM) 10 and ADAM17 [38] whilst
inhibiting the expression of the amyloidogenic beta- and gamma-secretases [39].
The above provides a framework (Fig. 1) to link a wide array of diverse bodies
of data on dementia pathophysiology, by highlighting the importance of the
astrocyte melatonergic pathway and how it can become desynchronized from NF-kB
and YY1 induced BACE1/amyloid-, resulting in excessive amyloid-
production and the maintenance of inflammatory interactions of astrocytes,
neurons, and microglia in the course of dementia pathophysiology.
3. Circadian Rhythm and Dementia
Recent data implicates decreased pineal melatonin coupled to sleep/circadian
disruption in Alzheimer’s disease pathoetiology, which is supported by data
showing a dramatic 10-fold decrease in pineal melatonin between adolescence and
the ninth decade of life [25], with consequences that include the suppression of
melatonin’s anti-inflammatory, antioxidant and mitochondrial optimizing effects
at night in dampening the consequences of day-time stressors and challenges,
including immune and glial cell reactivity [26]. The relevance of
night-time/sleep linked systemic processes is also indicated by the circadian
role of the gut microbiome. Butyrate is primarily produced during fasting [40],
which has important implications for the regulation of the glucocorticoid
receptor (GR) in the course of not only stress/HPA axis activation but also for
the cortisol awakening response (CAR). CAR is a dramatic and important, although
under-investigated, circadian rhythm, typically described ‘as preparing the body
for the coming day’. Gut microbiome-derived butyrate and CAR may be significantly
regulated by variations in circadian and local melatonin availability, which
butyrate and melatonin prevent GR nuclear translocation, with relevance to a host
of diverse medical conditions including cancer [41].
3.1 Circadian Rhythm and Gut Microbiome
A plethora of medical conditions are associated with alterations in the gut
microbiome, invariably involving decreases in the short-chain fatty acid,
butyrate [42]. As noted, butyrate is primarily induced during fasting, including
when asleep [40]. Pineal melatonin effects include the suppression of gut
permeability and associated decrease in gut dysbiosis, concurrently elevating
butyrate levels [43]. Melatonin is also very highly produced in the gut,
especially after feeding where is seems to increase the swarming of gut bacteria
in the presence of food [44, 45]. The gut microbiome and the timing of food intake
are integral aspects of the circadian rhythm.
The benefits of butyrate are mediated via a number of processes, including: (1)
activation of the G-protein coupled receptors (GPR), GPR-41, -43 and -109 [46];
(2) via histone deacetylase inhibition (HDACi) thereby allowing butyrate to
epigenetically regulate systemic and CNS cells [47]; (3) by optimizing
mitochondrial function, involving the upregulation of sirtuin-3 and the
deacetylation and disinhibition of PDC, thereby increasing ATP from OXPHOS and
the TCA cycle, coupled to decreased mitochondrial oxidant production [48, 49]. As
the upregulation of the mitochondrial melatonergic pathway is intimately linked
to mitochondrial optimization, as shown in intestinal epithelial cells [50],
butyrate upregulates the mitochondrial melatonergic pathway [50]. Factors
influencing the tryptophan-melatonin pathway in a given cell will therefore have
consequence for how the gut microbiome regulates that cell.
Stress/GR activation and proinflammatory cytokines can suppress pineal melatonin
[51], whilst increasing gut permeability/dysbiosis, thereby lowering butyrate
levels [52, 53]. Such data highlights the dynamic two-way interactions of systemic
processes and CNS processes over the circadian rhythm (see Fig. 1). Variations in
pineal melatonin, local tryptophan-melatonin pathway regulation and gut
microbiome-derived butyrate also interact to modulate the consequence of HPA axis
activation, including in the course of the cortisol awakening response (CAR).
3.2 HPA Axis and Cortisol Awakening Response
The HPA axis has been extensively investigated following its induction by
stress, including in Alzheimer’s disease pathoetiology, where heightened levels
of circulating cortisol are evident, as shown in a recent meta-analysis [54].
However, although of unknown physiological relevance, morning CAR does not seem
significantly altered in Alzheimer’s disease patients [54]. CAR drives a large
surge in cortisol release starting just before the end of sleep and lasting for
the first 30 minutes following awakening. The role of CAR is generally placed in
the vague context of ‘preparing the body for the coming day’, with hope that its
relevance may be better clarified by more rigorous methodology [55]. The effects
of CAR, and indeed stress driven HPA axis activation, is considerably complicated
by the diverse ways that glucocorticoid receptor (GR) can influence cell
processes. The GR is the main mediator of HPA axis and CAR effects, with the GR
being predominantly expressed in the cytoplasm in a complex with heat shock
protein (hsp)90 and p23. Upon activation, the GR is transported to the nucleus
where it induces genes containing the glucocorticoid receptor element (GRE) in
their promotor.
The availability of pineal and/or local melatonin is relevant to CAR effects,
with melatonin preventing GR nuclear translocation [56], including possibly via
the upregulation of bcl2-associated athanogene (BAG)1 [41]. Butyrate, via its
capacity as a HDACi, also suppresses GR nuclear translocation, involving
increased acetylation of the GR and hsp90 [57, 58]. Consequently, variations in
pineal and local melatonin as well as butyrate availability over the course of
sleep not only regulate inflammation, antioxidant status and mitochondrial
function, but also have direct impacts on GR nuclear effects during CAR. This may
be of some importance given the dramatic effects that cortisol can have on the
function of all immune and glial cells, in contrast to melatonin and butyrate
(see Table 1 in [41]), and therefore on patterned immune/glia responses for the
coming day. This would indicate that although CAR level/slope may not be
significantly different in Alzheimer’s disease [54], the dramatic decrease in
pineal melatonin and gut microbiome-derived butyrate will significantly regulate
CAR consequences in Alzheimer’s disease. As the different cells in a given local
microenvironment, including the tumor microenvironment [59], may have their
tryptophan-melatonin pathway differentially regulated, GR activation can have
distinct effects in the cells of a given microenvironment. This alters the nature
of the homeostatic interactions that occur in the course of CAR ‘preparing the
body for the coming day’. Such homeostatic alterations are proposed to contribute
to the pathoetiology of ‘autoimmune’/‘immune-mediated’ disorders [33], which
recent data indicates to include Alzheimer’s disease [60].
Notably, this is complicated by the diverse effects of GR activation. The GR has
a number of genomic and non-genomic effects, including via the induction of
intracellular signaling pathways following plasma membrane GR activation [61].
The GR may also interact with other transcription factors in the nucleus to
modulate diverse genes with no GRE in their promoter. GR effects, both in CAR and
stress/HPA axis activation, may also be further complicated by factors
upregulating BAG-1, as recently proposed for melatonin [62]. BAG-1 can not only
prevent nuclear translocation but can also chaperone the GR to mitochondria [63],
where it can be translocated over the inner and outer mitochondrial membranes (by
mitochondrial import inner membrane translocase subunit (TIM) 23 and
mitochondrial import outer receptor subunit (TOM) 20) allowing the GR to interact
with PDC and hsp60, with consequences for mitochondrial metabolism, including the
regulation of the mitochondrial melatonergic pathway [41].
3.3 Butyrate, Melatonin, ApoE4 and CAR Interactions in Mitochondria
Regulation
Importantly, the consequences of CAR/GR, butyrate and pineal/local melatonin
interactions are crucially determined by how they interact to regulate
mitochondrial function. This involves both the dampening of residual inflammation
and mitochondrial oxidant production, as well as how CAR may prime systemic and
CNS cells for the coming day. It is widely recognized that suboptimal
mitochondrial function is an integral aspect of dementia, both systemically [64]
and in brain cells [65, 66], with mitochondrial dysfunction evident in many of
dementia’s currently conceptualized ‘comorbidities’, such as obesity [67], T2DM
[68] and depression [69]. Mitochondrial function may be especially relevant in
reactive cells, namely immune and glial cells, including astrocytes, which have a
powerful role in the regulation of neuronal activity and survival [69].
There is a growing appreciation of the importance of astrocytes in the
regulation of a wide array of neuropsychiatric and neurodegenerative conditions
that have been classically conceptualized as ‘neuronal’ disorders, including
depression [69], Parkinson’s disease [70], amyotrophic lateral sclerosis (ALS)
[14], and multiple sclerosis [71]. The role of astrocytes in such conditions is
attributed to a wide array of processes including by astrocyte AhR nuclear
translocator (ARNT)-like 1 and BAG3 determining levels of -synuclein
and hyperphosphorylated tau, implicating the circadian rhythm and BAG regulation
in the modulation of neurodegenerative processes [3]. It is proposed here that
dysregulation of the astrocyte tryptophan-melatonin pathway is an early change in
the pathoetiology of Alzheimer’s disease.
Astrocyte Mitochondrial Tryptophan-Melatonin Pathway
TLR2/4 can be activated by numerous ligands, including LPS, hsp60, hsp90,
amyloid-, -synuclein, fibrinogen and high-mobility group box
(HMGB)1. Astrocyte TLR2/4 signaling increases NF-kB and YY1, thereby increasing
BACE1 and amyloid- production [72, 73]. This is parsimonious with
amyloid- as an endogenous antimicrobial [9]. As both NF-kB and YY1
upregulate the melatonergic pathway, as shown in other cell types [35, 36], the
concurrent/sequential release of melatonin following TLR2/4 activation will have
autocrine and paracrine effects that dampen and time-limit inflammatory responses
and raised oxidants. As cytokines and oxidants can induce further TLR2/4 ligands
as well as the nucleotide-binding domain, leucine-rich–containing family, pyrin
domain–containing-3 (NLRP3) inflammasome [74], the autocrine and intracrine
effects of astrocyte melatonin will prevent prolonged signaling via TLR2/4.
Melatonin suppresses the NF-kB and reactive oxygen species (ROS)-driven NLRP3
inflammasome [74], which may be partly mediated over the circadian rhythm by
pineal melatonin upregulating the 7 nicotinic acetylcholine receptor
(7nAChR) [75], which suppresses immune inflammation and is a
significant treatment target in Alzheimer’s disease [76]. As to whether local
paracrine melatonin release from astrocytes regulates local 7nAChR
levels will be important to determine. Such data highlights the potentially
important role of pineal and local astrocyte melatonin in the regulation of an
Alzheimer’s disease pathophysiological factor (NLRP3) [77] and protective factor
(7nAChR) [78]. As stress/GR activation can regulate 7nAChR
[79], the interactions of pineal melatonin’s circadian induction of the
7nAChR [80] with the wider night-time changes in morning CAR modulation
will be important to determine. Notably, the 7nAChR is also expressed
on the mitochondrial outer membrane where its binding by agonists suppresses
Ca influx and decreases cytochrome c release [81], suggesting that it may
linked to optimizing mitochondrial resilience under challenge [82] as well as
wider cellular and mitochondrial plasticity.
Astrocyte melatonin release has been long established [83], being regulated by
the main Alzheimer’s disease susceptibility gene, apolipoprotein (Apo)E4 [84].
ApoE4 is the major susceptibility gene for Alzheimer’s disease, with carriers of
two ApoE4 alleles having an 8-15-fold increase in Alzheimer’s disease
susceptibility [85]. ApoE is predominantly expressed in astrocytes in the brain,
where it regulates lipid metabolism, with ApoE4 increasing the unsaturated fatty
acid chains on triglycerides [86]. Whether the ApoE4 upregulation of astrocyte
mitochondrial melatonergic pathway is relevant to neuronal loss in dementia will
be important to determine, including whether there is any role for a heightened
dependence of astrocyte melatonin in ApoE4 carriers. ApoE4 effects include the
downregulation of astrocyte monoamine oxidase (MAO)-A and MAO-B, thereby
increasing the availability of serotonin as a precursor for the melatonergic
pathway [85]. This is one means by which astrocyte ApoE4 can increase astrocyte
melatonin production [85], thereby contributing to a role for enhanced astrocyte
melatonin in the maintenance of homeostatic interactions of astrocytes with
neurons and other brain cells. The suppressed capacity to induce astrocyte
melatonin over aging may therefore have more significant impacts on ApoE4
carriers, thereby indicating a more significant role for the greater drop in
local melatonin dampening of local inflammation in ApoE4 carriers.
As noted, heightened amyloid- levels are evident in other classical
neurodegenerative conditions, including glioblastoma [10], Parkinson’s disease
with Lewy bodies [13], tauopathies [87] and ALS [14]. The pathophysiologies of
all of these conditions implicate a significant astrocyte role [14, 88, 89, 90],
including via regulation by ApoE alleles [91, 92, 93, 94]. Clearly, it will be important
to clarify the nature of astrocyte ApoE in the regulation of the astrocyte
tryptophan-melatonin pathway over aging in the pathoetiology of dementia and
associated neurodegenerative conditions.
The loss of serotonergic neurons is intimately linked to the suppression of the
astrocyte tryptophan-melatonin pathway in dementia, with serotonergic neuronal
loss being a relatively early event in Alzheimer’s disease [95]. Decreased
serotonin levels are also evident in Alzheimer’s disease platelets, with platelet
serotonin inversely correlating with cerebrospinal fluid (CSF)
hyperphosphorylated-tau and amyloid- [96]. Such systemic suppression of
serotonin availability may be linked to decreased gut microbiome-derived
butyrate, which downregulates MAO-B, thereby increasing serotonin availability
[97]. Butyrate’s suppression of MAO-B, thereby enhancing serotonin availability
may be another route whereby butyrate upregulates the melatonergic pathway [50]
in astrocytes and other cells as well as modulating the capacity of pineal
melatonin at night to also upregulate the astrocyte melatonergic pathway. The
regulation of serotonin availability may therefore be another aspect of
gut-pineal interactions in the modulation of morning CAR activation of the GR.
Inflammation driven indoleamine 2,3-dioxygenase (IDO) and GR induced tryptophan
2,3-dioxygenase (TDO), by converting tryptophan to kynurenine decrease tryptophan
availability and thereby will also suppress serotonin availability.
Whether the role of autocrine melatonin in switching from an inflammatory
M1-like phenotype in macrophages [35] and microglia [80] to a quiescent,
pro-phagocytic M2-like phenotype is paralleled in astrocytes will be important to
determine. This would seem not unlikely, given the role of exogenous melatonin in
dampening astrocyte reactivity [98, 99]. The maintenance of astrocyte reactivity
is an important driver of microglia reactivity, which is also dampened by
autocrine and paracrine melatonin [100]. Such data highlights the importance of
maintained astrocyte reactivity in regulation of the wider CNS inflammatory
activity in dementia, as well as the role of astrocyte
TLR2-4/NF-kB/YY1/BACE1/amyloid- in driving the maintained
amyloid- production that further contributes to inflammation and
neuronal loss in the course of dementia. However, although previous data is
highly suggestive of a role of such processes, clearly the astrocyte
mitochondrial melatonergic pathway requires investigation. The proposed
sequential release of melatonin following
TLR2/TLR4/NF-kB/YY1/BACE1/amyloid- pathway in astrocytes (as well as
microglia and neurons) will dampen the maintained heightened inflammation that is
widely thought to underpin neuronal loss in dementia and across other
neurogenerative conditions, as indicated by the effects of exogenous melatonin
across diverse preclinical models [101, 102, 103]. Such studies also indicate that in
the course of dampening inflammatory signaling melatonin will have autocrine and
paracrine effects that optimize mitochondrial function, thereby acting on core
process that underpin the complex and dynamic flurry of cell fluxes that are
typical of the chaos of ‘endpoint’ disorders, such as the current classification
of later stage Alzheimer’s disease. The beneficial effects of pineal melatonin
and gut microbiome-derived butyrate may then be intimately linked to their
night-time capacity to induce the astrocyte mitochondrial melatonergic pathway,
thereby optimizing astrocytes in their regulation of neuronal activity and
survival. This would suggest that dementia, like cancer [26], may be importantly
determined by night-time, sleep linked processes, upon which the morning CAR
activation of the GR will act to regulate local microenvironment homeostasis. See
Fig. 2.
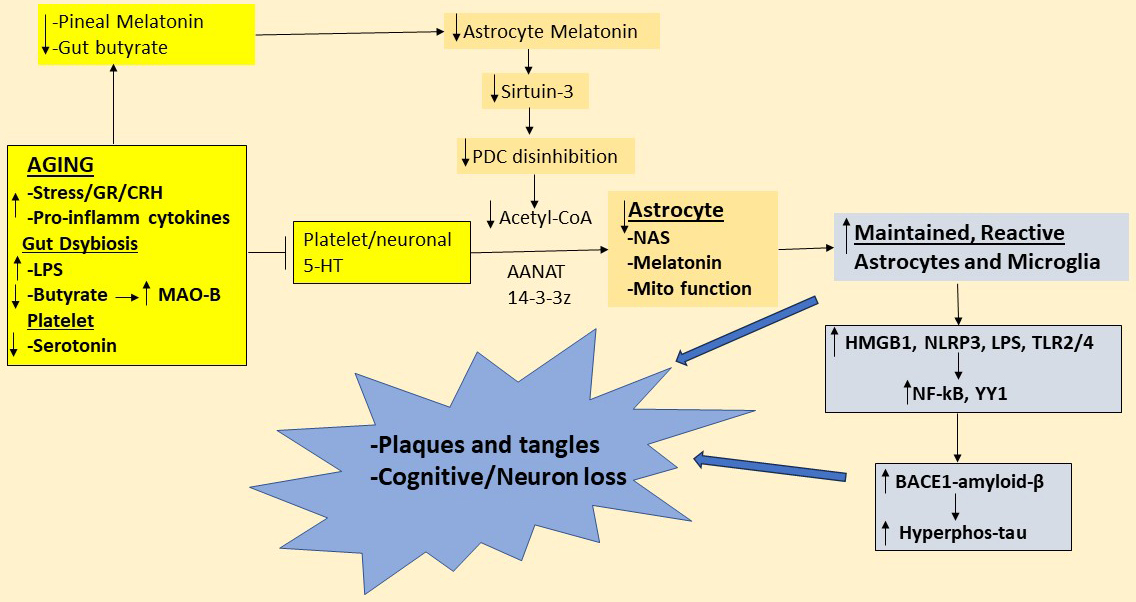 Fig. 2.
Fig. 2.
Shows how changes over aging suppress pineal melatonin and gut
microbiome-derived butyrate as well as platelet and neuronal serotonin (yellow
shade), which all contribute to suppress astrocyte (gold shade) melatonin and NAS
availability. This contributes to astrocyte mitochondrial dysfunction, with
enhanced astrocyte reactivity increasing microglia reactivity, thereby
contributing to maintained, raised levels of HMGB1 and LPS activation of TLR2/4
and increased NLRP3 inflammasome induced IL-1 and IL-18, further
contributing to the inflammatory milieu that enhance BACE1/amyloid- and
associated increase in hyperphosphorylated tau. The emergent plaques and tangles
in the ‘end-point chaos’ of later stage Alzheimer’s disease may therefore be
powerfully determined by factors, including systemic, that regulate the
availability of the astrocyte tryptophan-melatonin pathway. Abbreviations: 5-HT,
serotonin; acetyl-CoA, acetyl-coenzyme A; AANAT, aralkylamine
N-acetyltransferase; AhR, aryl hydrocarbon receptor; BACE1, -site
amyloid precursor protein-cleaving enzyme 1; CRH, corticotrophin-releasing
hormone; GR, glucocorticoid receptor; HMGB1, high-mobility group box; Hyperphos,
hyperphosphorylated; LPS, lipopolysaccharide; MAO-B, monoamine oxidase-B; Mito,
mitochondrial; NAS, N-acetylserotonin; NF-kB, nuclear factor
kappa-light-chain-enhancer of activated B cells; NLRP3, nucleotide-binding
domain, leucine-rich–containing family, pyrin domain–containing-3; PDC,
pyruvate dehydrogenase complex; TLR, toll-like receptor; YY1, yin yang 1.
3.4 The Aryl Hydrocarbon Receptor and Circadian Rhythm
Raised aryl hydrocarbon receptor (AhR) levels and activation are closely linked
to aging [104], including via disruption to the circadian rhythm [105]. As
highlighted in Fig. 1, AhR activation induces cytochrome P450 (CYP)1A2 and
CYP1B1, thereby O-demethylating melatonin to NAS as well as increasing melatonin
hydroxylation [106, 107]. The AhR is typically held in a cytoplasmic complex with
hsp90 and when activated by an AhR ligand translocates to the nucleus where it
forms a dimer with the AhR nuclear translocator (ARNT), thereafter inducing genes
with the xenobiotic response element (XRE) in their promotor. The AhR can also be
expressed on the mitochondrial membrane where it regulates Ca influx via
the voltage dependent anion channel (VDAC)1 [108] and interacts with the
mitochondria-located translocator protein kDa18 (TSPO) [109]. The diverse and
sometimes contrasting effects of the AhR may be partly determined by site of
translocation as well as by other alternatively sited mitochondria receptors,
such as the 7nAChR, TrkB and GR [110].
The AhR has numerous endogenous and exogenous ligands, including kynurenine
derived from pro-inflammatory cytokine induction of indoleamine 2,3-dioxygenase
(IDO) and cortisol/GR induction of tryptophan 2,3-dioxygenase (TDO). The array of
AhR ligands, site of expression, and the AhR regulation of the mitochondrial
melatonergic pathway allows AhR activation to have a complexity of consequences.
Such plasticity of AhR activation considerably complicates its role in aging
[111], whilst also highlighting the adaptability of the AhR in the regulation of
core cellular processes as determined over the course of evolution. As the
conversion of tryptophan to kynurenine by IDO and TDO also increases other
kynurenine pathway products, including neuroregulatory kynurenic acid and
quinolinic acid, AhR activation by kynurenine will be coupled to concomitant
AhR-independent effects of kynurenine pathway products on neuronal activity and
interarea communication across the brain. Given that 60% of brain kynurenine is
derived from the periphery [112], systemic processes that drive large kynurenine
increases, such as from white adipocyte (WAT) in obesity [113], can drive
significant changes in the CNS relevant to dementia pathophysiology, including
depriving tryptophan for the tryptophan-melatonin pathway, and kynurenic
acid/quinolinic acid neuroregulation as well as AhR activation.
By O-demethylating melatonin to NAS, thereby increasing TrkB activation, the AhR
is a significant contributor to Alzheimer’s disease pathophysiology. As noted,
NAS is a brain-derived neurotrophic factor (BDNF) mimic via its activation of the
BDNF receptor, TrkB [114]. TrkB activation of the TrkB-FL is a treatment target
for Alzheimer’s disease, given its trophic and metabolic benefits [115]. However,
in the presence of amyloid- the truncated TrkB-T1 is markedly increased
and significantly contributes to neuronal loss, as shown in Alzheimer’s disease
preclinical models [116, 117]. By increasing the NAS/melatonin ratio and
associated TrkB-T1 activation, the AhR and its ligands can contribute to
Alzheimer’s disease pathophysiology by a number of mechanisms. The loss of pineal
melatonin’s suppression of pro-inflammatory cytokines/IDO/kynurenine and
cortisol/GR/TDO/kynurenine will contribute to heightened AhR activation, thereby
increasing the NAS/melatonin ratio and heightened TrkB-T1 activation-linked
neuronal loss, especially in the presence of raised amyloid- levels. The
suppression of gut microbiome butyrate will also contribute to this via decreased
HDACi, thereby increasing GR nuclear translocation and TDO induction. As noted,
the marked suppression of pineal melatonin and gut derived butyrate will enhance
GR nuclear translocation, thereby modulating not only the HPA axis stress
response, but also the consequence of CAR as it ‘prepares the body for the coming
day’. Such processes allow the AhR to be intimately linked to the circadian
rhythm in the regulation of Alzheimer’s disease pathophysiology. See Fig. 1.
3.5 GR, MERTK, TrkB-T1, Melatonergic Pathway and Autoimmunity in
Alzheimer’s Disease
Interestingly, stress/GR activation of astrocytes induces MER proto-oncogene,
tyrosine kinase (MERTK), thereby driving astrocytes to phagocytose excitatory
synapses on to GABAergic neurons, which is mediated by the GR acting as a nuclear
transcription factor [118]. MERTK is one of the TAM receptors (Tyro3, Axl, and
Mertk), which are linked to immune regulation, cell differentiation and apoptotic
cell/debris clearance [119], with MERTK alleles associated with Alzheimer’s
disease risk, especially in females [120]. As to whether stress/GR-driven
increase in astrocyte MERTK, by driving astrocyte phagocytosis of excitatory
synapses onto GABAergic neurons, contributes to the increased glutamatergic
excitotoxicity in Alzheimer’s disease [121] requires investigation. As heightened
glutamatergic N-methyl-D-aspartate receptor (NMDAr) activation, including by
amyloid-, contributes to the raised TrkB-T1 levels in Alzheimer’s
disease [122], the loss of melatonin and butyrate suppression of GR nuclear
translocation will enhance GR-induced MERTK and GABAergic suppression and
heightened glutamatergic NMDAr activation, thereby increasing TrkB-T1 levels.
This indicates a role for circadian and systemic factors in the modulation of
substantial changes in how astrocytes regulate neuronal interactions and
activation. Notably, the GR can also increase BACE1 and amyloid- via
plasma membrane GR activation [123] as well as by regulating presenilin (PSEN)1
assembly on the endoplasmic reticulum (ER), thereby inducing amyloid-
accumulation on the ER mitochondrial associated membrane (MAM) [124]. The roles
of melatonin and butyrate in the modulation of these other routes of BACE1 and
amyloid- induction by stress/CAR activation of the GR have still to be
determined.
Whether the loss of night-time pineal melatonin and gut microbiome-derived
butyrate increases the morning CAR activation of astrocyte GR to not only
increase BACE1 and amyloid- but also contribute to MERTK induction and
heightened TrkB-T1 levels, especially in the presence of amyloid-, will
be interesting to determine. When this is coupled to an AhR-driven increase in
the NAS/melatonin ratio, thereby enhancing NAS activation of TrkB-T1 (on the
plasma and/or mitochondrial membranes), neuronal loss will be potentiated
[116, 117]. Overall, the circadian loss of night-time melatonin and gut
microbiome-derived butyrate will also be relevant to later stages of neuronal
loss in dementia, where heightened amyloid- and TrkB-T1 levels are
evident, as well as in the pathoetiology of dementia.
However, it is important to note the widespread mitochondrial dysfunction in
dementia cannot be simply understood as a consequence of various fluxes and
challenges, but rather mitochondrial function is a dynamic core aspect of
intercellular homeostatic interactions, with the suppression of the mitochondrial
melatonergic pathway a target for other cells in a given microenvironment when
the products of a challenged cell drive a prolonged maladaptive dyshomeostasis.
Such intercellular dyshomeostasis drives the pathoetiology of
‘autoimmune’/‘immune-mediated’ disorders [12, 33], where the regulation of the
mitochondrial melatonergic pathway in a ‘targeted’ cell (such as in pancreatic
-cells in T1DM [12]) is dynamically shaped by other cells in the local
microenvironment. This is most evident in the tumor microenvironment, where
cancers release kynurenine to activate the AhR to shape the metabolic function of
other tumor microenvironment cells in the course of generating ‘immune tolerance’
and shaping intercellular fluxes [59, 125].
The dynamic intercellular fluxes within a given microenvironment modulate
processes that shape mitochondrial function, including ROS production, thereby
driving ROS-dependent microRNAs (miRNAs) [126] that shape patterned gene
expression in an ever-changing dynamic over the circadian rhythm. Whether such
systemic and microenvironment determined mitochondrial ROS-driven miRNAs underpin
the increased TrkB-T1, especially in the presence of amyloid-, will be
important to determine over the course of dementia progression. How this is
coordinated with melatonergic pathway suppressing miRNAs, such as miR-7, miR-375
and miR-451 [127, 128, 129], may be of particular importance, given the use of
streptozotocin (a known melatonergic pathway inhibitor [130]) in preclinical
models of dementia [131] and autoimmune disorders [132]. Such suppression of
mitochondrial melatonin would overlap neuronal loss in Alzheimer’s disease with
that of Parkinson’s disease, where ‘autoimmune’/‘immune-mediated’ processes
involving CD8 t cell chemoattraction may be driven by decreased melatonin
regulation of PINK1/parkin-mediated mitophagy [133] and associated upregulation
of major histocompatibility complex (MHC)-1 [33], driving neuronal [134, 135] and
possibly microglia [136] destruction. This is supported by data showing the
presence of heightened CD8 t cells levels in the Alzheimer’s disease brain
in proximity to amyloid plaques [137, 138], which is strengthened further by data
showing CD8 t cells to drive neuronal loss in Alzheimer’s disease models
[134].
Such data indicates that the suppression of the mitochondrial melatonergic
pathway is a significant driver of dysregulated mitophagy via the loss of
melatonin’s regulation of PINK1/parkin/mitophagy [133], arising from systemic
(pineal melatonin, gut butyrate, CAR/GR, WAT kynurenine) changing the dynamic
interactions in a given microenvironment where prolonged inflammation and
microbial/viral-type signaling (TLR2/4/9) drives a dyshomeostasis that cannot be
dealt with by the immediately responding immune cells (astrocytes and microglia),
leading to the chemoattraction (via MHC-1) of CD8 t cells that drives cell
destruction (predominantly neurons but also glia). This has parallels to the
course of acute viral infection, as evident in the COVID-19 pandemic [139], where
the immune response is initially determined by ‘first responders’ (such as
neutrophils, macrophages and mast cells) but is taken over by the immune system
‘second wave’ (CD8 t cells and natural killer cells). Neuronal loss in
Alzheimer’s disease therefore has significant ‘autoimmune’/‘immune-mediated’
overlaps with neuronal loss in the Parkinson’s disease substantia nigra pars
compacta [33, 140].
Overall, the suppressed capacity to maintain the mitochondrial melatonergic
pathway in astrocytes may be an important initiator of dementia pathoetiology
with the subsequent protracted pathophysiology involving alterations in dynamic
mitochondrial homeostatic interactions in a given microenvironment. Systemic and
local microenvironment processes interact to suppress the mitochondrial
melatonergic pathway, thereby initiating the chemoattraction of CD8 t cells
and the ‘autoimmune’/‘immune-mediated’ destruction of neurons that underpin the
progressive cognitive loss in dementia. The above suggests changes in the nature
of Alzheimer’s disease pathophysiology over time, including in the nature of
immune involvement, rather than a static progression of the same
pathophysiological processes. This may be more akin to concepts of ‘staging’ in
neuropsychiatric conditions, such as bipolar disorder [141], likely in
association with distinct treatments at different physiologically determined
‘stages’ [142]. Such ever-changing processes ultimately culminate in the
‘end-point chaos’ of the currently defined late-stage Alzheimer’s disease. See
Fig. 3.
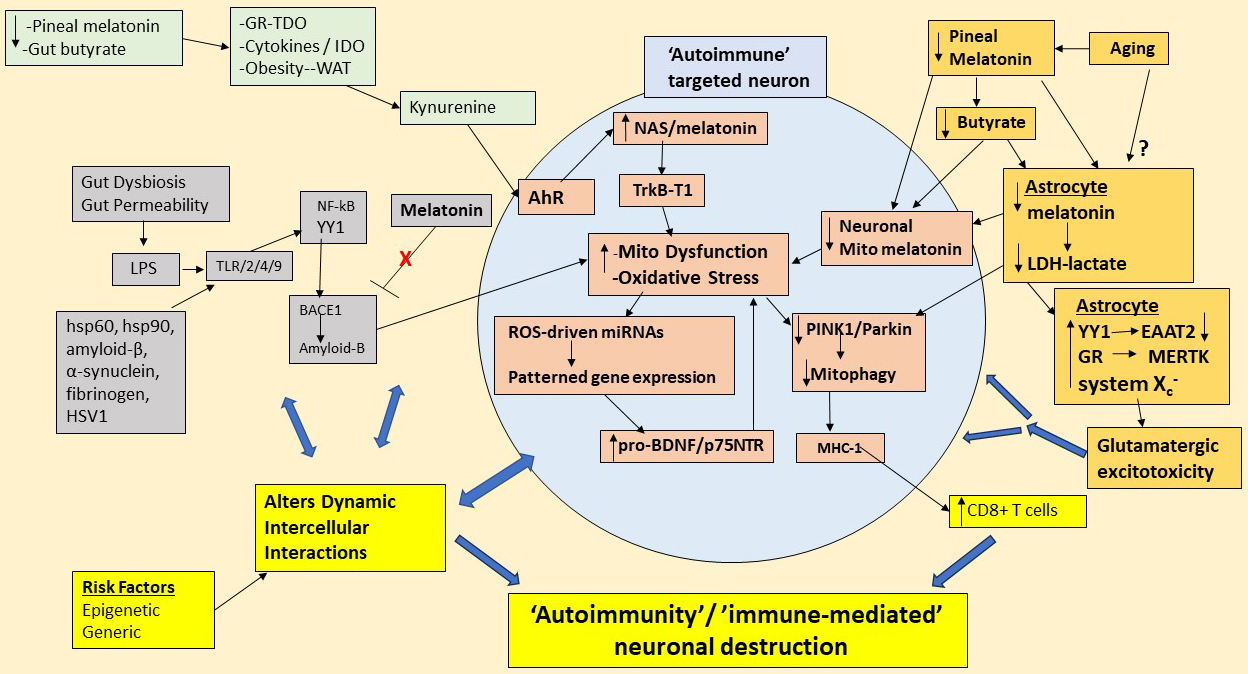 Fig. 3.
Fig. 3.
Shows how gut dysbiosis, gut permeability, pro-inflammatory
cytokines, GR, AhR, NAS/melatonin ratio, and decreased pineal melatonin act
primarily on astrocytes to decrease astrocyte melatonin release, thereby
suppressing the anti-inflammatory and mitochondria optimizing effects of
autocrine and paracrine melatonin. Consequently, astrocyte and neuronal BACE1
and amyloid- continue to be produced in association with raised tau
hyperphosphorylation, thereby driving the endpoint ‘chaos’ of plaques and tangles
that classically define Alzheimer’s disease. The decreased astrocyte
mitochondrial melatonin at night is at least partly mediated via a decrease in
pineal melatonin and gut microbiome-derived butyrate, although there may be more
aging-associated factors that also suppress the astrocyte tryptophan-melatonin
pathway (top right-hand, gold shade). The loss of pineal and astrocyte melatonin
suppresses lactate dehydrogenase (LDH) and therefore lactate production by
astrocytes, thereby depriving energy for neuronal mitochondria and decreasing
neuronal mitochondria melatonergic pathway induction. The suppression of
astrocyte melatonin maintains the NF-kB induction of YY1, thereby decreasing
EAAT2, which, like the increased System X and MERTK, enhances
glutamatergic excitotoxicity that raises neuronal apoptotic susceptibility and
contributes to spreading apoptotic susceptibility (gold shade). Gut derived LPS,
as well as other TLR2/4/9 ligands (grey shade) induce NF-kB and YY1 to increase
BACE1 and amyloid-, which in the absence of melatonin leads to prolonged
amyloid- production that exacerbates neuronal mitochondrial function.
Decreased pineal melatonin and gut butyrate will heighten the GR-TDO,
pro-inflammatory cytokine-IDO and white adipocyte (WAT) derived kynurenine to
activate the AhR. By increasing NAS and suppressing melatonin, the AhR will
contribute to neuronal susceptibility via oxidative stress induced TrkB-T1, which
NAS activates. Oxidative stress and neuronal mitochondrial dysfunction also
increase pro-BDNF and its ligand, p75, which also contributes to neuronal
vulnerability as well as dysregulating patterned immune responses. The neuronal
mitochondrial dysfunction and suppressed melatonin decrease
PINK1/parkin/mitophagy, and increase MHC-1, which chemoattracts CD8 t cells
that drive neuronal destruction (yellow shade). Such immune-mediated processes
will be contributed to, if not initiated by, alterations in the homeostatic
interactions in the neuronal microenvironment, which will be subject to
epigenetic and genetic risk factors (yellow shade). Abbreviations: AhR, aryl
hydrocarbon receptor; BACE1, -site amyloid precursor protein-cleaving
enzyme; BDNF, brain-derived neurotrophic factor; EAAT, excitatory amino acid
transporter; GR, glucocorticoid receptor; hsp, heat shock protein; HSV1, herpes
simplex virus; IDO, indoleamine 2,3-dioxygenase; LDH, lactate dehydrogenase; LPS,
lipopolysaccharide; MERTK, MER proto-oncogene, tyrosine kinase; MHC-1, major
histocompatibility complex-class 1; Mito, mitochondria; NAS, N-acetylserotonin;
NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; PINK1,
PTEN-induced kinase 1; ROS, reactive oxygen species; TDO, tryptophan
2,3-dioxygenase; TLR, toll-like receptor; TrkB-T1, tyrosine kinase receptor
B-truncated; WAT, white adipocyte; YY1, yin yang 1.
4. Hypothalamic Interactions with Circadian Rhythm and Systemic
Processes
As well as the gut microbiome, pineal melatonin, and CAR/HPA axis,
conceptualizing Alzheimer’s disease as a systemic disorder may require the
incorporation of wider regulators of circadian and systemic processes, including
the hypothalamus. The hypothalamus is classically determined by variations in
neuronal release of various peptides that regulate ‘basic animal’ processes, such
as reproduction, aggression, sex drive, appetite, thirst, and social bonding.
Although alterations in hypothalamic function have long been identified in
Alzheimer’s disease [143, 144], the relative paucity of amyloid- levels
and effects have led to this brain regulator of core systemic processes being
relatively little investigated out-with the hypothalamic role in initiating the
HPA axis [145]. However, hypothalamic cells, including tanycytes and astrocytes
are important regulators of many systemic processes linked to Alzheimer’s disease
risk, including obesity, T2DM and glycemic dysregulation [146]. Integrating
alterations in wider hypothalamic function may therefore be an overlooked aspect
of Alzheimer’s disease pathoetiology.
The most investigated nucleus of the hypothalamus is the paraventricular nucleus
(PVN), primarily due to PVN neurons initiating CAR and the HPA
axis/stress response linked to Alzheimer’s disease pathophysiology [147]. The
cortisol awakening response (CAR) is regulated by suprachiasmatic nucleus (SCN)
derived vasoactive intestinal peptide (VIP) projections to the PVN
neurons, with VIP increasing CRH release to initiate the stress/CAR circadian
rhythm [148]. The circadian timing of SCN release is determined by the
day, vs night, GABA uptake by the astrocyte GABA transporter (GAT)3, allowing
increased extracellular GABA at night to suppress VIP levels [149]. Such
astrocyte regulation of VIP may be of some importance, given preclinical data
showing chronic VIP administration from an early age to significantly decrease
amyloid- plaques and preserve susceptible brain areas against atrophy in
an Alzheimer’s disease preclinical model [150]. As well as being regulated by
astrocytes, VIP also regulates astrocyte function by increasing glycolysis-driven
lactate provision as an energy source for neurons where lactate is converted to
pyruvate [151]. The conversion of glucose to pyruvate in astrocytes is proposed
to favor the conversion of pyruvate to lactate and the relatively suppressed
utilization of pyruvate in the production of acetyl-CoA by the astrocyte PDC
under conditions of neuronal activity/challenge and hypoxia [151]. Whether this
is dependent upon the availability of the astrocyte mitochondrial melatonergic
pathway, including in the course of aging when the astrocyte mitochondrial
melatonergic pathway may be suppressed will be important to determine.
Preclinical data indicates that the genetic downregulation of astrocyte lactate
dehydrogenase generates memory deficits that are only evident in the aged [152].
Are variations in the astrocyte mitochondrial melatonergic pathway significant
regulators of the astrocyte-neuron lactate shuttle? This is indirectly supported
by data in Sertoli cells where melatonin increases lactate dehydrogenase mRNA and
protein levels [153], suggesting that lost astrocyte (and perhaps pineal)
melatonin suppresses pyruvate conversion to lactate, thereby depriving neurons of
their major source of metabolism. Is suppressed astrocyte melatonin (and/or
pineal melatonin) over aging thereby intimately linked to a decrease in astrocyte
lactate dehydrogenase, thereby increasing the availability of pyruvate for PDC
conversion to acetyl-CoA? Would this suggest that the maintenance of the
astrocyte mitochondrial melatonergic pathway carries precedence of astrocyte
lactate production for neurons?
Is suppressed astrocyte mitochondrial melatonergic pathway over aging associated
with a compensatory increase in the activity of the cystine-glutamate antiporter
(system X)? If so, the need for astrocytes to compensate melatonergic
pathway suppression by system X upregulation would be expected to
associate with heightened glutamatergic excitotoxicity in neurons. System
X is upregulated in many neurodegenerative disorders, including ALS,
Parkinson’s disease and Alzheimer’s disease [154]. These authors showed that TLR4
signaling by LPS increases system X in glia [154], which would seem
not unlikely to be further increased under prolonged TLR2/4 signaling that fails
to induce the NF-kB/YY1 induction of the astrocyte melatonergic pathway. In such
circumstances, the astrocyte GSH production would preserve astrocytes but would
not provide the autocrine and paracrine anti-inflammatory effects of melatonin.
As indicated above, heightened glutamatergic activity contributes to an
excitotoxicity driven neuronal loss in Alzheimer’s disease [121], with enhanced
glutamatergic NMDAr activation contributing to heightened TrkB-T1 levels,
decreased BDNF, and increased neurotoxic pro-BDNF release to activate heightened
p75 receptors in Alzheimer’s disease [122, 155], thereby accelerating
amyloid- production and cognitive loss, as shown in preclinical models
[156]. Such glutamatergic processes contribute to the emergence of seizures in
Alzheimer’s disease [7] and well as linking depression pathophysiology to
dementia [157]. Raised circulating pro-BDNF levels also alter immune cell
function, contributing to the immune dysregulation in Alzheimer’s disease and how
this overlaps depression with dementia [69, 158]. The suppression of the astrocyte
melatonergic pathway and associated system X upregulation will
further contribute to raised glutamatergic signaling, thereby spreading
excitotoxic signaling to other regions and layers within the cortex and limbic
system. YY1 upregulation will also exacerbate glutamatergic excitotoxicity via
the YY1 suppression of the astrocyte excitatory amino acid transporter (EAAT)2,
thereby increasing glutamate availability at the synapse [159]. As YY1 is highly
regulated by HDAC effects at the promotor of YY1 induced genes [160], the loss of
the HDACi capacity of gut microbiome-derived butyrate would be expected to
contribute to further EAAT2 suppression.
Although classically conceptualized as providers of lactate and trophic support
to neurons, it is important to note that astrocytes are functional immune cells.
As in any condition when immune cells become dysregulated, their homeostatic
interactions with other cells can dramatically change, as shown in COVID-19
fatalities that were primarily mediated by immune cell responses [161]. The above
would indicate that the suppressed mitochondrial melatonergic pathway (as well as
suppressed pineal melatonin) in Alzheimer’s disease drives compensatory processes
in astrocytes (such as system X upregulation and increased glutamate
release) that compromises neuronal survival. This seems driven by suppressed
astrocyte mitochondrial function inhibiting neuronal mitochondrial function via
decreased neuronal lactate availability for conversion to pyruvate and thereby
suppressing PDC/acetyl-CoA and mitochondrial melatonergic pathway induction in
neurons. As noted, the autocrine and paracrine effects of astrocyte melatonin
would suppress TLR2/4-BACE1-amyloid- as well as the hyperphosphorylation
of tau, thereby being the major driver of classically defined Alzheimer’s disease
pathophysiology. The above would suggest that amyloid- derived plaques
and hyperphosphorylated tau driven tangles are downstream consequences of
dysregulated glia mitochondrial melatonergic pathway dysregulation.
Amyloid- and hyperphosphorylated tau are therefore not useful treatment
targets but are simply downstream consequence of a dysregulated astrocyte
mitochondrial melatonergic pathway that contributes to wider pathophysiological
processes as regulated by alterations in the circadian rhythm and gut microbiome
(including suppressed EAAT2 and enhanced system X). Such
dysregulation may be passed on to other neurons via heightened glutamatergic
activity, in the presence of a suppressed astrocyte melatonergic pathway at sites
to which such overly excited neurons project. As in Parkinson’s disease, the
suppression of the melatonergic pathway will enhance mitophagy dysregulation,
driving an increase in oxidant-induced MHC-1, leading to
‘autoimmune’/‘immune-mediated’ destruction of neurons by infiltrating CD8 t
cells [33, 34, 140]. See Fig. 3.
The above provides a framework for linking and investigating wider
pathophysiological processes. The dramatic suppression of pineal melatonin in the
circulation and third ventricle over the course of aging is relevant to SCN
regulation, including the SCN regulation [162], and thereby to
PVN neuronal regulation and the CAR/HPA axis. Notably, PVN
neuronal activation is significantly suppressed by PVN oxytocin neurons, which
seems likely to be mediated by the release of oxytocin in dense core vesicles
that act to on oxytocin receptors on PVN astrocytes. This would parallel the
effects of PVN oxytocin neuronal projections to the central amygdala where
oxytocin acts on astrocyte oxytocin neurons to suppress CRH release and the
consequent CRH induction of the dynorphin/kappa-opioid receptor activation that
seems to underpin the dysphoria evident in mood and affective disorders as well
as in PCOS [163, 164].
This may have relevance to Alzheimer’s disease. Data shows melatonin, SCN
melatonin receptor (MT1r) and VIP to be significantly decreased in later stages
of dementia [165], implicating suppression of pineal melatonin with not only
alterations in GR nuclear translocation but also in the timing and amplitude of
the CAR/HPA axis. Oxytocin is popularly associated with social
processes/bonding/cognition [166], with social isolation being a susceptibility
and accelerating factor in Alzheimer’s disease [167]. Notably, the oxytocin
receptor is significantly regulated by HDAC [168], indicating that the loss of
gut microbiome-derived butyrate will impact on the level and influence of
astrocyte oxytocin receptors in the modulation of CRH.
5. Future Research and Treatment Implications
It is becoming increasingly clear in recent decades that understanding neuronal
loss in dementia has to progress from a conception of ‘good’ (BDNF) and ‘bad’
(plaques and tangles) to a conception that embraces and incorporates the
complexity of data pertaining to Alzheimer’s disease. The above goes some way to
incorporate most of the detailed data on Alzheimer’s disease pathophysiology. A
number of future research directions and treatment/prevention implications are
detailed below.
5.1 Future Research Implications
Whether local paracrine melatonin release from astrocytes regulates local
7nAChR levels, thereby impacting on immune/glia inflammation and
associated consequences on cognition will be important to determine.
Whether the suppressed capacity to induce astrocyte melatonin over aging has
heightened consequences for ApoE4 carriers will be important to clarify.
There is a growing appreciation of the role of platelets, and their regulation
by gut microbiome-circadian interactions, in the pathophysiology of an array of
diverse medical conditions, including Alzheimer’s disease, amyotrophic lateral
sclerosis, and cancer [169]. The role of gut microbiome-circadian interactions in
the modulation of Alzheimer’s disease pathophysiology will be important to
determine, including via the regulation of platelet serotonin as a precursor for
the mitochondrial melatonergic pathway.
The importance of the gut microbiome in Alzheimer’s disease is indicated by
clinical and preclinical data indicating that the increased risk of Alzheimer’s
disease in the partners of Alzheimer’s disease patients may be mediated
cohabitation linked similarities in their gut microbiomes [170].
Whether the role of autocrine melatonin in switching from an inflammatory
M1-like phenotype in macrophages [35] and microglia [80] to a quiescent,
pro-phagocytic M2-like phenotype is paralleled in astrocytes will be important to
determine.
A number of processes may contribute to the increased glutamate/GABA ratio in
Alzheimer’s disease, including MERTK phagocytosis of excitatory inputs to GABA
neurons, YY1 suppression of EAAT2, and increased System X. It will be
important to determine the relative influences of such processes, including
heightened glutamatergic activity induction of pro-BDNF, TrkB-T1 and the
p75 in driving neuronal loss and Alzheimer’s disease pathophysiology.
Whether the loss of night-time pineal melatonin and gut microbiome-derived
butyrate increases the morning CAR activation of astrocyte GR to not only
increase BACE1 and amyloid- but also contribute to MERTK induction and
heightened TrkB-T1 levels, perhaps especially in the presence of
amyloid-, will be important to determine.
The conversion of glucose to pyruvate in astrocytes favors the conversion of
pyruvate to lactate, with relatively little pyruvate utilized by the astrocyte
mitochondrial PDC, as evident in neurons under challenge, during neuronal
activity and in conditions of hypoxia [151]. Whether this is dependent upon the
availability of the astrocyte mitochondrial melatonergic pathway, including in
the course of aging when the astrocyte mitochondrial melatonergic pathway may be
suppressed will be important to determine.
Whether a suppressed astrocyte mitochondrial melatonergic pathway underpins
System X upregulation in a quest to regulate astrocyte antioxidant
status by GSH provision will be important to determine. Whether the loss of
astrocyte (and/or pineal) melatonin decreases astrocyte lactate dehydrogenase,
thereby decreasing the conversion of pyruvate to lactate will be important to
determine. As astrocyte lactate is converted to pyruvate in neurons, thereby
enhancing neuronal mitochondrial function, acetyl-CoA and neuronal mitochondrial
melatonergic pathway induction, the suppression of astrocyte melatonin may have
dramatic consequences for neuronal function, as well as the loss of astrocyte and
neuronal melatonin increasing levels of hyperphosphorylated tau. This is
important for future research to determine.
5.2 Treatment Implications
The above systemic conceptualization of Alzheimer’s disease and its emphasis on
astrocytes and the astrocyte melatonergic pathway as a crucial hub has a number
of treatment implications that will be better clarified when the astrocyte
melatonergic pathway is more extensively investigated. However, a number of
treatment targets are readily apparent.
(1) The utilization of melatonin and monitoring of the gut microbiome to
optimize butyrate production is likely to suppress Alzheimer’s disease
pathoetiology for those genetically at risk and in the early stages of mild
cognitive impairment. The utility of melatonin and butyrate is likely to wane as
dementia progresses, although this has still to be systematically investigated.
(2) A number of factors may act to suppress the astrocyte tryptophan-melatonin
pathway, including suppressed 14-3-3 isoforms, serotonin and acetyl-CoA. Whether
the adjunctive use of tryptophan supplements with melatonin and butyrate affords
any added protection will be important to determine, given their safety profiles
and ready availability.
(3) A number of AhR inhibitors show utility in Alzheimer’s disease preclinical
models, including green tea’s epigallocatechin gallate [171], curcumin [172] and
resveratrol [173], with efficacy often being modelled on diverse physiological
processes, such as the induction of sirtuin-1 [174], although all dampen
inflammatory processes in astrocytes [172, 175, 176], whilst also inhibiting and
regulating the AhR [177, 178, 179]. As well as helping to preserve melatonin levels
via AhR inhibition, these nutriceuticals can increase the tryptophan-melatonin
pathway via other mechanisms, including via the inhibition of MAO, thereby
increasing serotonin availability [180, 181, 182]. Increasing serotonin
availability from dorsal raphe neurons as well as platelets is likely to have
utility under conditions when the AhR is relatively suppressed [169].
(4) Although requiring more technical development, the utilization of
mesenchymal stem cell exosomes that target the astrocyte and/or pinealocyte
tryptophan-melatonin pathway would allow a more precise treatment focus on key
hubs in Alzheimer’s disease pathophysiology.
(5) It is important to mention that social processes and physical contact can
have important physiological consequences that can modulate Alzheimer’s disease
pathophysiology, as highlighted by the acceleration of dementia by loneliness,
with effects at least partly mediated via the regulation of the HPA axis [183].
(6) Some of the physiological consequences of social interaction may be mediated
via the upregulation of hypothalamic and amygdala oxytocin and the oxytocin
suppression of stress/dysphoria associated CRH via hypothalamic and amygdala
astrocytes [163, 164]. Recent work indicates that oxytocin intranasal
administration has a number of benefits, including cognitive in Alzheimer’s
disease patients [166].
6. Conclusions
There is a growing dissatisfaction with lack of progress in understanding
Alzheimer’s disease pathophysiology, and the consequent lack of any plausible
treatment. It seems clear that there is more to Alzheimer’s disease than
amyloid- and its annihilation by anti-amyloid antibodies [1]. Given the
increases in amyloid- in numerous other medical conditions and data
showing amyloid- to be an endogenous antimicrobial, it is clear that
amyloid- is part of a dysregulated inflammatory process. The data
reviewed above highlight the role of systemic processes, including the circadian
rhythm (pineal melatonin and cortisol awakening response), gut
microbiome/permeability (LPS and butyrate) and white adipocytes (kynurenine
activation of the AhR) that interact to modulate astrocyte function. As brain
‘immune type’ cells, astrocytes have a powerful role in the regulation of
neuronal survival and function, including by the provision of energy (lactate)
and antioxidants. Astrocyte dysregulation is therefore a major problem for
neuronal survival and function. Over the course of aging, there is a 10-fold
decrease in pineal melatonin, leading to the loss of its antioxidant,
anti-inflammatory and mitochondria optimizing effects, which has significant CNS
and systemic consequences. Astrocytes have long been known to constitutively
produce and release melatonin. Whether astrocyte melatonin is decreased, as in
the pineal gland, over aging is surprisingly unknown, especially as exogenous
melatonin has been extensively shown to prevent amyloid- induced
neuronal loss in preclinical models. The two transcription factors that
upregulate BACE1 and amyloid- production, NF-kB and YY1, have been shown
in other cell types to induce melatonin, suggesting that it may be the loss of
concurrent/sequential melatonin in astrocytes that underpins the prolonged
amyloid- production. Suppressed astrocyte melatonin would be compatible
with the System X upregulation in astrocytes to acquire antioxidant
support by glutathione synthesis, which has the unfortunate consequence of
increasing glutamatergic excitotoxicity, further contributing to a spreading
neuronal loss. The terminal process in neuronal death seems to arise as a
consequence of decreased astrocyte lactate provision and the incapacity of
neurons to optimize mitochondrial function, including the mitochondrial
melatonergic pathway. The loss of neuronal melatonin drives an oxidant-driven
decrease in mitophagy and increase in MHC-1, which chemoattracts CD8 t
cells, implicating ‘autoimmune’/‘immune-mediated’ processes as a final stage of
neuronal death in Alzheimer’s disease. This provides a number of viable research
and treatment targets, the investigation of which should clarify more appropriate
management that is targeted to core pathophysiological processes in both the
prevention and treatment of dementia.
Abbreviations
5-HT, serotonin; 5-HTTP, 5-hydroxytryptophan; 7nAChR, alpha 7nicotinic
acetylcholine receptor; AADC, aromatic-L-amino acid decarboxylase; AANAT,
aralkylamine N-acetyltransferase; acetyl-CoA, acetyl-coenzyme A; ACTH,
adrenocorticotropic hormone; AhR, aryl hydrocarbon receptor; ALS, amyotrophic
lateral sclerosis; AMPK, AMP-activated protein kinase; Apo, apolipoprotein; ASMT,
N-acetylserotonin O-methyltransferase; BAG-1, bcl-2 associated athanogene 1; BAT,
brown adipocyte; BDNF, brain-derived neurotrophic factor; CAR, cortisol awakening
response; CRH, corticotrophin releasing hormone; CSF, cerebrospinal fluid; CYP,
cytochrome P450; EAAT, excitatory amino acid; GPR, G-protein coupled receptors;
GR, glucocorticoid receptor; GRE, glucocorticoid receptor element; HDAC, histone
deacetylase; HMGB, high-mobility group box; HPA, hypothalamic-pituitary-adrenal;
hsp, heat shock protein; IDO, indoleamine 2,3-dioxygenase; LAT-1, large amino
acid transporter 1; MAO, monoamine oxidase; MERTK, MER Proto-Oncogene, Tyrosine
Kinase; MHC, major histocompatibility complex; NAS, N-acetylserotonin; NF-kB,
nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP,
nucleotide-binding domain, leucine-rich–containing family, pyrin
domain–containing-; OXPHOS, oxidative phosphorylation; PCOS, polycystic ovary
syndrome; PDC, pyruvate dehydrogenase complex; PINK1, PTEN-associated kinase 1;
PVN, paraventricular nucleus; SCN, suprachiasmatic nucleus; T1DM, type 1 diabetes
mellitus; TCA, tricarboxylic acid; TDO, tryptophan 2,3-dioxygenase; TIM,
mitochondrial import inner membrane translocase subunit; TOM, mitochondrial
import outer receptor subunit; TrkB-FL, tyrosine receptor kinase B-full length;
TrkB-T1, tyrosine receptor kinase B-truncated; VIP, vasoactive intestinal
peptide; WAT, white adipocyte.
Author Contributions
GA was responsible for the entire preparation of this manuscript.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of Interest
The author declares no conflict of interest.




