







1 Fondazione Italiana Fegato – ONLUS, Liver Research Center, 34149 Trieste, Italy
Academic Editor: Alessandro Poggi
Abstract
Discs large MAGUK scaffold protein 5 (DLG5) is a multi-domain member of membrane-associated guanylate kinase (MAGUK) family, which plays a major role in the maintenance of cell epithelial polarity being part of the SCRIB-LGL-DLG complex. Although polarity proteins have been generally considered tumor suppressors, recent discoveries led to reconsidering their role in cancer. This is also true for DLG5 in different cancer types, including hepatocellular carcinoma (HCC). In this cancer, DLG5 was negatively associated with malignant characteristics, however recent findings associated DLG5 expression with advanced stages of HCC. In vitro studies evidenced its possible role in sustaining cell growth and migration by the interaction with several intracellular pathways, such as Hippo, Hedgehog, and PI3K/AKT signaling pathways. In this review, we summarize the recent finding on the dual role of DLG5 and other polarity proteins in cancers. What emerges is a still undefined role of those proteins in cancers, especially in HCC, one of the most frequent cancers worldwide, where the function of DLG5 and other polarity proteins is still largely unexplored.
Keywords
- reviews
- DLG5
- polarity proteins
- EMT
- cancer
- liver cancer
- HCC
Discs large MAGUK scaffold protein 5 (DLG5) is a multi-domain member of the membrane-associated guanylate kinase (MAGUK) family, which plays a major role in the maintenance of cell epithelial polarity [1], a fundamental process for epithelial tissue integrity [2]. As a MAGUK protein, it carries the evolutionarily conserved adaptor domains src homology 3 (SH3), guanylate kinase (GUK), and multiple repetitions of the post-synaptic density protein 95/disc large tumor suppressor/zona occludens (PDZ) domain (Fig. 1) [1]. However, it is different from other family members in the presence of an N-terminal coiled-coil and a caspase recruitment domain CARD domains (Fig. 1) [3].
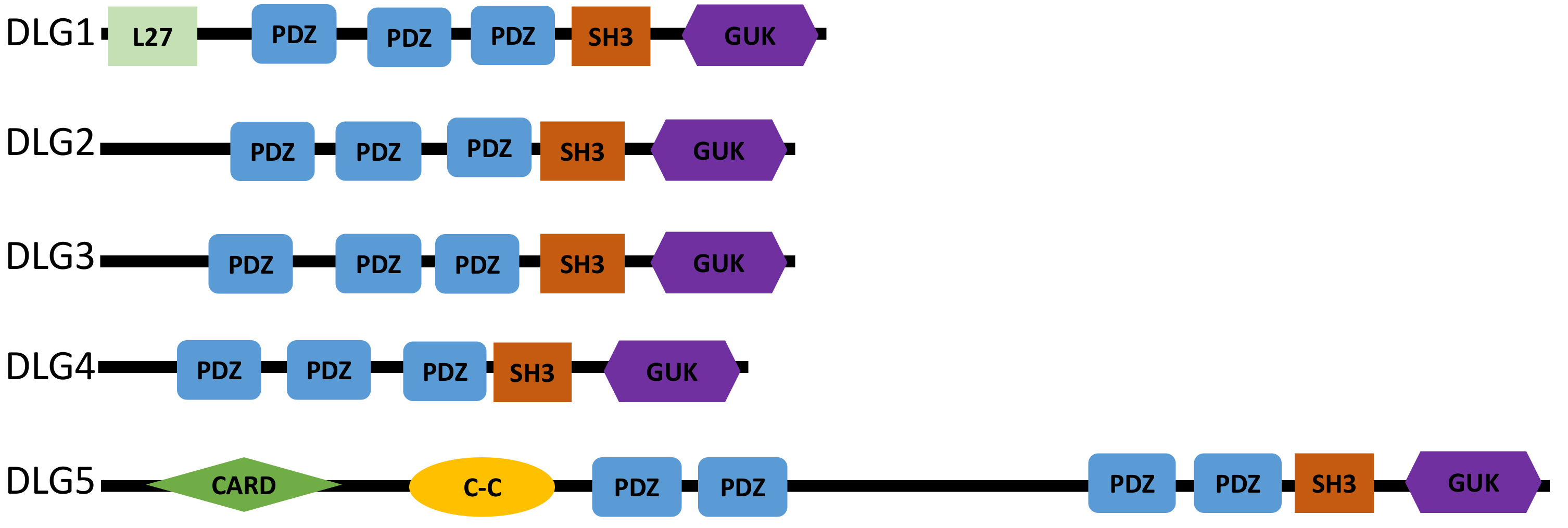 Fig. 1.
Fig. 1.Structure of the DLG human proteins. All the DLG proteins contain multiple PDZ domains, an SH3, and a GUK domain. DLG5 consists of an additional PDZ domain and an N-terminal coiled-coil and a CARD domain. Compared to the other DLG proteins, DLG5 has a longer protein sequence consisting of 1919 amino acids. C-C, N-terminal coiled-coil domain; PDZ, Post-synaptic density protein 95/disc large tumor suppressor/zona occludens domain; SH3, Src homology 3; GUK, Guanylate kinase domain.
DLG5 gene was firstly identified in humans in 1998 by Nakamura et al. [1], as a novel homolog of Drosophila tumor suppressor dlg and termed p-dlg. Since its discovery [1], it was clear the important role of DLG5 in maintaining the structure of epithelial cells and in transmitting extracellular signals to the membrane and cytoskeleton. Indeed, the pathogenic involvement of Dlg5 depletion has been investigated in knockout mice models, displaying epithelial polarity impairment and the presence of severe brain malformations and kidney disorders [3, 4]. In humans, DLG5 gene polymorphisms have been associated with an increased risk of Crohn’s disease [5, 6]. Since it is involved in cell proliferation pathways [1], functional and expression alterations of DLG5 have been related to cancer [7, 8, 9].
In mammals, three cell polarity complexes exist [10], the crumbs (CRB) [11] and partition defective (PAR) [12] complexes, which work for the maintenance of apical polarity, and the scribble (SCRIB)-LETHAL giant larvae homolog (LGL)-discs large homolog (DLG) complex (SCRIB-LGL-DLG complex) [13], that controls the basolateral polarity (Fig. 2). As part of the SCRIB-LGL-DLG complex, DLG5 is located at the basolateral membrane domain and is required for basolateral domain formation and maintenance (Fig. 2) [14]. Through the interaction with intracellular components, DLG5 acts as a scaffold protein involved in both epithelial cell structuring, adherens junction (AJ) formation, and intracellular signal transduction [14].
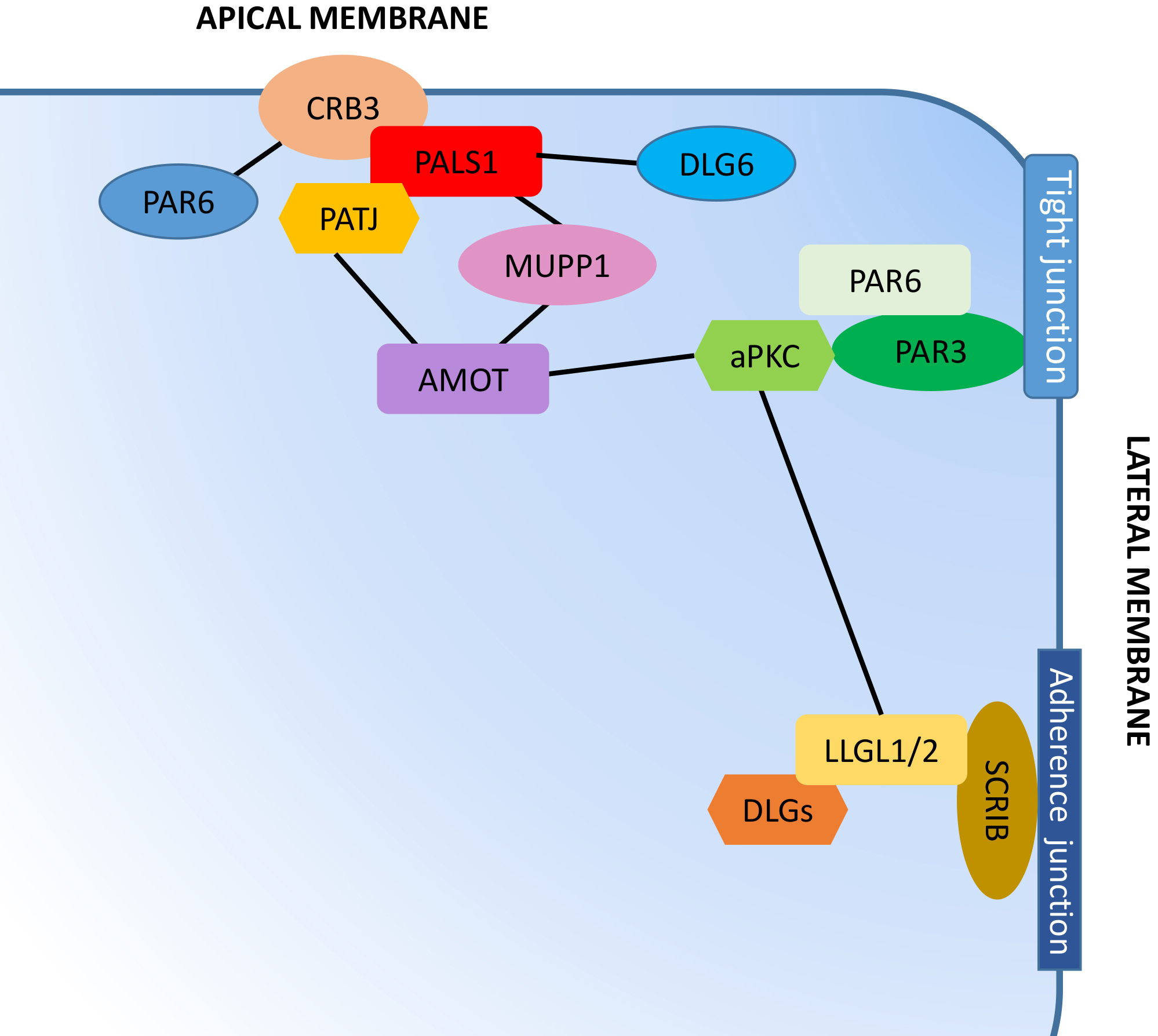 Fig. 2.
Fig. 2.Polarity protein complexes in human cells. The crumbs complex, consisting of crumbs3 (CRB3), protein associated with lin seven-1 (PALS1), and PALS1-Associated tight junction protein (PATJ), localizes at the apical membrane. The partitioning defective complex, consisting of partitioning defective 3 (PAR3), PAR6, and atypical protein kinase C (aPKC), is associated with a tight junction in the proximity of the apical membrane. The scrib complex, composed of scribble (SCRIB), Discs Large (DLG) proteins, and lethal giant larvae homolog (LGL) 1 or 2, localizes at the adherens junctions alongside the basolateral cell area. The dynamic composition of the individual complexes and the mutual antagonistic relationships, such as the LGL phosphorylation by aPKC that promotes its removal from the apical domains, is fundamental in determining the asymmetry, typical of apical-basal polarized cells. Black lines indicate the interaction between complexes and other proteins involved in cell polarity maintenance.
Studies on a DLG5–/– cell model revealed that DLG5 is required for the
membrane stabilization of N-cadherin and
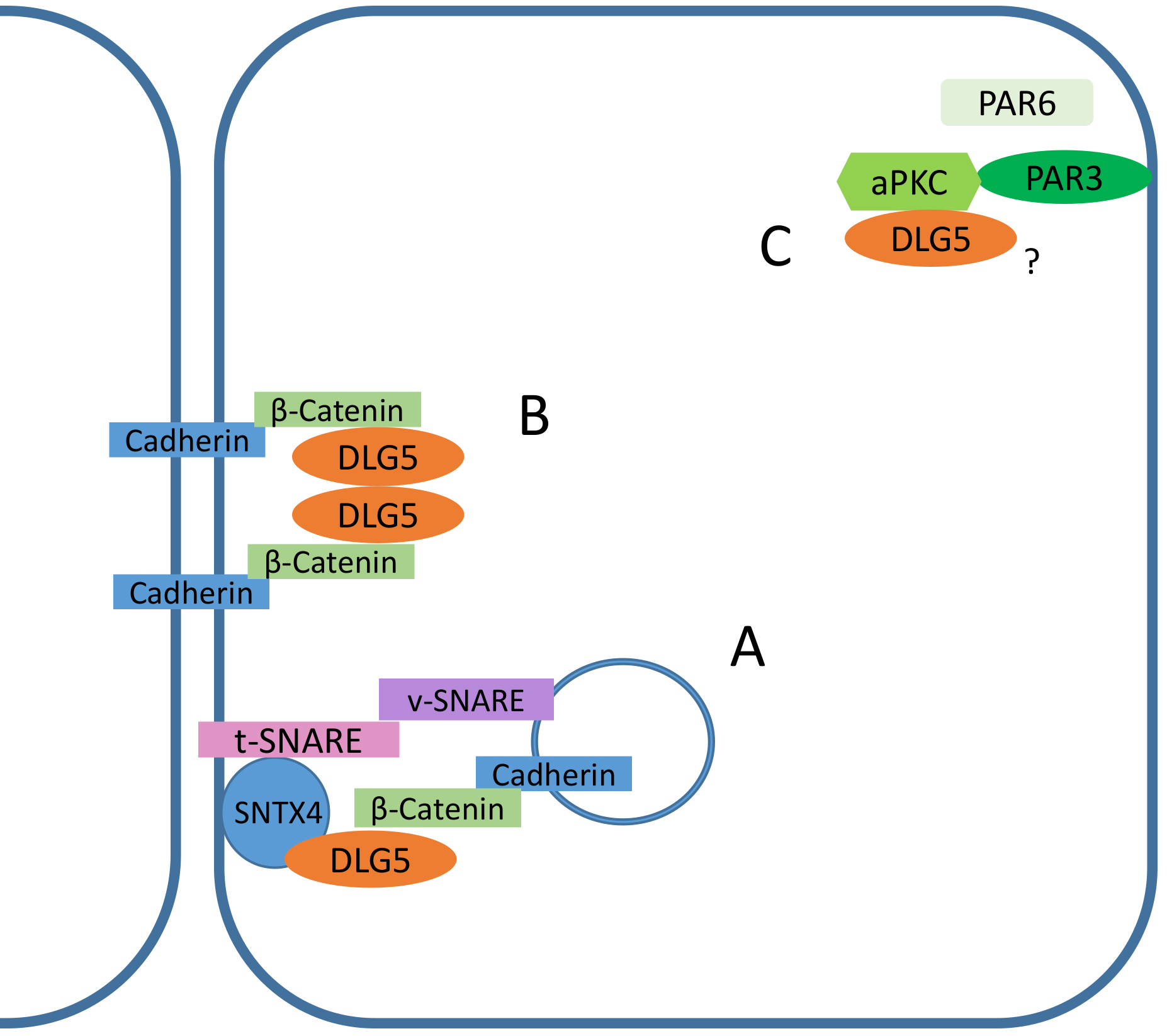 Fig. 3.
Fig. 3.Functional roles of DLG5 in epithelial cells. (A)
Although DLG5 is a member of the SCRIB-LGL-DLG complex deputed to the maintenance of the basolateral polarity, it can be found at the apical membrane where it facilitates the proper localization of atypical protein kinase C (aPKC), the only active enzymatic component of the PAR complex [15], necessary for the stabilization of tight junctions (Fig. 3) [16]. Indeed, in Dlg5–/– mice mutants, aPKC localized to the lateral membrane or cytoplasm, instead of the apical membrane of lung cells [15]. Since PAR3 in DLG5–/– lung epithelial cells correctly localizes to the apical membrane, it was hypothesized that DLG5 can work as an intermediator linking aPKC to the Par3-containing membrane domain [15].
Despite evidence linking DLG5 depletionto increased cell proliferation and epithelial-mesenchymal transition (EMT), some studies suggest a dual role for DLG5 in cancer. Studies on prostate cancer cells reported the concurrent involvement of DLG5 in cell proliferation induction and migration suppression, suggesting that DLG5 may act both as an oncogene and a tumor suppressor in the same cancer type (Table 1, Ref. [7, 8, 9, 17, 18, 19, 20, 21, 22, 23, 24, 25]). Increased DLG5 expression was detected in some advanced-stage hepatocellular carcinoma (HCC) and prostate cancer tissues [9, 26], suggesting a cancer-promoting role of DLG5. Supporting this hypothesis, the depletion of DLG5 in a model of lung carcinoma reduces cell resistance to etoposide and doxorubicin [17]. DLG5 overexpression was also observed in cells derived from pancreatic ductal adenocarcinoma (PDAC) and associated with an increase in cell growth [22]. In addition, DLG5 was also found to activate the Hedgehog (Hh) signaling through the binding of Smoothened (SMO) protein, promoting the activation of the transcription factor glioma-associated oncogene 2 (GLI2) [27], which may potentially result in the enhance of several carcinogenic pathways [18, 28]. Indeed, DLG5 promoted glioblastoma cell invasion and self-renewal by sequestering cullin3 (CUL3) and preventing GLI1 proteasomal degradation (Figs. 4,5), thus supporting the Hedgehog signaling [18].
| DLG5 expression | Cancer type | Effect in cancer | References |
| Down | Bladder cancer | Metastasis | [7] |
| Down | Breast cancer | Acquisition of stem cell characteristics, Tamoxifene resistance | [8] |
| Down | Breast cancer | Proliferation | [20] |
| Down | Hepatocellular carcinoma | Invadopodia formation | [19] |
| Down | Hepatocellular carcinoma | Cell proliferation | [23] |
| Down | Hepatocellular carcinoma | EMT | [24] |
| Down | Prostate cancer | Cell migration and invasion | [9] |
| Down | Squamous cell lung cancer | EMT, Poor prognosis | [25] |
| Up | Breast cancer and Hepatocellular carcinoma | Inhibit Hippo Pathway | [21] |
| Up | Glioblastoma | Cell vitality, invasion and self renewal | [18] |
| Up | Pancreatic cancer | Cell growth | [22] |
| Up | Non-Small Cell Lung Carcinoma | Etoposide and doxorubicin chemioresistance | [17] |
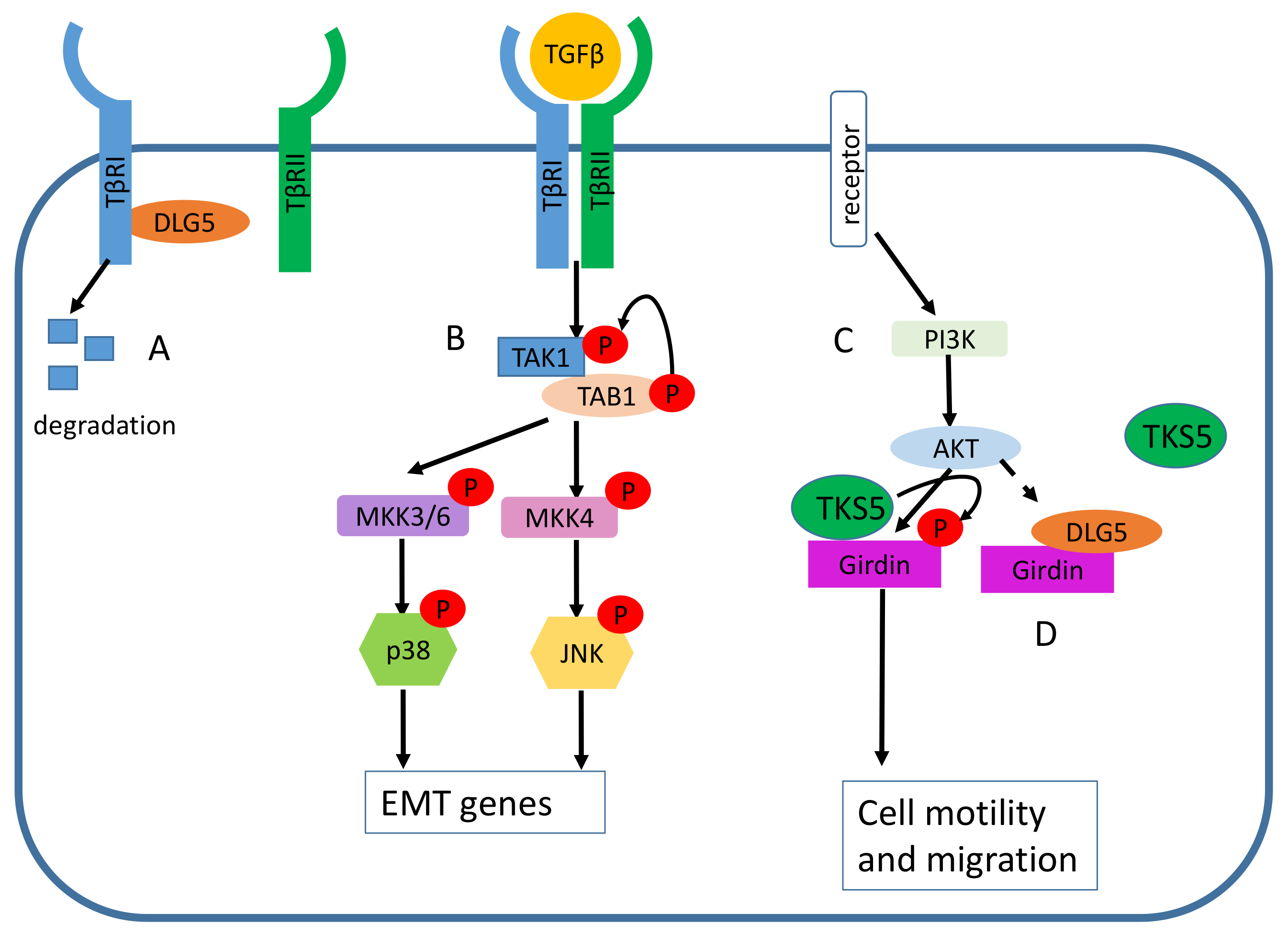 Fig. 4.
Fig. 4.The role of DLG5 in cancer prevention. (A) DLG5 induces the
degradation of the transforming growth factor-
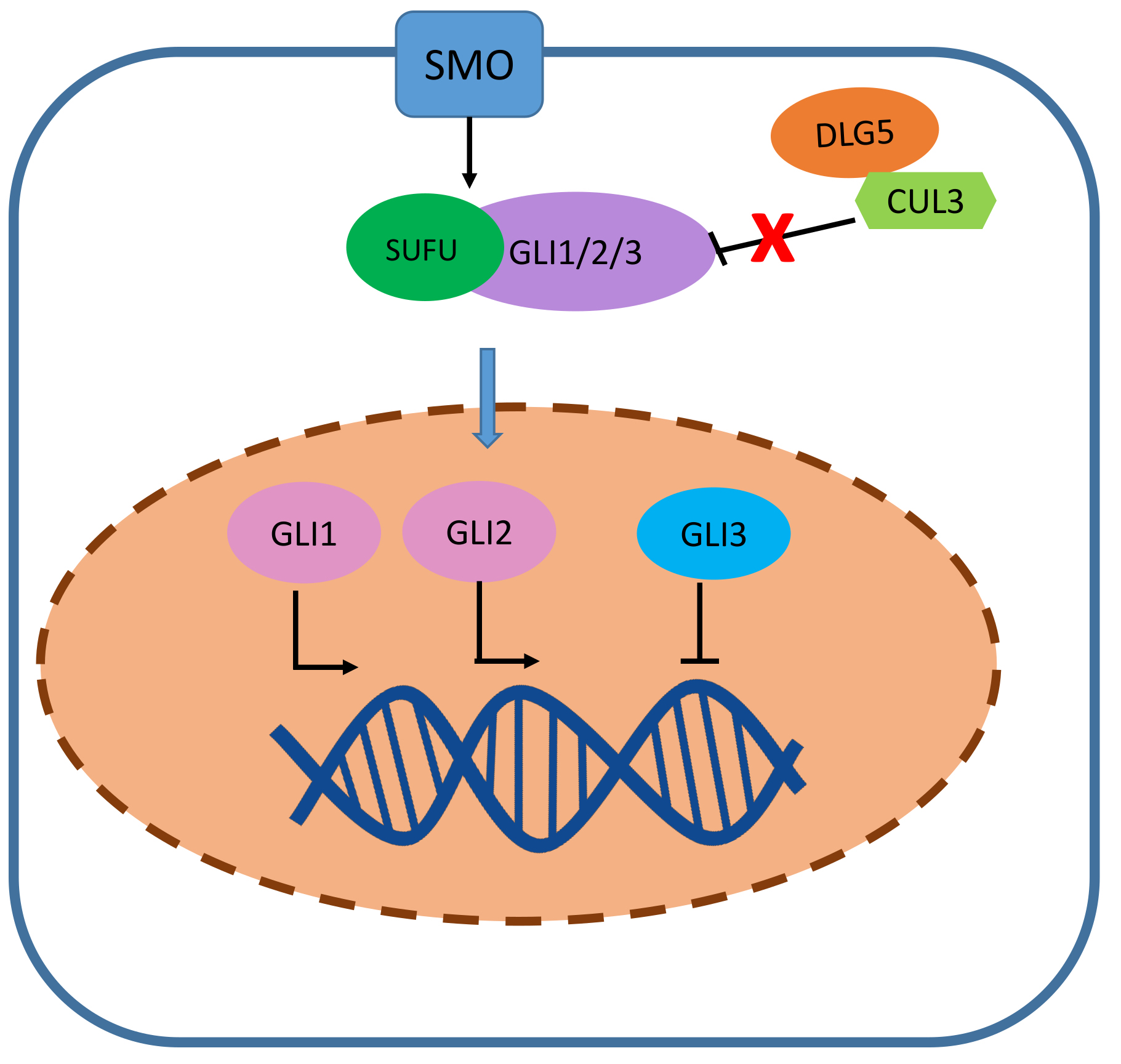 Fig. 5.
Fig. 5.The role of DLG5 in cancer promotion. In glioblastoma cells, by sequestering cullin 3 (CUL3), DLG5 prevents glioma-associated oncogene 1 (GLI1) proteasomal degradation, thus once the hedgehog pathway is activated GLI1/2 can migrate into the nucleus and promote the transcription of proliferation-related genes.
Dysregulation and miss-localization of polarity proteins is a common event in cancer that leads to a re-organization of the protein complexes and loss of epithelial polarity , determining the EMT and the alteration of tissue physiology [2]. Consistently, DLG5 is found to be downregulated in several solid cancer types, including hepatocellular carcinoma (HCC) [19], bladder [7], breast [8], and prostate cancer [9], with a consequent enhancement of multiple cancer processes, such as cell motility [7, 9, 19], proliferation [9, 20], and therapy resistance acquisition (Table 1) [8].
In bladder cancer tissues, the methylation-mediated silence of DLG5 expression
led to the promotion of cell invasiveness [7]. DLG5 loss detected in breast and
prostate cancer cell lines induced EMT associated with the down-regulation of the
epithelial markers, such as E-cadherin and tight junction protein 1 (TJP1), also
known as zona occludens 1 (ZO1), and with the increased expression of the
mesenchymal markers vimentin and N-cadherin [9, 20]. Sezaki et al.
[29, 30] revealed that the DLG5-mediated inhibition of EMT in a renal epithelial
cell line occurs via the degradation of the transforming growth factor-
DLG5 loss in a prostate cancer cell model promoted cell migration due to the Akt-mediated phosphorylation and activation of Girdin (Fig. 4) [9], a key component of the Phosphatidylinositol-4,5-Bisphosphate3-Kinase/serine/threonine-protein kinases (PI3K/Akt) signaling pathway, having a role in actin filament remodeling during cell motility, macrophage chemotaxis, autophagy, tumor angiogenesis [32, 33, 34].
DLG5 involvement in the suppression of the metastatic invasion was confirmed in a mouse model, in which the inoculation of Dlg5-silenced bladder cancer cells led to the onset of pulmonary metastasis [7].
As recently emerged, DLG5 plays a role in connecting cell polarity with intracellular pathways [21, 27], indeed some evidence linked DLG5 to the Hippo pathway. The Hippo tumor suppressor pathway plays an important role in regulating many biological processes, including cell proliferation, survival, differentiation, and organ size control [35]. In mouse embryos, the loss of Dlg5 leads to an impairment of brain development due to phosphorylation at the activation sites of mammalian ste20-like (MST) 1/2 kinases (MST1/2) and large tumor suppressor kinase 1/2 (LATS1/2) that, in turn, inhibits the transcription of proliferation genes by promoting the degradation of yes1 associated transcriptional regulator (YAP) [21]. Indeed, the phosphorylation of YAP on Serine 127 by LATS1/2 promotes its association with 14-3-3 protein and its subsequent cytoplasmic sequestration and inactivation [36]. The phosphorylation on Ser381 is recognized by Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase (BTRC), an F-box protein that participates in the ubiquitination and degradation of YAP [37]. During embryogenesis active cell proliferation is essential in ensuring the correct organ morphogenesis. Indeed, in normal conditions, DLG5 binds to MST1/2 through its third PDZ domain and inhibits its kinase activity by enrolling the microtubule affinity regulating kinase 3 (MARK3) (Fig. 6A) [21]. In this manner, YAP1 translocates into the nucleus, where it activates the DNA-binding transcription factor TEA Domain transcription Factor (TEAD) resulting in the transcription of tumor-promoting genes (Fig. 6A) [35]. The role in inhibiting the Hippo pathway was further proven in breast cancer (MDA-MB-231) and liver cancer (HepG2) cell lines by Kwan et al. [21]. The transient knockdown of endogenous DLG5 in MDA-MB-231 cells determined an increase in YAP phosphorylation and a loss of its predominately nuclear localization [21]. On the contrary, HepG2 resulted in a decrease in total YAP protein levels associated with a decrease in the expression of endogenous targets of the Hippo pathway, such as connective tissue growth factor (CTGF), cysteine-Rich heparin-binding protein 61 (CYR61), and ankyrin repeat domain 1 (ANKRD) [21]. This is consistent with the tumor-suppressing roles of the Hippo pathway where different cellular signals lead to persistent phosphorylation, and thus activation, of MST1/2 and LATS1/2 kinases that promote YAP degradation and prevent cell proliferation.
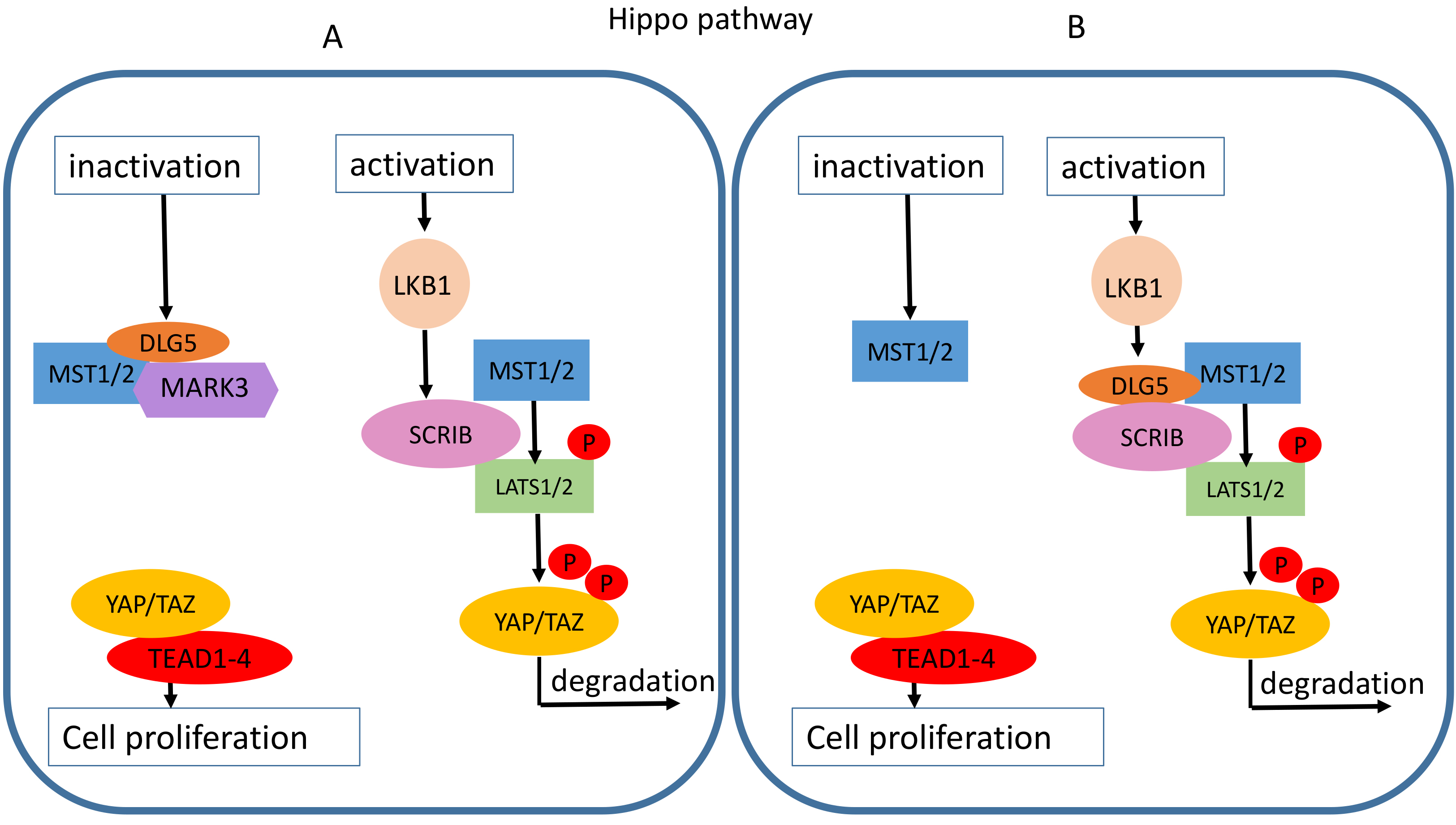 Fig. 6.
Fig. 6.Putative roles of DLG5 in the Hippo signaling pathway. (A) DLG5 induces cell proliferation by interfering with the phosphorylation cascade that leads to YAP/TAZ degradation. DLG5 blocks the large tumor suppressor kinase 1/2 (LATS1/2) phosphorylation by recruiting microtubule affinity regulating kinase 3 (MARK3) in the proximity of mammalian ste20-like kinase 1/2 (MAST1/2). (B) The DLG5 loss increase cell proliferation. When present DLG5 promotes scribble (SCRIB) interaction with MST1/2 and LATS1/2, and thus YAP phosphorylation and degradation.
Contrary to what was observed by Kwan et al. [21] in breast cancer and HCC cells, Liu J et al. [20] associated DLG5 loss with an increase of actively proliferating breast cancer cells. Indeed, the DLG5 silencing impaired the interaction of the epithelial polarity protein SCRIB [20], a downstream effector of Liver Kinase B1 (LKB1) [38], with MST1/2 and LATS1/2, preventing the phosphorylation of transcriptional coactivator YAP, consequently causing its nuclear accumulation and the downstream expression of genes promoting cell proliferation (Fig. 6B) [20]. DLG5 loss was associated with reduced SCRIB expression and mislocalization to the cytosol [20]. Thus in this model, DLG5 seems to indirectly participate in the hippo pathway regulation by recruiting SCRIB to the membrane proximity, instead of a direct binding to MST1/2 (Fig. 6B).
DLG5 depletion also correlated with nuclear localization of transcriptional co-activator with PDZ-binding motif (TAZ), also known as WW domain-containing transcription regulator 1 (WWTR1), a transcriptional coactivator negatively regulated by Hippo pathway, increasing the number and size of CD44+/CD24– breast cancer stem cells (BCSCs) and the gain of tamoxifen resistance [8].
Recent studies have explored the involvement of DLG5 in HCC (Table 2, Ref. [19, 21, 23, 24, 26]), one of the most frequent cancers worldwide [39]. Analysis of DLG5 expression in HCC tissue samples evidenced the higher expression in non-tumor tissues, meanwhile, only 28% of HCCs expressed DLG5, mainly at TNM stage I-II [19]. In addition, DLG5 levels were negatively correlated with multiple tumor nodule formations, tumor-associated angiogenesis, cancer stages, and stemness features. Consistently, DLG5 expression was positively related to the increase in overall and disease-free survival [19]. It was demonstrated that in HCC cell models (HepG2 and SMMC-7721), DLG5 undergoes continuous ubiquitination and proteasome degradation in a phospho-S730-dependent manner, through the interaction with beta-transducin repeat containing E3 ubiquitin-protein ligase (BTRC). When disruption of this process occurs, the accumulation of DLG5 leads to an inhibition of cell proliferation [23].
| DLG5 expression in HCC | References | |
| Up | Non-tumor tissues | [19] |
| Up | Advanced cancer stages | [26] |
| Negatively related to: | Tumor nodules formation | [19] |
| Vascular invasion | ||
| Stem-like features | ||
| Positively related to: | Overall survival | [19] |
| Disease-free survival | ||
| DLG5 function in HCC | References | |
| Inhibits: | Invadopodia formation | [19] |
| EMT | [24] | |
| Cell proliferation | [23] | |
| Hyppo pathway | [21] |
The increased levels of DLG5 in HCC cells can inhibit the formation of invadopodia [19], actin-rich membrane structures involved in extracellular matrix degradation [40], by binding Girdin and inhibiting its interaction with Tyrosine Kinase Substrate With Five SH3 Domains (TKS5) (Fig. 4). When a DLG5 suppression occurs in HCC, Girdin interacts with TKS5, promoting its phosphorylation and subsequent activation (Fig. 4), resulting in invadopodia formation [19, 41]. Another recent study identified a DLG5 regulatory pathway involving non-coding RNAs (ncRNAs) [24]. The long non-coding RNA (lncRNA) Transmembrane Phosphatase With Tensin Homology Pseudogene 1 (TPTEP1), was shown to be downregulated in the tumor compared to the adjacent non-tumoral tissue [24]. The lncRNA TPTEP1 sponges miR-454-3p, a microRNA (miRNA) able to promote proliferation, invasion, and EMT, by targeting DLG5. When lncRNA TPTEP1 is silenced, miR-454-3p exerts its tumor-promoting function by targeting DLG5, thus hampering its role in EMT inhibition [24].
A more recent analysis of the TCGA database associated DLG5 overexpression with advanced stages of HCC, suggesting a tumor-promoter function [26]. Supporting this hypothesis, Kwan J et al. [21] underlined the role of DLG5 in the inhibition of the Hippo signaling pathway, with possible implications for cell proliferation promotion (Table 2).
Despite little evidence in the literature, DLG5 may assume a relevant role as a predictor for overall survival and therapy response, especially in consideration of its roles in multiple aspects of cancerogenesis. In several tumors, including HCC, DLG5 depletion has been largely associated with the promotion of cell invasion and metastasis onset, being therefore related to poor prognosis [7, 9, 24, 25].
At present, it is worth noticing that the ambivalence of DLG5 role in cancer complicates the prediction of its impact on disease promotion and clinical outcome, also entangling its potentiality as a therapeutic target. However, evidence started emerging on its involvement in therapy resistance in cancer. The overexpression of DLG5 was associated with acquired etoposide chemoresistance in a cell model of NSCLC [17]. Concordantly, DLG5 was up-regulated in NSCLC patients nonresponding to anti-PD-1 therapy and associated with reduced overall survival [42], suggesting its possible value as a prognostic predictor in those patients. On the contrary, the downregulation of DLG5 in breast cancer was related to the acquirement of tamoxifen resistance [8]. Overall, the involvement of DLG5 dysregulation in therapy resistance and further major cancer-promoting processes highlights its potential value as a prognostic biomarker in cancer, paving the way to further studies.
Cell polarity is defined as the asymmetrical organization of cell shape, membrane, and intracellular components that differentially regulates multiple cell functions, including material secretion and absorption, migration, cell division, and barrier preservation [43, 44]. Epithelial cell polarity is characterized by a spatial and functional membrane division into apical and basolateral domains, which is necessary for the epithelial homeostasis and tissue polarized functions, such as the directional transport of molecules across the epithelium [45, 46]. Polarity proteins aggregate into multi-protein complexes responsible for the establishment and maintenance of cell polarity [2]. In cancer, those proteins can be aberrantly expressed, mutated, or re-localized within the subcellular compartments [2, 10, 47] resulting in cell and tissue architecture abnormalities that disrupt tissue homeostasis and sustain cell proliferation and invasiveness [46, 48]. However, the role of cell polarity proteins in cancer remains controversial and inconsistent conclusions still exist about their tumor-promoting role in several cancers.
In early studies in Drosophila melanogaster, epithelial polarity proteins showed a clear role in suppressing uncontrolled proliferation [1, 47, 48]. This role was further confirmed in subsequent studies assessing the tumor suppressor role of these protein complex members [49, 50, 51]. However, some contrasting roles were recently evidenced for cell polarity proteins in human cancers. The downregulation of polarity proteins, such as PAR-3, CRB3, and (LLGL Scribble Cell Polarity Complex Component 1) LLGL-1, have been linked to proliferation, migration, and invasiveness in multiple cancer types [49, 50, 51]. Contrarily, the up-regulation of other cell polarity proteins sustained cancer growth. That was the case of DLG5, whose over-expression was observed in HCC and glioblastoma [18, 26], DLG1 that was positively correlated to the progression of low-grade cervical lesions [52], DLG3, which was significantly higher in cell lines with high metastatic potential and in HCC tissues, being associated to poorer survival [53], SCRIB, that was overexpressed in colorectal cancer and associated with malignant characteristics [54], and Par-6 Family Cell Polarity Regulator Beta (PARD6B), whose overexpression in HCC was determined from alterations in the enhancer region [55].
Considering all these evidences, Saito et al. [47] reinterpreted the involvement of polarity proteins in cancers, hypothesizing that, besides their role in regulating cell polarity, they can work as important players in the maintenance of cancer cell fitness and in regulating several pathways, including proliferation [56], apoptosis [57], and acquisition of stem features [58]. What emerges is a new scenario in which polarity proteins acquire a cell type-, time- and subcellular localization-specific function. Thus, cancer cells re-direct polarity proteins to other compartments where they can exert other functions. For example, polarity proteins are re-assigned to regulate front-rear polarization and directional cell migration, or confined in the cytoplasmatic compartment where they cooperate in several cancer-related pathways [13, 59]. Indeed, in vitro studies highlighted the role of cytoplasmic SCRIB in enhancing cell invasiveness by stimulating the secretion of Secreted Protein Acidic And Cysteine Rich (SPARC) [60], an inducer of extracellular matrix remodeling and cell motility [61], through the activation of the transcription factors specificity protein 1 (SP1) and activator protein 1 (AP1) [60]. In HCC tissue, cytoplasmic SCRIB expression was inversely related to E-cadherin expression and positively correlated with the level of tumor undifferentiation and poor prognosis [60]. Analogously, high levels of mislocalized SCRIB in breast cancer affected Phosphatase And Tensin Homolog (PTEN) subcellular localization and Akt/mTOR/S6kinase signaling pathway, increasing breast cancer cell proliferation [62]. However, in many studies, it is not clear if the cytoplasmic accumulation is a cause or an effect of carcinogenesis and the role of these proteins in this subcellular compartment remains to be clarified [63, 64, 65].
The disruption of a static epithelial phenotype toward the gain of a motile mesenchymal polarity is strongly related to the acquirement of a tumor invasive and metastatic potential [66]. Migrating mesenchymal cells are indeed characterized by the front-rear polarity, in which proteins are asymmetrically distributed between the front and the back of the cells, thus regulating their directional motility. Interestingly, despite their substantial morphological and functional differences, apical-basal and front-back polarities still share several constituents required for their formation and maintenance [13], further complicating the comprehension of their involvement in cancer progression. Indeed, during the switch from an epithelial to a mesenchymal phenotype, cell adhesion complexes are inhibited and panels of pro-migrating genes are activated, while some epithelial polarity components are preserved and switch into novel pathways promoting invasiveness [13, 67, 68]. These controversial roles of some polarity proteins in cancer generated conflicting observations, as in the case of DLG5 in HCC, which is inversely correlated with cancer progression but shows an increased expression in advanced cancer stages. Therefore, the knowledge about the functions of DLG5 and other polarity proteins in cancer is still poor and needs to be broadened.
CT and DP conceptualized the manuscript. CA and DP collected the information and wrote the manuscript. CT and DP revised the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.