


1 Department of Biochemistry and Physiology, Physiology Section, Faculty of Pharmacy and Food Sciences, University of Barcelona, 08028 Barcelona, Spain
2 Laboratory of Cellular and Molecular Pathology, Institute of Biomedical Sciences, Faculty of Health Sciences, Universidad Autónoma de Chile, 3460000 Talca, Chile
3 Department of Pharmacology, Toxicology and Therapeutic Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, 08028 Barcelona, Spain
4 Institut de Neurociences (INUB), University of Barcelona, 08028 Barcelona, Spain
5 Biomedical Research Networking Centre in Neurodegenerative Diseases (CIBERNED), 28001 Madrid, Spain
6 Department of Pharmacy, Pharmaceutical Technology and Physical Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, 08028 Barcelona, Spain
7 ACE, Alzheimer Center Barcelona, Universitat Internacional de Catalunya (UIC), 08017 Barcelona, Spain
8 Unit of Biochemistry and Pharmacology, Faculty of Medicine and Health Sciences, University of Rovira i Virgili, 43003 Reus (Tarragona), Spain
9 Departamento de Biología Celular y Molecular, Centro Universitario de Ciencias Biológicas y Agropecuarias (CUCBA), Universidad de Guadalajara, 44100 Zapopan, Mexico
10 Department of Cellular Biology, Physiology and Immunology, Faculty of Biology, University of Barcelona, 08028 Barcelona, Spain
Academic Editor: Graham Pawelec
Abstract
The increases in population ageing and growth are leading to a boosting in the
number of people living with dementia, Alzheimer’s disease (AD) being the most
common cause. In spite of decades of intensive research, no cure for AD has been
found yet. However, some treatments that may change disease progression and help
control symptoms have been proposed. Beyond the classical hypotheses of AD
etiopathogenesis, i.e., amyloid beta peptide (A
Keywords
- dementia
- clinical studies
- novel therapies
- neuroinflammation
- synapses
Alzheimer’s disease (AD) is the most important neurodegenerative disorder. It is
closely related to the aging process, and it is associated with a functional
cognitive decline that ultimately causes patients’ death. It was first described
in 1906 by Alois Alzheimer, a German psychiatrist who reported the case of a
51-year-old woman with cognitive impairment, disorientation, delirium, and other
behavioral changes [1]. Although qualitatively described at the beginning of the
twentieth century, the molecular identities of the two main neuropathological
hallmarks of AD, i.e., the amyloid beta peptide (A
Despite the existence of several descriptive hypotheses trying to explain the
causes of AD, its etiology remains unknown. Beyond the microscopic finding of
senile A
On another front, the potential pathologies that coexist with AD during aging inflict additional damage to the brain, hence increasing the risk of dementia [15]. If the risk of suffering more than one disease increases with age, mixed pathologies may be a predominant cause of dementia in the elderly. In this sense, comorbidities such as type 2 diabetes mellitus (T2DM) may explain the higher risk of dementia in the elderly [16]. Likewise, the specific combinations of mixed pathologies and their relative contribution to the loss of cognition can vary widely for each individual. Clearly, a better understanding of the role of these mixed pathologies in AD may allow to unveil the brain alterations involved in age-related synaptic and neuronal loss leading to cognitive impairment. Moreover, from a therapeutic point of view, a combined therapy could be necessary to stop AD progression, as well as different pathologies associated with aging [17].
In the present review, we focus on therapeutic strategies designed to mitigate
the risk and modify the evolution of AD. Specifically, we describe drugs under
clinical study targeting A
| Row | Ref. | Phase clinical trials | Biopharma name | Main findings |
| 1 | [18] | Aducanumab FDA | Biogen | Immunotherapy. Causes reduction in amyloid plaques, which is very likely to cause a reduction in clinical decline due to AD. |
| Approval | ||||
| 2 | [19] | Donanemab (Phase 3) | Eli Lilly | Immunotherapy. Humanized IgG1 antibody directed at an N-terminal pyroglutamate A |
| NCT04437511 | ||||
| NCT04640077 | ||||
| 3 | [20] | Lecanemab (Phase 3) | BioArctic Neuroscience | Immunotherapy. Humanized IgG1 antibody which selectively binds strongly to soluble A |
| 4 | [21] | Gamunex (Phase 2/3 trial) | Grifols Biologicals Inc. | Immune Globulin Intravenous (IGIV) infusion administered in participants with mild to moderate AD. Decreases A |
| 5 | [22] | Azeliragon (Discontinued) | Pfizer, TransTech Pharma, Inc., | RAGE Inhibitor. RAGE is involved in amyloid transport into the brain. |
| NCT03980730 | vTv Therapeutics LLC | |||
| 6 | [24] | Tideglusib (Phase 2) | Zeltia Group | Non-ATP-competitive glycogen synthase kinase 3 (GSK-3) inhibition. |
| 7 | [25] | Lithium (Phase 2) | Unknown | Reduces cognitive and functional decline in amnestic MCI and modifies Alzheimer’s disease-related CSF biomarkers. Under research. |
| NCT01055392 | ||||
| 8 | [26] | TRx0237 (Phase 3) | TauRx Therapeutics Ltd | Prevents tau aggregation and attenuates downstream pathological consequences of aberrant tau. |
| NCT03446001 | ||||
| 9 | [27] | Tilavonemab or ABBV-8E12 (Discontinued) | AbbVie, C2N Diagnostics, LLC | It binds to tau’s N-terminus. Under research. |
| NCT02880956 | ||||
| 11 | [27] | Gosuranemab or BIIB092 (Discontinued) | Biogen | Humanized IgG4 monoclonal anti-tau antibody. Neutralizes toxicity of tau at the preclinical level. |
| NCT03352557 | ||||
| 12 | [23] | Masitinib (Phase 3) | AB Science | Inhibits the protein tyrosine kinase c-kit, PDGF and FGF receptors, and fyn and lyn kinases. Under research. |
| NCT01872598 | ||||
| 13 | [28] | ALZT-OP1 | AZTherapies, Inc. | Combination regimen of cromolyn (designated ALZT OP1a) and ibuprofen (designated ALZT OP1b). Ibuprofen is a nonsteroidal anti-inflammatory drug and cromolyn acts as an anti-inflammatory compound by suppressing cytokine release and decreased soluble A |
| NCT02547818 | ||||
| 14 | [29] | GV-971 | Shanghai Green Valley Pharmaceuticals | Mixture of acidic linear oligosaccharides derived from brown algae. Restores the gut microbial profile to normal and decreases microglial activation, brain A |
| 15 | [30] | Atuzaginstat (Phase 2/3) | Cortexyme, Inc. | Protease inhibitor targeting the lysine proteases of the periodontal pathogen Porphyromonas gingivalis. which is involved in AD. Under research. |
| NCT03823404 | ||||
| 16 | Liraglutide (N/A) | Novo Nordisk A/S | Analog of glucagon-like peptide 1. GLP-1 prevented the decline of cerebral glucose metabolism in AD patients. Under research. | |
| NCT01469351 | ||||
| 17 | [31] | Metformin (Phase 2/3) | A drug widely used to treat Type 2 diabetes. Previous studies reported positive results in preclinical models of AD. Under research. | |
| NCT04098666 | ||||
| 18 | [32] | NE3107 (Phase 3) | BioVie Inc | Decreases inflammation and improves insulin function in human and preclinical models. Under research. |
| NCT04669028 | ||||
| 19 | [33] | Losartan (Phase 2/3) | Merck | The study aims to evaluate the effects of aerobic exercise training and intensive vascular risk reduction (Losartan + amlodipine) on cognitive performance in older adults who have high risk for AD. Under research. |
| NCT02913664 | ||||
| 20 | [34] | Troriluzole (Phase 2) (Discontinued) | Biohaven Pharmaceuticals | Prodrug formulation of riluzole. Inhibits voltage-gated sodium channels, therefore decreasing glutamate levels at the synaptic cleft. |
| NCT03605667 | ||||
| 21 | [35] | Blarcamesine (Anavex 2-73) (Phase 2/3) | Anavex Life Science Corp. | Binds sigma-1 receptor in the high nanomolar and the muscarinic receptor in the low micromolar range. Under research. |
| NCT03790709 | ||||
| 22 | [36] | Levetiracetam (Phase 3) | UCB S.A. | Anti-convulsant drug and a modulator of the synaptic vesicle protein modulator SV2A. Under research. |
| NCT03486938 | ||||
| 23 | [37] | Icosapent ethyl (IPE), | Formulation of omega-3 fat ethyl eicosapentaenoic acid (ethyl-EPA) purified from fish oil. Under research. | |
| Ethyl eicosapentaenoic acid (E-EPA), | ||||
| AMR101, Miraxion | ||||
| (Phase 2/3) | ||||
| NCT02719327 |
AD can be classified into early-onset familial AD (or autosomal dominant AD, EOAD) which affects 5–10% of patients, and late onset Alzheimer’s disease (sporadic AD, LOAD) affecting 90–95% of AD patients [38].
EOAD is caused by mutations in genes encoding amyloid precursor protein (APP),
presenilin (PSEN)1 and PSEN2. Regarding APP, it is a type I membrane protein that
can be cleaved through three distinct pathways,
Unlike EOAD, LOAD is considered a multifactorial disease in which over 20
genetic risk sites could be involved, including clusterin, Sortilin-1 related
receptor, TREM2 and ApoE genes, among others [42]. The ApoE gene stands as the
most important one. It encodes the major apolipoprotein of the central nervous
system, playing key roles in lipid transport, growth, repair, reorganization, and
maintenance of neurons [43]. Hence, ApoE facilitates cellular uptake of
lipoproteins by binding to members of the LDLR family and participates in the
activation of signaling pathways involved in the modulation of lipid homeostasis.
Two amino acid substitutions at positions 112 and 158 lead to three possible
isoforms of ApoE, namely ApoE2, ApoE3 and ApoE4, which are encoded by three
common alleles (
The treatments of AD fall into two main categories: symptomatic and disease-modifying. The purpose of symptomatic treatments is cognitive improvement or control of neuropsychiatric symptoms, without having an impact on the biological causes leading to neuronal death. By contrast, disease-modifying treatments are designed to induce neuroprotection through changing the neuropathology of AD, often acting on a variety of intermediate mechanisms. Unfortunately, most therapeutic agents developed in the last 15 years have failed. In the European Union, only four symptomatic treatments have been approved so far, three cholinesterase inhibitors (donepezill, galantamine and rivastigmine) and an n-methyl-D-aspartate receptor antagonist (memantine), which was approved more than a decade ago.
More recently, two new drugs are being administered with the aim of slowing the
evolution of AD. These are sodium oligomannate (approved in China) and adacunumab
(approved by the FDA). These drugs are indicated for the treatment of a moderate
stage of AD, and are mainly based in the cholinergic hypothesis, initially
proposed by Davies and Maloney in 1976 [48]. Choline acetyltransferase is a key
enzyme for the synthesis of acetylcholine. Davies and Maloney compared the
activities of key enzymes involved in the synthesis of different
neurotransmitters, including acetylcholine,
Cholinesterase inhibitors inhibit the activity of the enzyme acetylcholinesterase (AChE), which hydrolyzes the neurotransmitter acetylcholine left over in the synaptic space, producing choline and acetic acid. This inhibition decreases acetylcholine elimination, hence leading to an increased availability of the neurotransmitter in the synaptic space. Although this has been shown to be effective in improving the cognitive function of patients with mild to moderate AD [50], no evidence indicates that these drugs slow progression of mild cognitive impairment (MCI) in non-demented patients, and it is clear that they do not prevent the development of dementia.
Currently, three cholinesterase inhibitors (rivistagmine, donepezil and galantamine) are being used for AD treatment. Rivastigmine selectively inhibits both cortical AChE in the central nervous system and butyrylcholinesterase (BuChE), which is documented as the predominant cholinesterase in many key regions affected in AD, including the hippocampus, thalamic nuclei and the amygdala [51]. Donepezil is a reversible and highly selective inhibitor of AChE which has also been found to be effective in treating cognitive impairment in patients with mild to moderate AD [52]. Indeed, Birks and Harvey [53] reported that a 10 mg/day treatment of donepezil during 52 weeks improved cognitive function, daily living activities performance and behavior in AD patients with mild, moderate or severe dementia. Finally, galantamine is characterized by two pharmacological mechanisms that involve the inhibition of AChE and the binding to nicotinic acetylcholine receptors in order to modulate allosterically the actions of ligands [54]. It also has very little activity in inhibiting BuChE. The clinical benefits of galantamine in AD patients have been recently demonstrated by Xu and colleagues [55], who reported a significant beneficial effect on cognitive improvement evaluated with the Mini-Mental State Examination. This improvement on cognitive decline, although modest, was persistent. Since the authors confirmed the usefulness of galantamine in the treatment of AD, studies assessing a combinatory therapy of galantamine with other drugs would be interesting.
Learning and memory processes involve long-term potentiation (LTP), a persistent and rapid increase in synaptic transmission mediated by the neurotransmitter glutamate through the NMDA receptor [56]. Indeed, NMDA receptors are abundant in the pyramidal cells of the hippocampus and cortex (areas involved in cognition, learning, and memory). However, high glutamate levels are associated with neurotoxicity through the activation of the NMDA GluN2B extrasynaptic receptors, leading to long-term depression (LTD), spine shrinkage and synaptic loss through a mechanism known as excitotoxicity [57]. In addition, these receptors also induce an increase in intracellular calcium and mitochondrial alterations, as well as an increase in reactive oxygen species (ROS) and nitric oxide (NO) production, which also contribute to neuronal cell death [58].
Memantine is a non-competitive, moderate-affinity NMDA receptor antagonist thought to decrease glutamate-induced excitotoxicity while allowing the physiological actions of glutamate on learning and memory [59]. Nevertheless, a study conducted by Peters and colleagues [60] reported that a combined treatment of galantamine plus memantine did not offer additional cognitive or functional advantages in patients with mild-to-moderate AD patients as compared with MCI patients who received galantamine alone. However, both drugs should not be discarded in a future disease-modifying strategy for AD.
Despite being such an important disease, the number of drugs in development for AD is much lower than in other diseases with a higher therapeutic arsenal. This reflects the fact that AD’s biology is poorly understood, and the availability of biomarkers is a very limited. Moreover, the duration of clinical trials for assessing AD treatments is very long, which increases the risk of failure.
In any case, we may wonder why the treatments in development are failing or are
not effective. Based on numerous trials of failed drugs in patients with AD, a
plausible explanation could be that A
The “amyloid cascade hypothesis” is, probably, the most accepted
pathophysiological hypothesis in AD. As already explained, it proposes that an
altered cleavage of APP by
Based on the amyloidogenic hypothesis of AD, the elimination of oligomers
(soluble) and plaques (insoluble) with monoclonal antibodies could be able to
decrease the progression of the disease [18]. Monoclonal antibodies develop an
immune response against these A
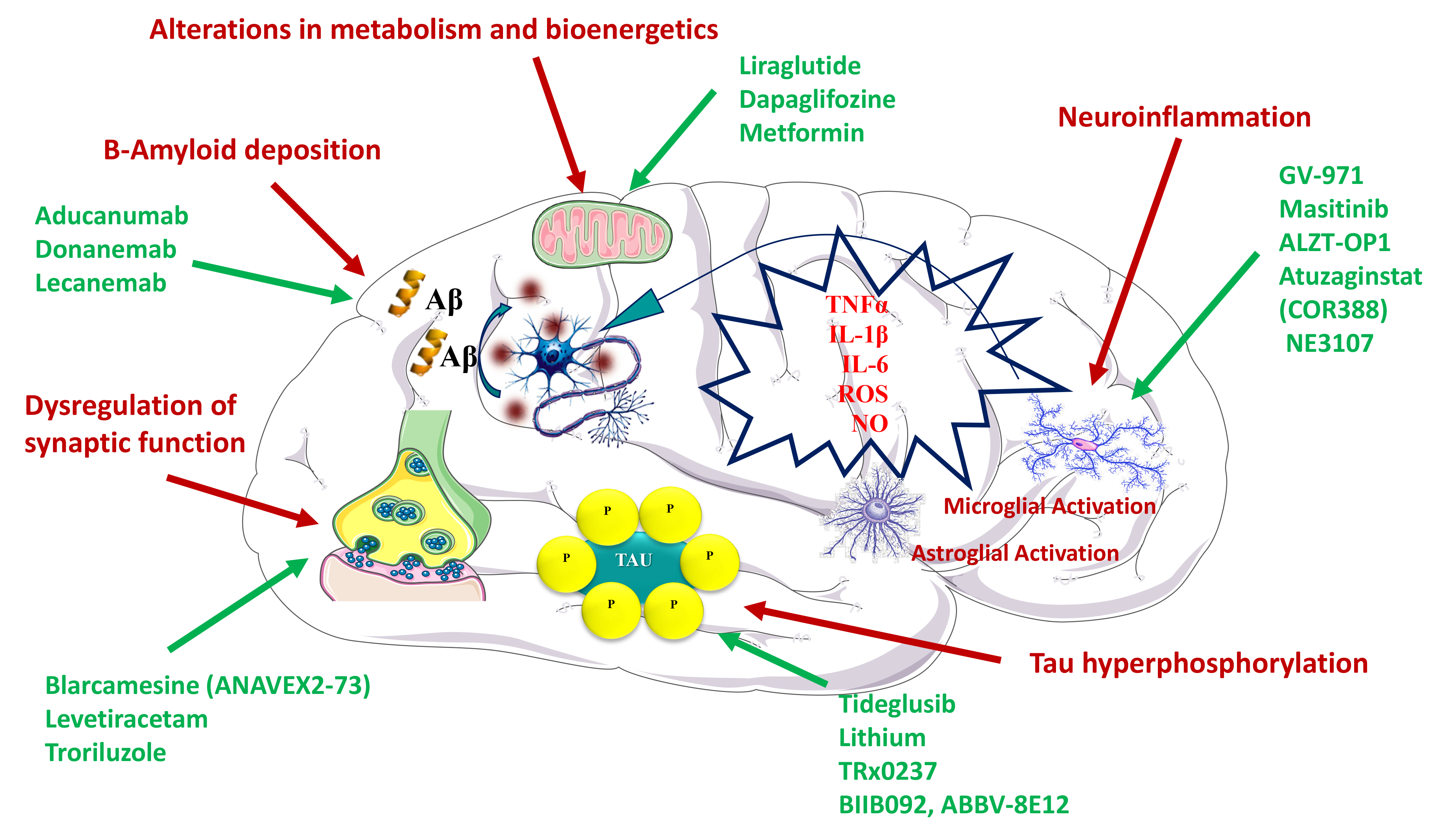 Fig. 1.
Fig. 1.Mechanisms proposed to explain the efficacy of different drugs in the treatment of Alzheimer’s disease. Pathophysiological mechanisms are depicted in red, whereas the specific therapeutic agents are depicted in green.
In turn, donanemab, which targets A
A
On another front, the receptor for advanced glycation end products (RAGE) not
only binds and transports A
As already mentioned, the fact that most drugs aiming to promote amyloid
clearance have failed, irrespective of whether they are monoclonal antibodies or
vaccines, challenges the amyloid cascade hypothesis. Moreover, some studies show
that patients treated with these drugs eliminate amyloid from the brain but do
not improve cognitively [74, 75]. For that matter, a more reconciling A
Tau is a microtubule-associated protein that can be found both in the CNS and at
the peripheral level. In a physiological context, tau exists as a soluble protein
that, apart from promoting correct assembly for the stabilization of the
microtubular structure, also plays a role in the balance of axonal transport and
synapses [76]. Tau is regulated both by normal homeostatic responses and by
stress responses, through a series of post-translational modifications such as
glycosylation, ubiquitination, glycation, nitration, oxidation and
phosphorylation, the last one being the most important one [77]. Tau
phosphorylation is regulated by several kinases, including glycogen synthase
kinase 3 (GSK3
Given that the severity of tau pathology is more correlated with the progression
of cognitive impairment than A
In turn, the second-generation tau aggregation inhibitor LMTX (TRx0237) has been extensively investigated and it is the only drug in this group that reached clinical phase III. Hence, two clinical phase III trials showed that a LMTX monotherapy seems to slow cognitive decline in patients with mild to moderate AD (NCT01689246, NCT01689233). Consequently, additional clinical phase III studies are underway (NCT03446001) [26].
In addition to these strategies, relatively new approaches based on tau have also been investigated; however, some of them did not show results in clinical trials and have been discontinued. These include inhibition of acetylation and deglucosylation of tau (MK-8719 and ASN120290, respectively), monoclonal antibodies against tau (Gosuranemab or BIIB092, Tilavonemab or ABBV-8E12) and reduction of tau levels by an antisense oligonucleotide (ASO) (IONIS-MAPTRx) [27].
The notion that inflammation and the immune responses play a fundamental role in the pathophysiology of AD is supported by many studies [83]. Much research has been focused on the involvement of astrocytes, microglia and CNS mast cells, but also on the impact of systemic inflammation caused by intestinal and gut microbiota. These have led to the development of different drugs targeting the inflammatory process that could result in neurodegeneration, including tyrosine kinase inhibitors that modulate mast cells (masitinib), monoclonal antibodies that regulate microglial activity as daratumumab (against CD38), AL002 (against TREM2) or AL003 (against SIGLEC-3), and curcumin, which also has antioxidant properties. The use of montelukast (a leukotriene receptor antagonist) and intestinal microbiota transplants, among others, are also being studied.
Microglia are CNS-resident phagocytes that play a vital role in maintaining
neuronal plasticity and synapse remodeling [84, 85]. They have been shown to be
involved in the maintenance of neural networks by releasing neurotrophic factors
such as BDNF, but also contribute to brain homeostasis by exerting synaptic
elimination [86]. Microglia can be activated by the accumulation of proteins that
act as a pathological trigger, producing the migration of the microglial cells
and the onset of an innate immune response. Indeed, microglia actively
participate as supportive cells by engulfing A
On another front, mast cells (MCs) have an important physiological function and
are involved in immunity and inflammation, especially in allergic inflammation.
Upon activation, MCs increase the synthesis and secretion of many inflammatory
mediators, such as TNF-
Masitinib is currently one of the most promising research drugs targeting inflammatory diseases such as rheumatoid arthritis, intestinal disease, asthma and mastocytosis. However, a potential new application of masitinib in neurodegenerative diseases, such as AD and multiple sclerosis (MS), has emerged. Masitinib inhibits c-kit tyrosine kinase, a surface receptor expressed by MCs and many other cells, which plays a prominent role as a regulator of the migration of neuronal stem and progenitor cells to areas of brain injury [101]. Hence, masitinib is thought to exert a neuroprotective effect through its activity on MCs and other non-neuronal cells of the CNS, with a subsequent modulation of inflammatory and neurodegenerative processes. It is currently in the phase 3 of a clinical trial in patients with mild to moderate AD (ClinicalTrials.gov Identifier: NCT01872598). Masitinib is administered as adjunctive therapy to patients who have been treated for a minimum of 6 months with a stable dose of a cholinesterase inhibitor (donepezil, rivastigmine or galantamine) and/or memantine. So far, the company has reported some positive results from this study. Masitinib is also capable of blocking the Src TK Fyn family in a nanomolar range. Fyn is a cytoplasmic tyrosine kinase (TK) belonging to the Src kinase (SFK) family involved in multiple CNS transduction pathways, including synaptic transmission, myelination, axon guidance, and formation of oligodendrocytes [102]. Shirazi and Wood reported that that Fyn is upregulated in the brain of AD patients and demonstrated the presence of a Fyn phosphorylation site in the tau-matched helical filament, supporting a role of Fyn in the neuropathogenesis of AD [103]. Therefore, treatment with masitinib may provide two benefits in the pathology of AD: (i) a reduction in neuroinflammation by modulating the mast cell-glia axis and (ii) a cognitive enhancement through a Fyn inhibition.
After many failures with monotreatments, the combination of cromolyn and
ibuprofen (ALZT-OP1) has been proposed as a suitable treatment for LOAD [28].
Cromolyn acts as a modulator of MCs and microglia, as well as an A
XPro1595 is a second-generation inhibitor of tumor necrosis factor
(TNF
From a different point of view, some studies have proposed an association
between intestinal microbiota and AD, in which dysbiosis cause neuroinflammation
induced by activation of the microglia [111]. In this regard, one of the latest
interesting drugs is sodium oligomannate (GV-971), a derivative of brown marine
algae based on a mixture of acidic linear oligosaccharides ranging from dimers to
decamers [112]. It was developed by Green Valley for reconditioning the
intestinal microbiota and treating mild-moderate AD. A preclinical study showed
that oral administration of GV-971 for one month markedly altered the composition
of the microbiota and reduced the concentrations of phenylalanine and isoleucine
in a murine model of AD [113]. These was paralleled by a reduction in microglial
activation, as well as in Th1 responses and cytokines in the brain.
Simultaneously, oligomannate treatment reduced A
Beyond the relation between intestinal microbiota and AD, recent studies also support an association between periodontitis, a chronic inflammatory oral disease, and neurodegeneration [115]. It is known that there is a relationship between host immune responses and pathogenic burden of microbial biofilm [116, 117], which is made up of several microorganisms, such as Porphyromonas gingivalis, and their toxic products, such as fimbrins, gingipain and lipopolysaccharides (LPS). Both periodontopathogenic bacteria and their products can enter the bloodstream, promoting the expression of inflammatory mediators that can damage other organs, including the brain. Interestingly, gingipains are proteases that generate pathogenicity factors such as Arg-gingipain (Rgp) and Lys-gingipain (Kgp), which can damage tau [30, 118]. In this regard, atuzaginstat (COR388) is an inhibitor of gingipains, which is being tested in a phase II/III clinical study for AD treatment (NCT03823404) (Fig. 1).
It is widely known that glucose is the main source of energy of the mammalian brain, and several pathologies of the CNS are a consequence of disturbed central or peripheral glucose energy metabolism [119]. In this sense, impaired glucose metabolism has been shown to be related with AD pathophysiology. Hence, alterations in brain glucose uptake have been described in patients with initial symptoms that precede the development of AD [120]. Moreover, it seems that insulin improves glucose uptake in the brain by increasing the activation of hippocampal insulin receptor, which plays a key role in synaptic plasticity, learning and memory [121]. Therefore, the rescue of cerebral insulin signaling and glucose metabolism constitutes a tempting goal to treat neurodegeneration.
A priori, many drugs used to treat T2DM could be recycled to also treat AD, including 116 (glucagon-like peptide 1 receptor agonist), dapaglifozine (SGLT2 inhibitor) and metformin, among others. Regarding the latter, metformin is currently the first-line treatment for T2DM, widely prescribed due to its safeness and virtual absence of side effects. Preclinical results in murine models of AD showed that metformin delayed the progression of cognitive impairment [122]. In turn, Luchsinger and colleagues reported that metformin treatment significantly improves the total recall in the selective reminding test, but not the Alzheimer Disease Assessment Scale-Cognition (ADAS-Cog), in a study with patients with amnestic MCI (ClinicalTrials.gov Identifier: NCT00620191). The authors concluded that additional clinical studies with a larger number of patients are necessary to demonstrate a greater efficacy of the drug. Other studies evaluating the efficacy of metformin on AD biomarkers and cognitive ability (ClinicalTrials.gov Identifier: NCT01965756) and for dementia prevention (NCT04098666) have been conducted. The results of the study NCT01965756 showed that metformin improved executive function and there was a trend in improving the learning process, memory and attention [31]. Regarding the study NCT04098666, no results have yet been published.
NE3107 is another drug which also reached the phase III (Clinicaltrials.gov: NCT04669028). Previous studies have shown that this drug has a dual therapeutic effect, presenting both neuroinflammatory and antidiabetic properties [32]. Hence, NE3107 has been reported to inhibit the inflammatory ERK pathway and to the improve insulin signaling. This dual effect makes this drug interesting in the treatment of AD (Fig. 1).
On a different note, intranasal administration of insulin glulisine is also being evaluated with the aim to increase insulin signaling in the brain [123]. Promising results have been published regarding intranasal insulin administration in patients with amnestic MCI and AD (ClinicalTrials.gov Identifier: NCT01767909). However, more studies are needed to better characterize the neuroprotective effect of insulin and brain insulin receptor stimulation in neurodegeneration.
An association between cognitive impairment in non-demented individuals and cardiovascular risk factors has been reported [124]. Hence, the inflammatory process associated with aging, atherosclerotic cardiovascular diseases and dementia could share common molecular mechanisms. In this sense, the control of cardiovascular risk factors could be an appropriate strategy to reduce or prevent the incidence of dementia [125].
Following this line of thought, drugs acting on cardiovascular and
cerebrovascular dysfunctions, BBB and neurovascular unit, hypertension,
atherosclerosis, amyloid cerebral angiopathy and lymphatic/glyphic system
dysfunction could be useful for AD treatment. For instance, losartan, an
antihypertensive drug, reduced plasma and brain A
Previous studies have reported a dysregulation of synaptic functions in AD [131, 132]. In fact, synaptic loss and dysfunction is strongly correlated with the cognitive decline observed in AD patients [133]. Indeed, surviving neurons in AD’s neurodegenerative process have been shown to lose synapses, and synaptic dysfunction has been shown to precede amyloid plaque deposition, as LTP impairment is present in early stages in the hippocampus of AD mice [134, 135, 136]. Hence, the loss of synaptic homeostasis or the integrity of the neural network would precede neuronal death and be key to AD development, an idea that fits with the proposed theories and clinical manifestations. Consequently, memory deficits in AD may even begin two decades before the first symptoms appear. Synapse degeneration is thought to begin in dendritic spines and, specifically, with a decrease in the number of molecules that regulate spinal signaling.
It has been shown that glutamate is involved in the development of dendritic spines [137]. On this basis, some drugs targeting neurotransmitters and mechanisms of neurogenesis are being studied (Fig. 1). An example is troriluzole, also known as BHV4157 (proriluzole), which is a prodrug of riluzole. Riluzole inhibits voltage-gated sodium channels and reduces synaptic glutamate by increasing its uptake and inhibiting its release [138]. Indeed, riluzole reduces synaptic glutamate levels by increasing the expression and function of glial glutamate transporters responsible for synaptic glutamate clearance [34]. Its efficacy in the treatment of AD is currently being evaluated in phase 2–3 clinical trials (Clinicaltrials.gov NCT03605667), although so far, the results do not seem to be encouraging.
In turn, blarcamesine (ANAVEX2-73) is an agonist of the Sigma-1 receptor (S1R)
and modulates cholinergic muscarinic receptors in mice [135, 139, 140]. It has
been reported that an oral dose (30 or 50 mg) of blarcamesine, followed by an
intravenous (IV) dose (3 or 5 mg) in a second period have suitable safety and
tolerability in patients with mild-to-moderate AD in a Phase IIa clinical study
[35]. Furthermore, phase IIb/III clinical studies consisting in 48 weeks of
daily treatment with blarcamesine or placebo, and primary outcomes of ADAS-Cog
and ADCS-ADL (Activities of Daily Living) evaluations are being conducted
(ClinicalTrials.gov Identifier: NCT03790709). Additionally, in preclinical models
of Rett syndrome (RTT), a neurodevelopmental disorder associated with increased
risk of cognitive impairment, blarcamesin improved calcium homeostasis, which
favors an improvement in mitochondrial and synaptic functions in all brain
regions [135]. Besides that, the anticonvulsant levetiracetam has also shown
potential as synapse modulator against neurodegeneration. Levetiracetam is an
antiepileptic drug that binds to SV glycoprotein 2A (SV2A), a constituent of
synaptic vesicle membranes at presynaptic terminals, involved in vesicle
trafficking and exocytosis [141]. SV2A is expressed in excitatory and inhibitory
synapses in the brain, including the hippocampus, and alterations in this protein
have been associated with AD [142, 143]. Interestingly, Rao and Savas [36]
reported that levetiracetam lowers A
Currently, a phase II-III study is evaluating the efficacy of AGB101, a proprietary extended-release formulation of levetiracetam, on slowing cognitive and functional impairment in patients with MCI (ClinicalTrials.gov Identifier: NCT03486938). No results have been reported yet.
The fact that diet quality and composition play a role in virtually all health conditions, affecting incidence, complications, management, recovery and quality of life is well supported by mounting evidence [144]. AD is not an exception, and currently several supplements and dietary strategies claim to promote cognitive enhancement. However, to date, no evidence-based product on the market has demonstrated a clear capacity to prevent or slow AD’s progression.
Omega-3 polyunsaturated Fatty Acids (
Despite the numerous failures in the development of therapies that modify the
evolution of AD, current technological progress in the fields of biomarkers
assays and genetics presents an unprecedented opportunity to reshape AD
therapeutic strategies towards the medicine of precision. As with other diseases,
an early detection of AD is essential. Hence, assessing the blood levels of AD
biomarkers such as tau and amyloid in asymptomatic patients could be beneficial
as an early approximation [149]. In symptomatic patients, positron emission
tomography and magnetic resonance imaging approaches to assess A
Undoubtedly, the trends in therapeutic strategies for AD will involve an increase in the diversity of non-amyloid or tau targets, including inflammation, insulin resistance, synapse and neuronal protection, cardiovascular factors, neurogenesis and epigenetic interventions. Indeed, some authors consider that AD should no longer be considered a brain disease, since its development is also attributed to peripheral factors as, for instance, intestinal dysbiosis. Hence, the increasing knowledge of the mechanisms involved in AD may favor the development of novel therapies based, for example, on the reconstitution of the intestinal microbiota. In this sense, sodium oligomannate opens a promising new therapeutic line of disease-modifying therapies worthy of future research. In passive immunotherapy, the FDA-approved aducanumab stands out, but anti-tau treatments still need to demonstrate its clinical efficacy. Also, an increase in the number of candidate drugs for disease modification treatments is expected, as well as a focus on potential combinatory multidrug strategies.
JO, ME, AMAC, ESL, assisted in the preparation of the figures, designed the concept of the manuscript, provided supervision. JO and AC wrote the manuscript. MC, TE, CBZ, GGC, MEUG, EV, JF and CA critically reviewed the manuscript and substantially contributed to the writing and content of the manuscript. All authors provided their contribution in writing the manuscript, critically reviewing the completed manuscript, and approved the submitted version of the manuscript.
Not applicable.
The authors would like to thank Anton Kabanen for English checking and the Reviewers for their opinions and suggestions.
Jordi Olloquequi and Miren Ettcheto are Serra Húnter fellows. Amanda Cano acknowledges the support of the Spanish Ministry of Science, Innovation and Universities under the grant Juan de la Cierva (FJC2018-036012-I). Authors acknowledge the support of Spanish Ministry of Economy and Competitiveness under project SAF2017-84283-R, and Biomedical Research Networking Centre in Neurodegenerative Diseases (CIBERNED) under the project CB06/05/0024. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The authors declare no conflict of interest. AC is serving as the guest editor and the editorial board member of this journal. We declare that AC had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to GP.