





1 Department of Gynecology and Obstetrics, Key Laboratory of Obstetrics & Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, Development and Related Diseases of Women and Children Key Laboratory of Sichuan Province, West China Second Hospital, Sichuan University, 610041 Chengdu, Sichuan, China
†These authors contributed equally.
Academic Editor: Chuanjin Wu
Abstract
Conventional treatments for ovarian cancer, including debulking cytoreductive surgery combined with carboplatin/paclitaxel-based chemotherapy, are insufficient, as evidenced by the high mortality rate, which ranks first among gynecological tumors. Therefore, there is an urgent need to develop new and effective treatment strategies. Recent evidence has shown that metabolic processes and cell behaviors in ovarian cancer are regulated by intracellular factors as well as metabolites in the tumor microenvironment (TME), which determine occurrence, proliferation, and metastasis. In this review, we describe the comprehensive landscape of metabolic cross-talk between ovarian cancer and its TME with a focus on the following four aspects: (1) intracellular metabolism based on the Warburg effect, (2) metabolism in non-tumor cells in the ovarian TME, (3) metabolic communication between tumor cells and non-tumor cells in the TME, and (4) metabolism-related therapeutic targets and agents for ovarian cancer. The metabolic cross-talk between ovarian cancer and its microenvironment involves a complex network of interactions, and interrupting these interactions by metabolic interventions is a promising therapeutic strategy.
Keywords
- review
- ovarian cancer
- tumor microenvironment
- metabolites
- cancer therapies
Ovarian cancer is the most lethal gynecological tumor [1] arising from common anatomical sites within the ovary. According to histology, it can be divided into many types, among which epithelial ovarian cancer (EOC) is dominant in the clinical setting [2, 3]. Owing to the lack of typical symptoms and effective diagnostic methods in the early stage, only 16.3% of cases are discovered and diagnosed at FIGO stage I. The large number of cases diagnosed at an advanced stage with metastasis at distant sites leads to a poor 5-year survival rate (49.1%) [4]. The current first-line standard therapy for ovarian cancer is debulking cytoreductive surgery combined with carboplatin/paclitaxel-based chemotherapy, and maintenance therapy with bevacizumab and poly ADP-ribose polymerase inhibitor (PARPi) is also used to improve progression-free survival [5]. Although ovarian cancer is sensitive to chemotherapy, drug resistance is extremely frequent [6]. After relapse, treatment outcomes are often poor [7, 8]. A more detailed understanding of the mechanisms underlying the oncogenesis and development of ovarian cancer may provide a basis for more effective monitoring and treatment.
In the past few decades, the vital role of the tumor microenvironment (TME) in tumorigenesis has been revealed. The extracellular matrix (ECM), which consists of several secreted molecules including inflammatory cytokines and chemokines, and the stromal cells, including cancer cells, endothelial cells (ECs), cancer-associated fibroblasts (CAFs), pericytes, and immune cells were most studied in TME [9]. The physiological characteristics of the TME (e.g., hypoxia, extracellular pH, and elevated interstitial fluid pressure) also show profound differences from those of normal human tissues [10]. Numerous studies have shown that the TME plays an important role in the proliferation, metastasis, and chemoresistance of ovarian cancer by metabolic remodeling [9, 11, 12]. The metabolism and behavior of ovarian cancer are regulated by intracellular factors in cancer cells and metabolic products of the TME [13, 14]. There is extensive bidirectional communication between tumor cells and the TME.
In-depth research on the TME has resulted in the continuous development of immunotherapies. A large number of new drugs have emerged, e.g., antiangiogenic agents, PARPi, tumor-intrinsic signaling pathway inhibitors, selective estrogen receptor downregulators, and immune checkpoint inhibitors (ICIs) [2, 15, 16]. Here, we review metabolic interactions between ovarian cancer cells and the TME at both the molecular and cellular levels. In particular, we describe metabolism in ovarian cancer cells as well as various non-tumor cells of the TME, physiological properties of the TME, and metabolic cross-talk. Finally, we describe recently developed treatment strategies targeting this metabolic cross-talk as well as general approaches (e.g., single-cell sequencing technology) with the potential to accelerate research in this area. Research in this area has the potential to provide a novel approach for cancer therapy aimed at altering metabolism in the TME and tumor cells.
In various cancer cells, glycolysis is enhanced and the oxidative phosphorylation (OXPHOS) capacity is reduced. As a solid tumor, intracellular metabolism in ovarian cancer is consistent with the Warburg effect. That is, even under aerobic conditions, ovarian cancer cells prefer cytoplasmic glycolysis to OXPHOS of the TCA cycle in mitochondria to provide adenosine triphosphate (ATP), nucleotides, lipids, and amino acids for growth [17, 18, 19].
The rapid proliferation of ovarian cancer cells leads to an imbalance in the intake and consumption of oxygen and nutrients. Malignant tumor cells usually have a much higher glycolysis rate than that of normal tissues and preferentially obtain glutamine [20]. Under adverse conditions for metabolic processes in the TME (hypoxia, acidosis, and low glucose), precancerous cells gain the Warburg phenotype by transcriptional reprogramming to adapt to the harsh environment [21]. The TCA cycle has also been recognized as “tumor-promoting” in recent years. Glycolysis and metabolites in the TCA cycle play vital roles in tumor growth and metastasis [22]. Response to metabolic stress in TME, ovarian cancer cells get an increasingly glycolytic and flexible metabolic phenotype [23]. Under hypoxic conditions, glucose uptake, lactate secretion, cellular respiration, and ATP synthesis are reduced, and the most aggressive cells are assembled into spheroids by the downregulation of respiratory function, which increases the invasive capacity [24]. This mutually reinforcing effect leads to further disease progression. Ovarian cancer cell metabolism based on the Warburg effect is summarized in Fig. 1.
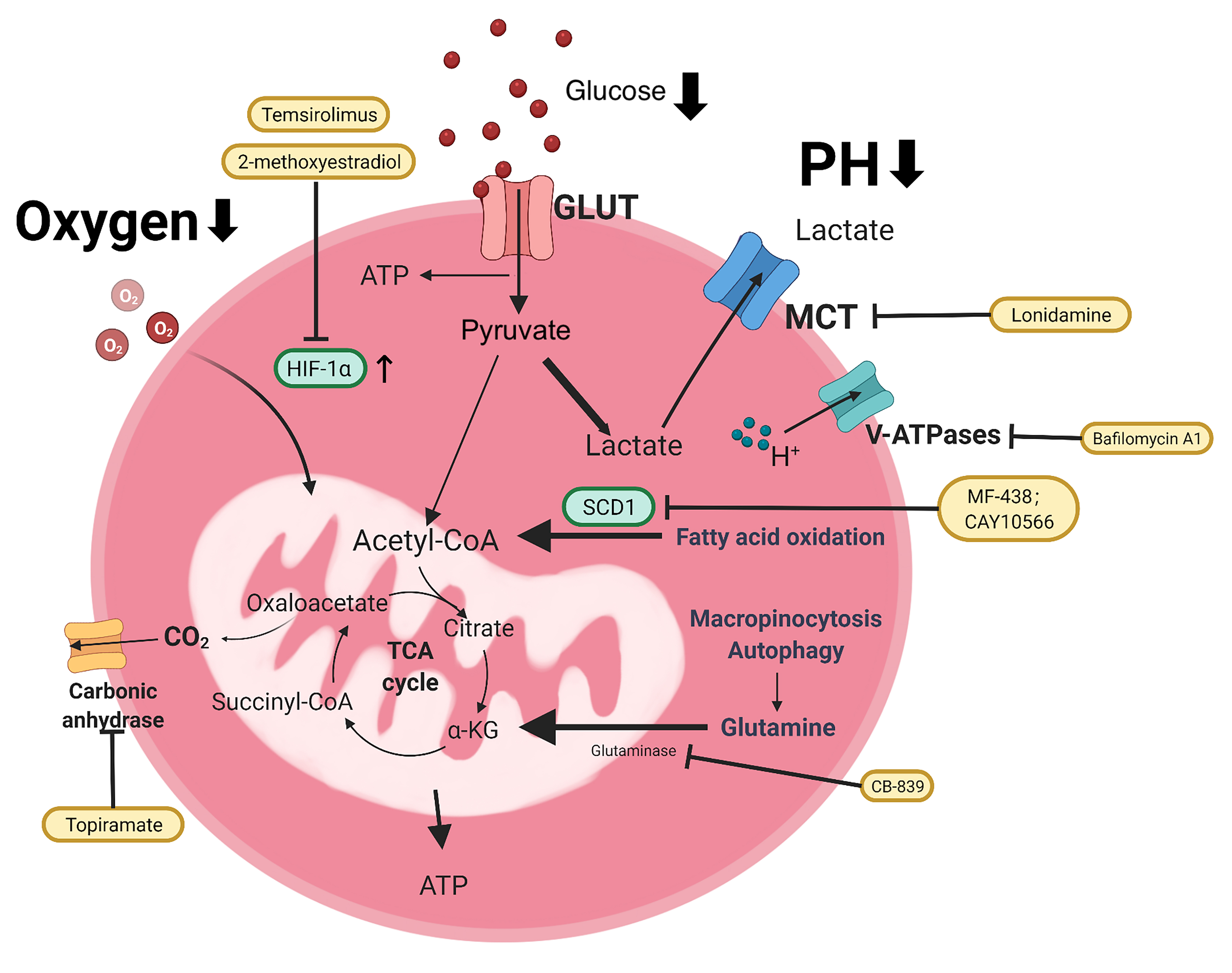 Fig. 1.
Fig. 1.Metabolism of ovarian cancer cells based on Warburg effect. Ovarian cancer cells take up oxygen and nutrients (glucose, glutamine, etc.) from the extracellular matrix, mainly through glycolysis, rather than through the TCA cycle of OXPHOS in the mitochondria, which provides ATP for cancer cell growth, even under aerobic conditions. The main processes are represented by thick arrows. The metabolic inhibitors are shown in the yellow box. GLUT, glucose transporters; MCT, monocarboxylate transporters; SCD1, stearoyl-CoA desaturase; V-ATPase, Vacuolar H+- ATPase.
Hypoxia is an important feature in the TME of solid tumors. With the increasing volume of ovarian cancer itself, the tumor tissue expands away from blood vessels containing nutrients and oxygen, and this lack of blood supply leads to further hypoxia and even necrosis in the central area [24, 25]. The adaptation of tumor cells to the hypoxic environment is the basis for tumor cell survival and proliferation. Tumor cells in the hypoxic region consume glucose by anaerobic glycolysis and release lactic acid, which is then uptaken by tumor cells in the adjacent oxygen-generating tumor region as a raw material for TCA circulation [26]. Multicellular aggregation and hypoxia were reported to reduce the response to the metabolic drugs AICAR (AMPK agonist) and metformin (ETC complex I inhibitor) as well as the chemotherapeutic drugs cisplatin and paclitaxel [23]. In addition, cysteine, which is commonly found in the ascites of patients with advanced ovarian cancer, was revealed to promote ovarian cancer cell adaptation to the hypoxic environment and contributes to platinum-based chemotherapy resistance [27]. Accordingly, hypoxia is an important direction for the development of tumor-targeted therapies.
Hypoxia-inducible factor 1 (HIF-1) is a major regulator of cellular responses to
hypoxia. HIF-1 belongs to the basic helix-loop-helix Per–Arnt–Sim (PAS) protein
family [28, 29]. It consists of a hypoxia-regulated
Acidosis in the TME in ovarian cancer develops through various pathways. The acidification of the TME provides a more hostile environment compared to that of non-neoplastic cancers, favoring tumor survival and growth. The production of a large amount of lactic acid in glycolysis and CO2 from the pentose phosphate pathway coupled with the incomplete vasculature around the tumor tissue causes the accumulation of catabolites and contributes to the low pH environment [35, 36]. Lactic acid is regulated by monocarboxylate transporters (MCTs) on the cell membrane [37]. The expression of MCT4 is abnormally increased in hypoxic tumor areas, and lactate is transported out of the cell through MCT4. In tumor regions with normal oxygen supply, the expression of MCT1, which transports lactate from extracellular to intracellular compartments, is increased [38]. In ovarian cancer, MCT1 may be positively correlated with lactic acid [39, 40]. Furthermore, extracellular acidosis leads to reprogramming to a highly plastic tumor phenotype, involving stem-related marker expression, high clonogenicity, and transdifferentiation ability, which can promote invasion, tumor progression, and metastatic disease [41].
Transporters and pumps which contribute to H+ secretion are important targets for tumor therapy [42, 43]. As a classical transport channel, proton pump vacuolar H+–ATPases (V-ATPases) consume ATP to pump protons out of the cell or organelles to avoid self-acidosis [44]. H+ excreted by tumor cells follows a concentration gradient into normal cell tissues. The substantial accumulation of H+ in normal cells activates enzymatic cascade reactions, leading to necrosis or apoptosis, which is conducive to tumor spread and metastasis [42]. Protein carriers, such as V-ATPases, which maintain the extracellular acidic microenvironment and thereby promote tumor malignancy, are also potential therapeutic targets worthy of further attention [45, 46]. Kulshrestha et al. [47] found that targeting the V-ATPase isoform can restore cisplatin activity in drug-resistant ovarian cancer via the ERK/MEK pathway. The application of specific V-ATPase inhibitors, such as bafilomycin and proton pump inhibitors, can reduce tumor acidity, reduce tumor metastasis, influence the survival of tumor cells, and prevent chemotherapy tolerance [42, 43, 44, 45, 48, 49]. Furthermore, various membrane transporters form structural and functional complexes with carbonic anhydrases (CAs), known as transporters [50]. The CA inhibitor topiramate (TPM) can inhibit the proliferation of ovarian cancer cells and induce G1 phase cell cycle arrest, cell stress, and apoptosis via the AKT/mTOR and MAPK pathways. It also exerts anti-metastatic effects by reducing the adhesion and invasion of ovarian cancer cells and affecting the expression of key regulators of the epithelial-mesenchymal transition (EMT) [51]. In tumor cells, these bicarbonate transporters are thought to play roles in pH regulation and cell migration and might become new drug targets for cancer therapy [50, 52, 53].
Ovarian cancer tumor cells consume glucose for growth and survival and are much more sensitive to “glucose deprivation” than are normal cells in the body [54]. Glucose absorption occurs through glucose transporters (GLUTs). GLUT1 and GLUT3 are highly expressed in ovarian cancer. GLUT1 is ubiquitously distributed in cells, and GLUT3 shows a high affinity for glucose and may promote uptake in tumors with strong glucose needs [55]. Related study has confirmed that GLUT1 expression is variable among different types and stages of ovarian cancer. The mean expression level of GLUT1 is higher in high grade serous ovarian cancer (HGSOC); meanwhile, it was also significantly higher in advanced (III/IV) ovarian cancer than in early (I/II) disease [56].
Pyruvate kinase M2 (PKM2), an isoform of pyruvate kinase (PK), regulates glycolysis and OXPHOS by controlling pyruvate production in the final step of glycolysis [57]. PKM2 activity is reduced in tumor cells, indirectly facilitating the redirection of glycolytic intermediates to the biosynthesis of other biomolecules and promoting tumor anabolism [58]. PKM2 is expressed only in the cytoplasm of ovarian cancer cells [59]. PKM2 inhibitors can significantly inhibit glycolysis and disrupt the Warburg effect according to the glucose consumption level of cells, thereby inhibiting ovarian cancer progression and cell migration in vitro [57, 60]. Targeting PKM2 is a promising treatment strategy for patients with ovarian cancer [61].
Lactate dehydrogenase A (LDHA) functions in the final step of glycolysis, converting pyruvate to lactate. Patients with ovarian cancer often have significantly higher serum LDH levels than normal due to upregulation of LDHA gene expression [62]. This high level of LDHA expression contributes to drug resistance in ovarian cancer. Xiang J found that the high level of LDHA affected the inhibitory effect of PARP inhibitors on wild-type BRCA ovarian cancer and that inhibition of LDHA significantly promoted the inhibitory effect of PARP inhibitors on A2780 and SKOV3 cells [63].
The cellular composition of the TME varies among tumor types and includes stromal and immune cells. Stromal cells are composed of vascular endothelial cells, fibroblasts, and adipocytes [64]. Herein we review metabolic processes in various non-tumor cells in the TME of ovarian cancer. Schematic of the composition of ovarian cancer microenvironment was shown in Fig. 2.
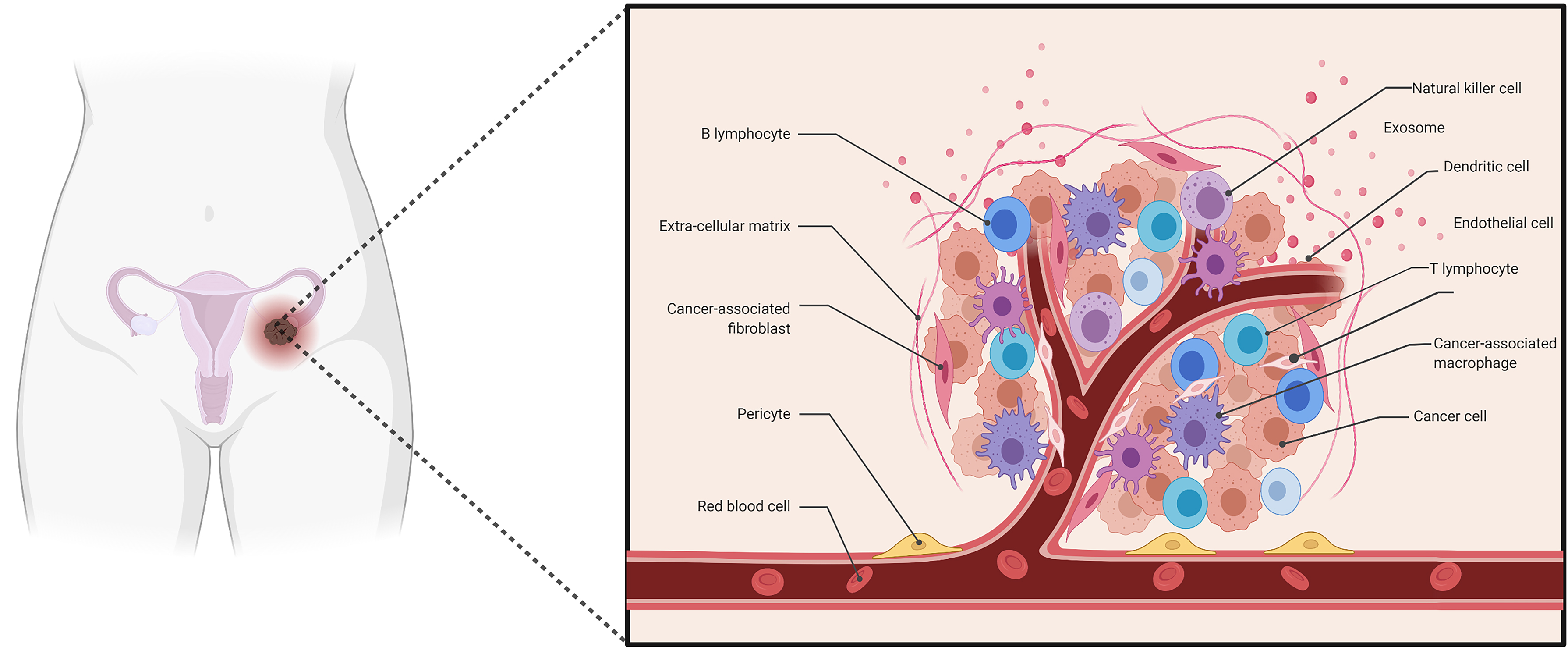 Fig. 2.
Fig. 2.Schematic of the composition of ovarian cancer microenvironment. The composition of the ovarian cancer microenvironment includes ovarian cancer cells, stromal cells, immune cells, pericytes and extracellular matrix (ECM), consisting of inflammatory cytokines, chemokines and other secreted molecules.
Endothelial cells, which form a monolayer to line blood vessels, not only separate circulating blood from tissues but also offer maintain metabolic homeostasis, deliver immune cells, and participate in the formation of new blood vessels [65]. In addition, tumor cells connect to ECs, altering the endothelial barrier and thereby facilitating cancer cell migration, invasion, and metastasis [9]. To meet tumor metabolic demands, ECs have to adapt to the environment. The upregulation of the glucose transporter GLUT1 in the tumor endothelium [64, 66] and the ability of tumor-derived VEGF to activate phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) expression [67] suggest that ECs have enhanced lactate dehydrogenase B expression and are likely to further boost glycolysis. To guarantee efficient energy generation essential for tumor cell motility and invasion, ECs co-compartmentalize glycolytic enzymes with actin-rich areas in invading structures, such as filopodia. Although the functional significance of EC glutamine metabolism and the level of importance of the two GLS isoforms (GLS1 vs. GLS2) are unknown, blocking GLS causes decreased EC proliferation and increased senescence [68], implying that glutamine plays a critical role in tumor vessel sprouting.
CAFs, derived from different tissue-resident fibroblasts in the TME, have the ability to produce a wide range of extracellular components, such as growth factors, cytokines, and ECM components, all of which contribute to tumor progression, metastasis, neoangiogenesis, ECM remodeling, and immunosuppression [65].
Adipocytes are unique in the TME because they can manage the energy balance and store excess energy as fat by secreting metabolites, enzymes, hormones, growth factors, and cytokines. Metalloproteases, such as MMP-1, MMP-7, MMP-10, MMP-11, and MMP-14, are secreted by adipocytes and play a crucial role in ECM changes [65]. In ovarian cancer, the presence of adipose-derived stem cells (ADSCs) in the TME and their transformation into CAFs are essential to promote growth, survival, EMT, and the acquisition of a tumor stem cell-like phenotype [69].
Generally speaking, immune cells in TME include macrophages, dendritic cells, neutrophils, mast cells, myeloid-derived suppressor cells (MDSCs), and lymphocytes [9]. They play vital roles in both tumor progression and tumor suppression through various signalling molecules, such as EGF, VEGF, IFNs, and ILs.
‘T cells exhaustion’ has been observed in a variety of tumors, which characterized by a non-responsiveness and loss of the effector function state caused by constant antigen exposure, and exhibit the upregulation of the expression of the immune-inhibitory programmed death receptor 1 (PD1) and the cytotoxic T-lymphocyte antigen-4 (CTLA4) [70]. On the one hand, hypoxia stimulates PD-L1 expression in tumor cells, tumor-associated macrophages (TAMs), and MDSCs, leading to T cell incompetence via the PD-L1/PD-1 axis; on the other hand, the hypoxic and acidic environment stimulates the immunosuppressive properties of regulatory T cells (Tregs) and TAMs, which actively inhibit the functions of T cells [71]. During activation of T cells, metabolic reprogramming imprints distinct functional differentiation. Aerobic glycolysis promotion is observed during differentiation to effectors [70]. In addition, with PD-1 ligation, activated T cells are unable to engage in glycolysis or amino acid metabolism which decrease the anti-tumor ability [72]. Due to the up-regulation of glycolysis, large amounts of lactic acid is accumulated in TME. lactic acid suppresse the proliferation of Th1 and Th17 cells and their anti-tumor effect [73].
Glycolysis, however, is not the only process required for T-cell activation. c-Myc-dependent glutaminolysis is also required for appropriate T-cell effector function because it leads to the creation of nucleotides and polyamines, which are required for cell proliferation [70]. Accumulation of the dicarbonyl radical methylglyoxal, a by-product of glycolysis produced by cysteine-sensitive amine oxidase, leads to a metabolic phenotype of MDSCs and MDSC-mediated paralysis of CD8+ T cells; the neutralization of dicarbonyl compound activity overcomes MDSC-mediated T cell suppression and improves the efficacy of cancer immunotherapy in conjunction with checkpoint inhibition in a mouse cancer model [74].
Tregs are a group of tumor-infiltrating immunosuppressive CD4+ T cells that rely
primarily on fatty acid oxidation (FAO) for energy, enabling survival in the
nutrient-depleted TME [75]. In the oxygen-depleted TME, HIF-1
Additionally, T cells can interact with fibroblasts to impact ovarian tumor
chemotherapeutic responses. It has been shown that gamma-glutamyltransferase
(GGT) is activated by CD8+ T cell-derived IFN
In terms of non-cellular components of the TME, the ECM, which is composed of collagen, fibronectin, elastin, and laminin, provides a physical scaffold for cells but also plays a vital role in promoting tumor metastasis [65]. Additionally, exosomes, including proteins, RNA, DNA, and lipids, which are derived from cellular parts of the TME, are the bridge between cancer cells and stromal cells [9]. Non-tumor cell metabolic interactions in the ovarian cancer microenvironment was shown in Fig. 3.
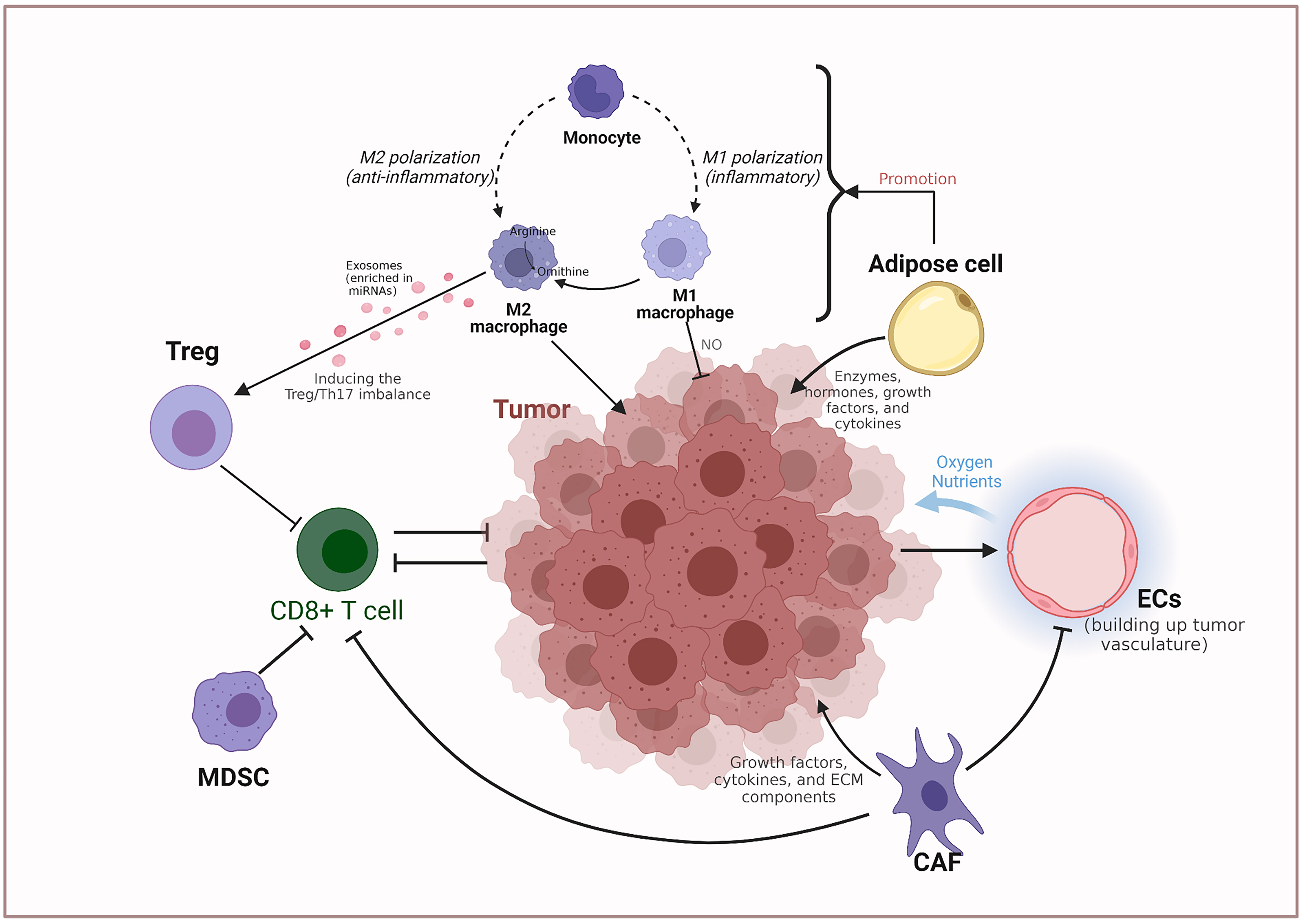 Fig. 3.
Fig. 3.Non-tumor cell metabolic interactions in the ovarian cancer microenvironment. CAF, Cancer-associated fibroblast; EC, Endothelial cell; MDSC, Myeloid-derived suppressor cell; NO, nitric oxide; Th17, IL-17-producing T helper; Treg, Regulatory T cell.
There is exchange and competition between the metabolism of tumor cells and non-tumor cells in ovarian cancer. This relationship will be discussed, with a focus on glucose metabolism, amino acid metabolism, lipid metabolism, and macromolecules (exosomes).
The abnormally high metabolic rate and nutrient depletion of tumor cells may
affect competition with neighboring T cells, leading to a decrease in the glucose
concentration in the ECM, which may lead to T cell metabolic depletion and
consequently affect the function of T cells [80, 81]. Song et al. [82]
have shown that ovarian cancer ascites restrict glucose uptake, thereby causing
CD4+ T cell IRE1
In the TME, lactic acid exerts multiple effects on various cell types, including effects on local pH and regulatory effects on cell metabolism and/or the redox status. The reverse Warburg effect complements the Warburg effect in terms of energy metabolism, where the interaction between cancer cells and adjacent stromal fibroblasts relies on lactate shuttle (MCT1 and MCT4), through which high-energy metabolites are transported from cells exhibiting aerobic glycolysis to OXPHOS-increasing cancer cells [87, 88]. Lactate secreted by cancer cells increases the synthesis of IL-17A, which inhibits the anti-tumor processes mediated by T cells [73]. Moreover, lactate can recruit and polarize immunosuppressive cells, such as regulatory T cells, TAMs, and MDSCs, further suppressing antitumor immune responses [89]. Lactic acid also blocks the differentiation of dendritic cells, limits their function, and leads to tumor escape from immune monitoring [90]. Tumor cell-derived lactic acid promotes the differentiation and polarization of TAMs, which has an important signaling role in the induction of polarization and the subsequent promotion of tumor growth [91]. M2-like TAMs produce immunosuppressive cytokines (such as IL10), chemokines, enzymes, and exosomes, which promote tumor invasion and immunosuppression, and enable cancer cells to survive, proliferate, and metastasize [92]. Decreased lactic acid levels can improve the efficacy of anti-tumor immunotherapy [93].
Glutamine is a dispensable amino acid in the human body [94]. However, for proliferative tumors, it is the second most essential nutrient after glucose and is stably converted to glutamate by phosphate-dependent glutamine enzymes (GLS) located in the inner membrane of mitochondria [95, 96, 97]. In low glutamine conditions, glutamine dependence drove CAFs migration toward free glutamine and facilitate the invasion of tumor cells [98]. Cancer cells influence CAFs metabolism to maximize Gln synthesis, and in a symbiotic manner (sustaining nucleotide generation and OXPHOS), CAF-derived Gln influences cancer cell metabolism and growth [13, 99]. Shen et al. [100] have shown that GLS inhibitor therapy effectively treats chemotherapy-resistant ovarian cancer, especially in cases with high GLS expression. Therefore, blocking glutamine secretion by CAFs or increasing glutamine decomposition in tumor cells is a potential target for the treatment of ovarian cancer.
Omental adipose stromal cells in the TME also communicate with ovarian cancer cells via arginine metabolism. Ovarian cancer cells use omental adipose stromal cell-secreted arginine and, in turn, secrete citrulline in the TME by the conversion of L-arginine to nitric oxide (NO) and citrulline by inducing nitric oxide synthase (NOS). NO promotes glycolysis and tumor cell proliferation, while citrulline is captured by stromal adipocytes and is converted into arginine. Secreted arginine is used by tumor cells to form a symbiotic cycle of metabolism [101, 102]. Arginine succinate synthase 1 (ASS1) is a rate-limiting enzyme in arginine synthesis in the urea cycle. Cells lacking ASS1 expression show arginine auxotrophy and sensitivity to arginine depletion. Tumors lack ASS1 and cannot synthesize endogenous arginine, which is completely dependent on exogenous sources. In ovarian cancer cell lines, ASS1 silencing resulted in resistance to platinum-based drugs and sensitivity to arginine deficiency [103]. T cells are sensitive to low-arginine environments. Intracellular L-arginine concentrations directly affect metabolic processes and the survival capacity of T cells. Therefore, supplementation with arginine and the prevention of arginine degradation are emerging strategies for enhancing the function of T cells [104, 105]. In addition to these, the indoleamine 2,3-dioxygenase (IDO) pathway is activated in ovarian cancer [106]. IDO promotes the local suppression of effector T cells by metabolically depleting tryptophan from the TME, producing tryptophan metabolites and generating broader systemic tolerance by activating circulating Treg cells [107]. The inhibition of targets in the IDO pathway may be an effective strategy for the treatment of ovarian cancer.
Fatty acids are required for the malignant progression of cancer, and
rate-limiting enzymes for lipid metabolism may be therapeutic targets [108].
Ovarian cancer cells prefer to metastasize to the omentum, which is rich in fat
cells and provides fatty acids for tumor cell growth [109]. Adipose cells and
adipose tissue directly mediate the tumor-promoting effects of ovarian cancer.
Fatty acids are used in the synthesis of biological macromolecules via
Extracellular vesicles (EVs) are released by tumor cells or non-tumor cells. EVs mediate the transfer of proteins, lipids, and nucleic acids between tumor or non-tumor cells to alter the function and phenotype of recipient cells. Molecules that were not previously thought to be exchanged between cells (such as mRNAs and microRNAs) are transferred by EVs too [12]. Paracrine secretion is a channel for macromolecular communication between ovarian cancer cells and the TME, such that ovarian cancer cells and CAFs maintain a glycolytic phenotype by the paracrine long noncoding RNA LINC00092 [112]. Moreover, ovarian cancer cells secrete lipid membrane vesicles (known as exosomes), which contribute to the invasive phenotype of ovarian cancer cells by activating intracellular signaling pathways that generate tumor-associated myofibroblasts from MSCs in the tumor mesenchyme [113]. Ovarian cancer cells regulate fibroblast behavior by releasing EVs that induce phenotypic and functional changes in normal stromal fibroblasts, resulting in a CAF-like state, and EVs released from a highly aggressive cell line (SKOV3) appear to be more effective in stimulating certain processes than those of a less aggressive cell line (CABA I) [114]. In a ovarian cancer mouse model, ovarian cancer cells secrete EVs carrying ARG1; these EVs are taken up by dendritic cells, inhibit the proliferation of antigen-specific T cells, and promote ovarian cancer progression [115]. Autophagy may be an important mechanism underlying the transport of nutrients from CAFs to tumor cells [116]. Increasing evidence strongly suggests that the molecules carried by EVs, like exosomes, promote the development of platinum resistance in EOC tissues, which is conducive to the adaptive pathological response of cancer cells to drugs [117].
Metabolic interactions between ovarian cancer cells and various non-tumor cells present in the TME are summarized in Fig. 4.
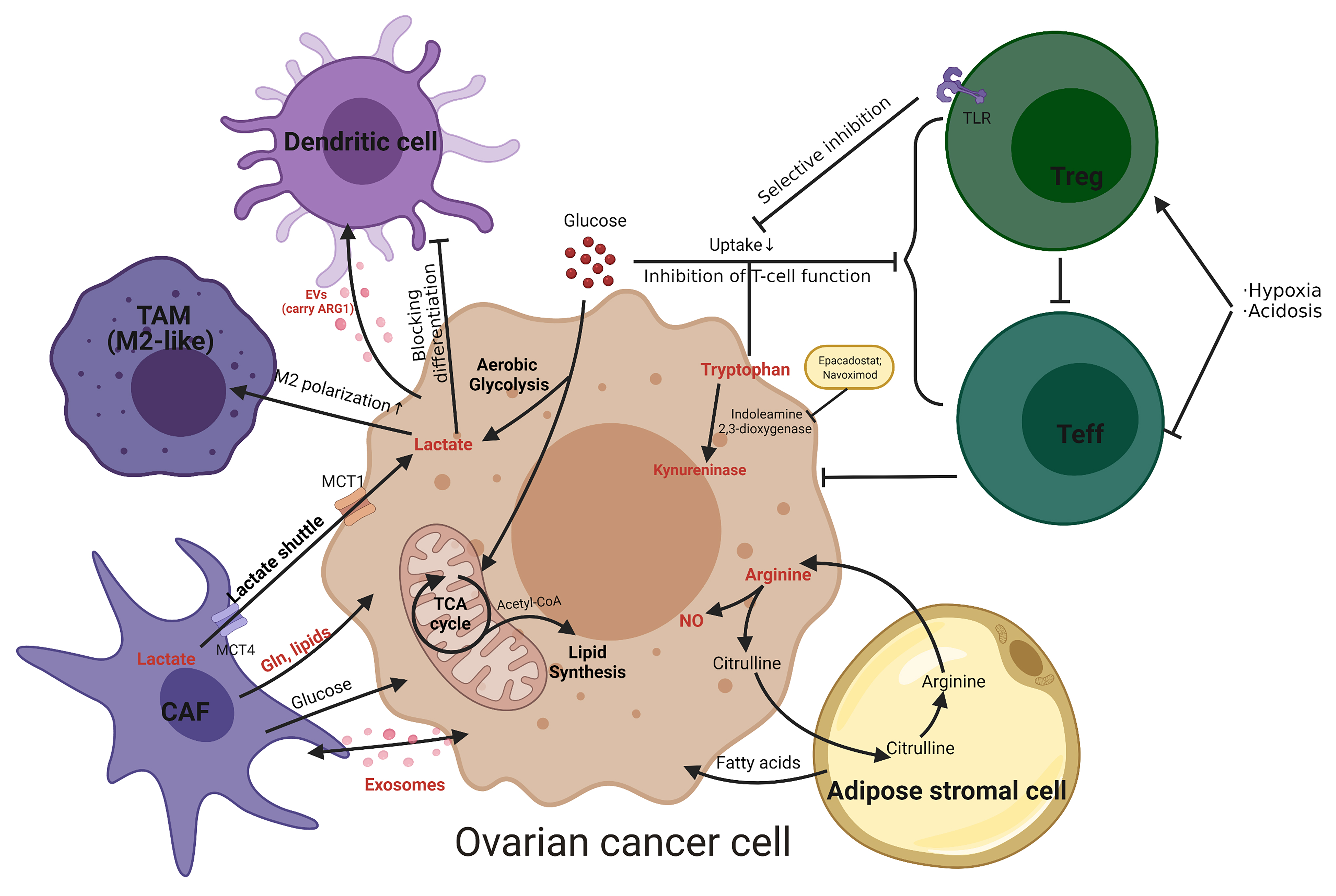 Fig. 4.
Fig. 4.Metabolic interactions between ovarian cancer cells and non-tumor cells present in the TME. Namely tumor-associated macrophages (TAM), cancer-associated fibroblasts (CAF), adipose stromal cells, effector T cells (Teff), regulatory T cells (Treg) and dendritic cell. Metabolites are shown in red. The metabolic inhibitors are shown in the yellow box. Gln, glutamine; NO, nitric oxide; MCT, monocarboxylate transporters; TLR, Toll-like receptors; EVs, extracellular vesicles; ARG1, arginase 1.
As mentioned above, there is a close metabolic interaction between ovarian cancer and the corresponding TME. Knowing more about the connecting between metabolism and ovarian cancer microenvironment is important for us to find new target for ovarian cancer therapies. Many studies have found that interrupting this metabolic exchange can effectively stop the progression of cancer, thus becoming a new idea for the treatment of ovarian cancer [118]. In what follows, we would summarize the effects of metabolic interventions on ovarian cancer cells and the TME (Table 1, Ref. [48, 49, 51, 100, 119, 120, 121, 122, 123, 124, 125]). Studies of the metabolic link between ovarian cancer and the TME are ongoing, and clinical trials of novel drugs targeting metabolic targets are scarce.
| Target | Metabolic agent | Effections on tumor | Stage and subjects | Side effect | Refs |
| V-ATPases | bafilomycin A1 | Inhibition proliferation and migration | Preclinical study in cells | / | [49] |
| Proton pump | esomeprazole (EMSO) | Increasing the sensitivity to PTX, reducing autophagy; promoting apoptosis. | Preclinical study in cells | / | [48] |
| Carbonic anhydrase (CA) | Topiramate (TPM) | Inhibition proliferation; induce apoptosis; anti-metastatic | Preclinical study in cells | / | [51] |
| HIF-1 |
2-methoxyestradiol | Anti-angiogenesis and growth | Hoosier OncologyGroup trial in patients with recurrent, platinum-resistant EOC. | fatigue (78%) nausea (78%), no drug-related serious adverse event occurred | [121] |
| mTOR |
Temsirolimus | Limiting the proliferation and progression of ovarian cancer, enhancement of bevacizumab viability | Phase I study in patients with gynecologic cancers (54% weith ovarian cancer) | thrombocytopenia (10%), mucositis (2%), hypertension (2%), hypercholesterolemia (2%), fatigue (7%), elevated aspartate aminotransferase (2%), and neutropenia (2%) | [119] |
| MCTs |
Lonidamine | Inhibition of the growth and DNA repair process of ovarian cancer cells and enhancement of cisplatin/carboplatin/paclitaxel viability | Phase II study in patients with advanced ovarian cancer | myalgia (9%), gastric pain (6%) , vomiting (6%) | [120] |
| Glutaminase | CB-839 | Induction of oxidative and replication stress, acting synergistically with PDL1 | Preclinical study in cells and mouse model | / | [100, 122] |
| IDO |
Epacadostat (INCB024360); Navoximod (GDC-0919) | Improving the activity of chemotherapy agents | Epacadostat: phase II study in patientsNavoximod: phase I study in patients | Epacadostat: fatig ue (36.4%) immune-related adverse events: primarily rash (18.2%) pruritus (9.1%) Navoximod: fatigue (22%) rash (22%) | [123, 124] |
| SCD1 |
MF-438; CAY10566 | Induction of lipid oxidation, ferroptosis and apoptosis in ovarian cancer cells | Preclinical study in cells and mouse model | / | [125] |
Both in vitro and in vivo studies have shown that glycolysis
plays a crucial role in EOC. When glycolysis is inhibited in vitro,
growth, invasion, migration, and viability of EOC cells are restricted.
Similarly, when glycolysis is disrupted in vivo, tumor growth or volume
is significantly reduced [126]. Therapeutic strategies targeting glycolysis focus
on several key enzymes and regulators, including HIF-1
ICI works by interfering with T cell surface PD1 and programmed death ligand-1 (PDL1); however, single-agent treatment with ICI has shown little survival benefit in ovarian cancer and is limited by acquired resistance and treatment failure due to the immunosuppressive TME [17]. Glutamine blockade suppresses the metabolic program of cancer cells and enhances the anti-tumor immune response [129] thus, glutaminase inhibition alone or in combination with immune checkpoint blockade is an effective therapeutic strategy. Ovarian clear cell carcinoma (OCCC) with ARID1A mutations is dependent on glutamine metabolism, and ARID1A inactivation upregulates GLS1 to increase glutamine utilization and metabolism via the TCA cycle [122]. The glutaminase inhibitor CB-839 inhibits the growth of ARID1A mutants in situ and in patient-derived xenografts and acts synergistically with anti-PDL1 antibodies in a conditional inactivation Arid1a-driven genetic mouse model of OCCC [100, 122]. In addition, the IDO pathway, an important pathway in tryptophan metabolic communication, is a key regulator of immune tolerance in ovarian cancer. The IDO1 enzyme inhibitor epacadostat (ClinicalTrials.gov NCT01685255) was generally well tolerated in an open-label, phase II clinical trial [123]. The most common treatment-emergent adverse event was fatigue (epacadostat, 36.4%). Immune-related adverse events, observed with epacadostat only, were primarily rash (18.2%) and pruritus (9.1%) [123]. The combination of the IDO inhibitor navoximod (GDC-0919) and the PD-L1 inhibitor atezolizumab also demonstrated an acceptable safety, tolerability, and pharmacokinetic profile in an interventional clinical trial (ClinicalTrials.gov NCT02471846) in patients with advanced cancer, including ovarian cancer [124]. The most common treatment-related adverse events in this trial were fatigue (22%) and rash (22%) [124].
With respect to lipid metabolism, Stearoyl-CoA desaturase (SCD1, SCD), which is highly expressed in ovarian cancer cells, is the rate-limiting enzyme that catalyzes the synthesis of monounsaturated fatty acids [125]. The inhibition of SCD1 induces lipid oxidation and cell death and significantly enhances the anti-tumor effects of ferroptosis inducers [125], suggesting that SCD1 is a promising therapeutic target.
The metabolic agents discussed in this review for the treatment of ovarian cancer are summarized in Table 1.
In the past decade, extensive research has focused on the TME and tumor metabolism. Remarkable progress has been made in our understanding of the complex network of interactions between cancer cells and the TME. The concept of metabolomics and the development of related technologies have contributed to a deeper insight into cancer metabolism [130]. The TME is clearly not just a silent bystander but acts as a double-edge sword in cancer-related processes. It contributes to physical strain, oxidative stress, nutritional shortages, competition, hypoxia, and immune surveillance [131]. Conversely, it can actively promote cancer progression; in the early stage of tumor growth, cancer cells and components of the TME form a complex and mutually reinforcing connection that promotes cancer cell survival, local invasion, and metastatic spread. The TME reinforces angiogenesis to reintroduce oxygen and nutrition delivery while also removing metabolic waste to overcome the hypoxic and acidic microenvironment. Tumors are surrounded by a variety of adaptive and innate immune cells that can promote and prevent growth [65].
Ovarian cancer cells alter the TME by consuming and secreting metabolites (glucose, lactic acid, amino acids, fatty acids, biomolecules, etc.), in turn affecting the phenotype of tumor cells [21]. This intricate metabolic interaction shapes the unique metabolic properties of the TME that maintain tumor growth and enable immune invasion. However, this complementary cause-effect relationship remains to be demonstrated.
The recent development of single-cell sequencing technology has provided a new perspective on cancer etiology, progression, and drug resistance. Single-cell sequencing provides a method to identify subpopulations of cancer cells in individual patients, that is, molecular classification at the single-cell level [132, 133, 134]. Winterhoff et al. [132] identified two major cell subpopulations in high-grade plasmacytoid ovarian cancer based on gene expression patterns: (1) an epithelial cell group with increased proliferation-related gene expression and (2) a stromal group characterized by increased expression levels of genes related to the ECM and EMT. Hu et al. [133] revealed a strong association between different secretory cell populations and high-grade plasmacytoid ovarian cancer subtypes by a combined analysis of single-cell data and tumor expression data and identified a link between the subtypes with high EMT scores and outcomes. Studies on the single-cell landscape of ovarian cancer can lay a foundation for accurate prognostic and therapeutic stratification. In parallel, single-cell spatial biology is another approach to study metabolic communication in ovarian cancer and its TME. Zhang et al. [135] used lipid droplet dynamics evaluated by stimulated Raman scattering imaging to quantify lipid metabolism in ovarian cancer. Zhu et al. [136] developed a SpatioImageOmics (SIO) pipeline to identify lipids associated with ovarian tumor microenvironment-related prognostic biomarkers. These studies clearly demonstrate the advantages of single cell spatial biology over conventional approaches for understanding the heterogeneity of ovarian cancer. This approach has implications for precision therapy and may be a direction for future research [136].
To comprehensively target tumor metabolism for anticancer therapy, it is necessary to identify targets related to the autonomous metabolism of ovarian cancer cells as well as the metabolism of non-tumor cells in the TME. Metabolic therapy is a new paradigm in tumor treatment. The application of metabolic enzyme inhibitors in ovarian cancer treatment can lead to the development of candidate drugs for ovarian cancer. In parallel with the targeted modulation of metabolic enzymes at the tumor site, integrative strategies aimed at altering systemic metabolic levels and the metabolic effects of diet are promising [130, 137]. By modifying metabolite concentrations and reshaping the TME, chronicization of ovarian cancer is expected to achieve treatment goals. Drugs that target metabolism can effectively inhibit the survival and growth of ovarian cancer cells at the cellular level; at the same time, their effects on normal cells and methods to specifically target ovarian cancer are important future directions in the area of metabolic therapy.
AA, Arachidonic acid; ADSC, Adipose derived stem cell; AKT, Protein kinase B; ARG1, Arginase 1; ASS1, Arginine succinate synthase 1; ATP, Adenosine triphosphate; CA, Carbonic anhydrase; CAF, Cancer-associated fibroblast; CTLA4, Cytotoxic T-lymphocyte antigen-4; DNA, Deoxyribonucleic acid; EC, Endothelial cell; ECM, Extracellular matrix; EGF, Epidermal growth factor; EMSO, Esomeprazole; EMT, Epithelial-mesenchymal transition; EOC, Epithelial ovarian cancer; EV, Extracellular vesicle; FAO, Fatty acid oxidation; FoxP3, Forkhead box protein P3; GGT, Gamma-glutamyltransferase; GLS, Glutaminase; GLUT, Glucose transporter; Gln, Glutamine; GSH, Glutathione; HIF-1, Hypoxia-inducible factor 1; HRE, Hypoxic regulatory element; ICI, Immune checkpoint inhibitor; IDO, Indoleamine 2,3-dioxygenase; IFN, Interferon; IL, Interleukin; iNOS, Inducible nitric oxide synthase; LDHA, Lactate dehydrogenase A; MAPK, Mitogen-activated protein kinase; MCT, Monocarboxylate transporter; MDSC, Myeloid-derived suppressor cell; MMP, Matrix metalloproteinase-1; MSC, Mesenchymal stem cell; mTOR, Mammalian target of rapamycin; NCD, NanoCrystal® dispersion; NO, Nitric oxide; NOS, Nitric oxide synthase; OCCC, Ovarian clear cell carcinoma; OXPHOS, Oxidative phosphorylation; PAS, Per/Arnt/Sim; PARPi, Poly ADP-ribose polymerase inhibitor; PD1, Programmed death receptor 1; PDL1, Programmed death ligand-1; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; PI3K, Phosphatidylinositol 3 kinase; PK, Pyruvate kinase; PKM2, Pyruvate kinase M2; RNA, Ribonucleic Acid; PPI, Proton pump inhibitor; PTX, Paclitaxel; SCD1, SCD, Stearoyl-CoA desaturase; TAM, Tumor-associated macrophage; Teff, Effector T cell; Th17, IL-17-producing T helper; TLR, Toll-like receptor; TME, Tumor microenvironment; TPM, Topiramate; Treg, Regulatory T cell; V-ATPase, Vacuolar H+–ATPase; VEGF, Vascular Endothelial Growth Factor; 2ME2, Panzem.
XZ and XL designed the study. XJZ and YN provided help and advice on grammar. YL and XL wrote and revised the manuscript. YN and XTZ help to design the figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We acknowledge BioRender for design support for the figures.
This research was funded by the National Natural Science Foundation of China, grant number No. 81902662; National Natural Science Foundation of China, grant number No. 81821002; Sichuan Science and Technology Program, grant number 2021YJ0011; Foundation of Development and Related Diseases of Women and Children Key Laboratory of Sichuan Province grant number No. 2022003 and The APC was funded by West China Second Hospital.
The authors declare no conflict of interest.