








1 Department of Life Sciences, Yeungnam University, 38541 Gyeongsan, Gyeongsangbuk-do, Republic of Korea
Academic Editor: Kevin Cianfaglione
Abstract
Background: Cardamine flexuosa is considered to be two
separate species in the Cardamine genus based on their geographical
distribution: European C. flexuosa and Eastern Asian C. flexuosa. These
two species have not shown any morphological differences to distinguish each
other. Recently, the Eastern Asian species has been regarded as Cardamine
occulta by their ecological habitats. Therefore, we are interested in analyzing
the C. occulta chloroplast genome and its characteristics at the
molecular level. Methods: Here, the complete chloroplast (cp) genome of
C. occulta was assembled de novo with next-generation sequencing
technology and various bioinformatics tools applied for comparative studies.
Results: The C. occulta cp genome had a quadripartite
structure, 154,796 bp in size, consisting of one large single-copy region of
83,836 bp and one small single-copy region of 17,936 bp, separated by two
inverted repeats (IRa and IRb) regions of 26,512 bp. This complete cp genome
harbored 113 unique genes, including 80 protein-coding genes, 29 tRNA, and four
rRNA genes. Of these, six PCGs, eight tRNA, and four rRNA genes were duplicated
in the IR region, and one gene, infA, was a pseudogene. Comparative
analysis showed that all the species of Cardamine encoded a small
variable number of repeats and SSRs in their cp genome. In addition, 56
divergences (Pi
Keywords
- Cardamine
- C. occulta
- chloroplast genome
- sequence divergence
- phylogeny
- hotspot regions
The chloroplast genomes have a stable and straightforward genetic structure, haploid, and are generally uniparentally transmitted [1]. This organelle is involved in plant cells for nitrogen fixation, photosynthesis, biosynthesis of starch, fatty acids, essential amino acids, and pigments [2, 3]. The cp genomes of flowering plants usually have a typical circular structure, 107–280 kb in length, that consists of a large single-copy (LSC) and a small single-copy (SSC) region, which are separated by two large, inverted repeats (IRs) region [4, 5]. Owing to the maternal inheritance characteristics, the nucleotide substitution rate of the cp genes is lower than that of nuclear genes but higher than that of the mitochondrial genes. Nevertheless, the rate of plastome genome evolution appears to be taxon and gene-dependent [6]. Therefore, techniques for analyzing the molecular phylogeny of plants are strongly dependent on plastome genome sequence data [7]. Thus far, more than 6100 land plant chloroplast genomes are available at the NCBI organellar genome database, which can be used for comparative studies to resolve the phylogenetic implications of the controversial clade. In addition, recent studies showed that the cp genome encompasses various polymorphic regions at both coding and non-coding regions generated through genomic expansion, contraction, inversion, indel, or genome rearrangement that could be used widely as an effective tool for plant phylogenomic analyses [8].
The genus Cardamine (bittercress) is one of the largest genera of the family Brassicaceae and is distributed widely across all the continents except Antarctica [9]. This genus comprises more than 200 exceptionally complex species and remains controversial and unresolved in many circumstances [10]. Among the Cardamine taxa, Cardamine flexuosa With. is distributed in Europe and Eastern Asia [10]. Moreover, the two taxa have not shown any morphological differences that can be used to distinguish these species [11]. Until 2006, the Eurasian taxa, C. flexuosa, is considered a single species [10]. From 2006 onwards, the C. flexuosa was considered two different species based on their location [12]. Recent studies showed that these two species differed by their ecological habitats and reported that the Eastern Asian C. flexuosa species should be considered C. occulta [9]. Differences were also found in the ploidy level. The tetraploid species C. flexuosa originated from Europe, whereas the octoploid C. occulta Hornem. is from Eastern Asia and introduced to other continents [11, 13]. The diploid species C. amaraeformis and C. hirsuta are the parental species for C. flexuosa. In contrast, the tetraploidy C. scutata (Diploid species C. amaraeformis and C. parviflora as the parents) and C. kokaiensis (Diploid species C. parviflora as the parent) are the parental for C. occulta [9].
Lihova et al. [10] reported that the populations of Eastern species C. occulta distinguished from the European species C. flexuosa based on the phylogenetic studies of internal transcribed spacer (ITS) region of rDNA and the trnL-trnF region of cpDNA. From the biogeographical perspective of C. flexuosa, the diploid parental species C. amaraeformis is currently absent from Eastern Asia, whereas the tetraploids C. scutata and C. kokaiensis are distributed in Eastern Asia [14]. Previous studies suggested that the morphologically close species of C. amaraeformis are C. torrentis Nakai, C. amariformis Nakai, and C. valida, present in Eastern Asia [15, 16]. Therefore, the diploid C. amaraeformis may have had a significantly broader dispersal area in the past, reaching easternmost Asia and contributing to multiple polyploidization events there [17]. A previous study characterized the cp genome of the parental species C. amaraeformis for C. occulta [18]. Therefore, the present study is interested in characterizing the complete chloroplast genome sequence of C. occulta (Asian C. flexuosa With.), and phylogenetic studies were carried out to resolve this issue. Moreover, there are no extensive comparative studies of the Cardamine genera at the whole plastome level. Therefore, this study compared the cp genome of C. occulta with other fourteen species of the Cardamine genomes and identified hotspot regions that could help develop the molecular markers to distinguish the controversial Cardamine species. Overall, this study will provide valuable information for understanding the evolutionary relationship of C. occulta in the Cardamine clade.
The fresh young leaves of Cardamine occulta were collected from
Cheongok Mountain, Bonghwa-gun, South Korea (geospatial coordinates:
N37
The online program Dual Organeller GenoMe Annotator (DOGMA) was accomplished to annotate the chloroplast genome sequence of C. occulta [23]. The initial annotation, putative starts, stops, and intron positions of homologous genes were improved by comparing with the closely related species of Cardamine. The transfer RNA genes were confirmed using the tRNAscan-SE version1.21 with default settings [24]. A circular cp genome map of the C. occulta was produced using the OrganellarGenome DRAW (OGDRAW) program [25].
The mVISTA program in the Shuffle-LAGAN model was applied to analyze the cp genome of C. occulta with 14 other closely related cp genomes of Cardamine genus, applying C. occulta annotation as a reference [26]. The boundaries between the IR and SC regions of all the genera of Cardamine were also compared and investigated.
The genetic divergence was investigated by extracting and aligning the
protein-coding genes, intergenic and intron-containing regions of 15
Cardamine species cp genome individually using Geneious Prime
(Biomatters, New Zealand). The genetic divergence among the Cardamine
species was estimated using nucleotide diversity (
The cp genome of C. occulta was compared with the other 14 species of
Cardamine cp genomes to determine the synonymous (K
Positive selection analysis was carried out based on the substitution analysis
of the Cardamine species. The site-specific model was applied to
estimate the non-synonymous (K
The program REPuter was used to determine the presence of repeat sequences in the Cardamine cp genomes, including forward, reverse, palindromic, and complementary repeats [30] The following parameters were used to detect repeats in REPuter: (1) Hamming distance 3, (2) minimum sequence identity of 90%, (3) and a repeat size of more than 30 bp. In addition, Phobos software v1.0.6 was used to find the SSRs in Cardamine cp genomes; parameters for the match, mismatch, gap, and N positions were set at 1, –5, –5, and 0, respectively [31]. Only one IR region was used in the repeat and SSR marker analyses.
This study used the cp genomes of 38 Brassicaceae species and two outgroup species for phylogenetic analysis based on 68 homologous CDs, LSC, SSC, and IR regions and the whole genomes separately. The 39 completed cp genome sequences were downloaded from the NCBI Organelle Genome Resource database (Supplementary Table 1). For ML analysis, the aligned protein-coding gene sequences were saved in PHYLIP format using Clustal X v2.1. Phylogenetic analysis was analyzed using the maximum likelihood (ML) method and the GTRGAMMA model using RAxML v. 8.2.X with 1000 bootstrap replications [29]. The same five individual data sets were also performed by Bayesian Markov chain Monte Carlo (MCMC) inference using the MrBayes v3.2.6 [32, 33] phylogenetic tree in Geneious Prime v2022.0.2. The gamma model of rate variation and the HKY85 substitution model were used for this analysis.
The complete Cardamine occulta chloroplast genome showed a quadripartite structure comprised of 154,796 bp, including a small single-copy (SSC) region of 17,936 bp and a large single-copy (LSC) region of 83,836 bp, which were separated by a pair of inverted repeats (IRa and IRb) of 26,512 bp (Fig. 1; Table 1). The average GC content of the cp genome was 36.3%. The IR regions had the highest GC content (42.4%), followed by the LSC (34%) and SSC regions (29.2%). The C. occulta cp genome encoded 113 unique genes: 80 protein-coding genes, 29 tRNAs and four rRNAs. Among the 113 genes, fourteen contained one intron (eight protein-coding and six tRNA genes), and three encoded two introns (clpP, ycf3, and rps12). The rps12 gene was a trans-spliced gene with its 5’- end exon located in the LSC region and its intron 3’-end exon duplicated in IR regions. In addition, 18 genes were duplicated in the IR regions (Supplementary Table 2).
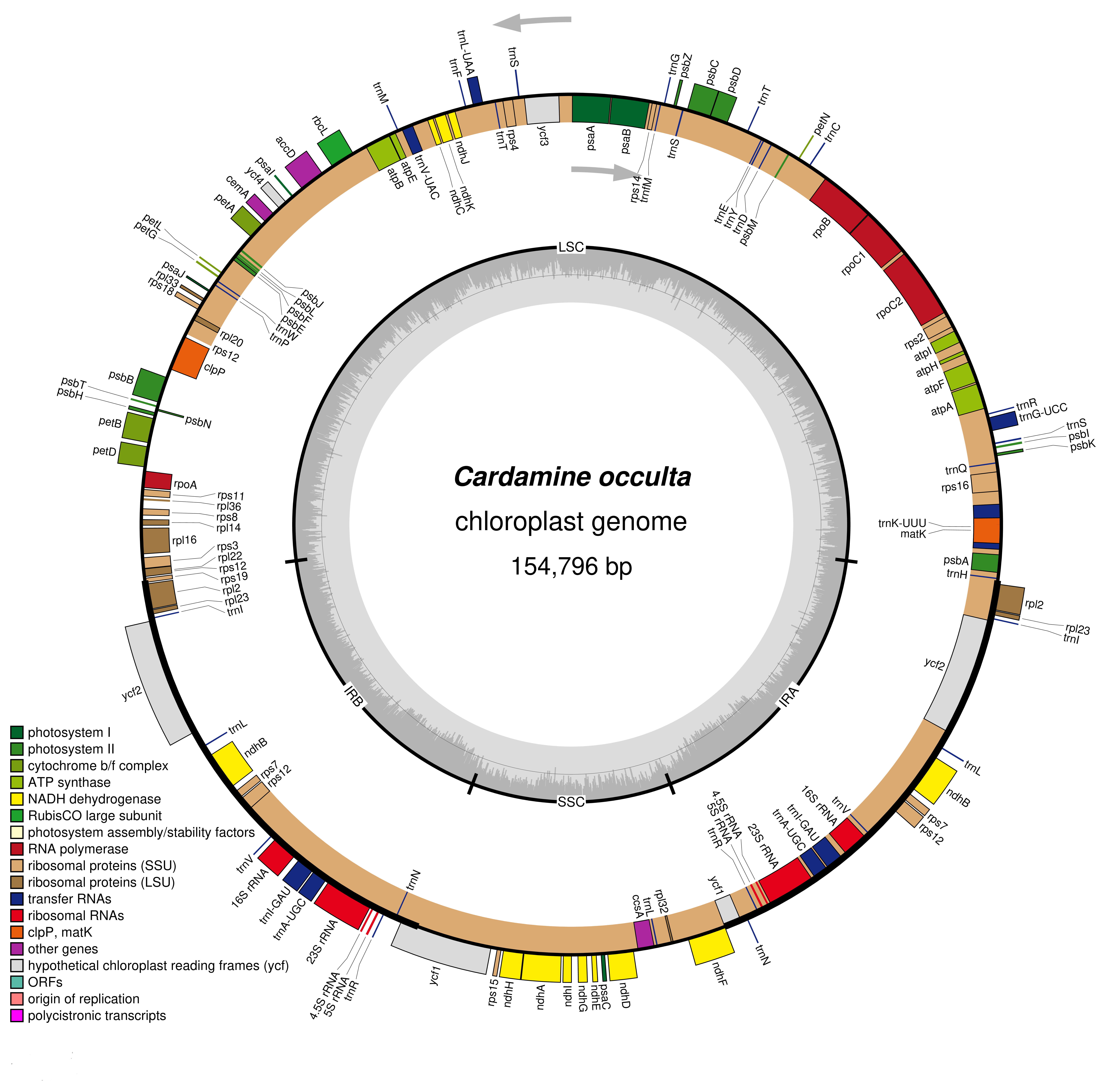 Fig. 1.
Fig. 1.Gene map of Cardamine occulta. Genes lying outside the outer circle are transcribed in a counter-clockwise direction, and genes inside this circle are transcribed in a clockwise direction. The colored bars indicate known protein-coding genes, transfer RNA genes, and ribosomal RNA genes. The dashed, dark grey area in the inner circle represents the GC content, and the light grey area implies the genome AT content. LSC, large single-copy; SSC, small single-copy; IR, inverted repeat.
| Genome features | Cardamine occulta |
| Total length (bp) | 154,796 |
| LSC length (bp) | 83,836 |
| SSC length (bp) | 17,936 |
| IR length (bp) | 26,512 |
| GC content (%) | 36.3 |
| Total genes | 131 |
| Genes duplicated in the IR region | 18 |
| Protein-coding genes | 80 |
| tRNA genes | 29 |
| rRNA genes | 4 |
The cp genome border LSC-IRb and SSC-IRa of C. occulta were compared with the other fourteen species of Cardamine genera (Fig. 2). The intact copy of the rps19 gene was distributed in the LSC/IRb border of all Cardamine species and dividends 106 bp to 135 bp in the IRb region resulting in the rpl2 gene being situated in the IRb region. Similarly, the pseudogene, ycf1, and ndhF are present in the IRa/SSC border of all the Cardamine cp genomes that exhibit overlap. The overlap of these two coding regions was conserved from 30–192 bp in the border of IRa/SSC of their cp genomes. In all the species of Cardamine genera cp genomes, the SSC-IRb junction contains the full-length ycf1 genes, whereas the IRa/LSC junction encodes the fragmented rps19 and trnH genes.
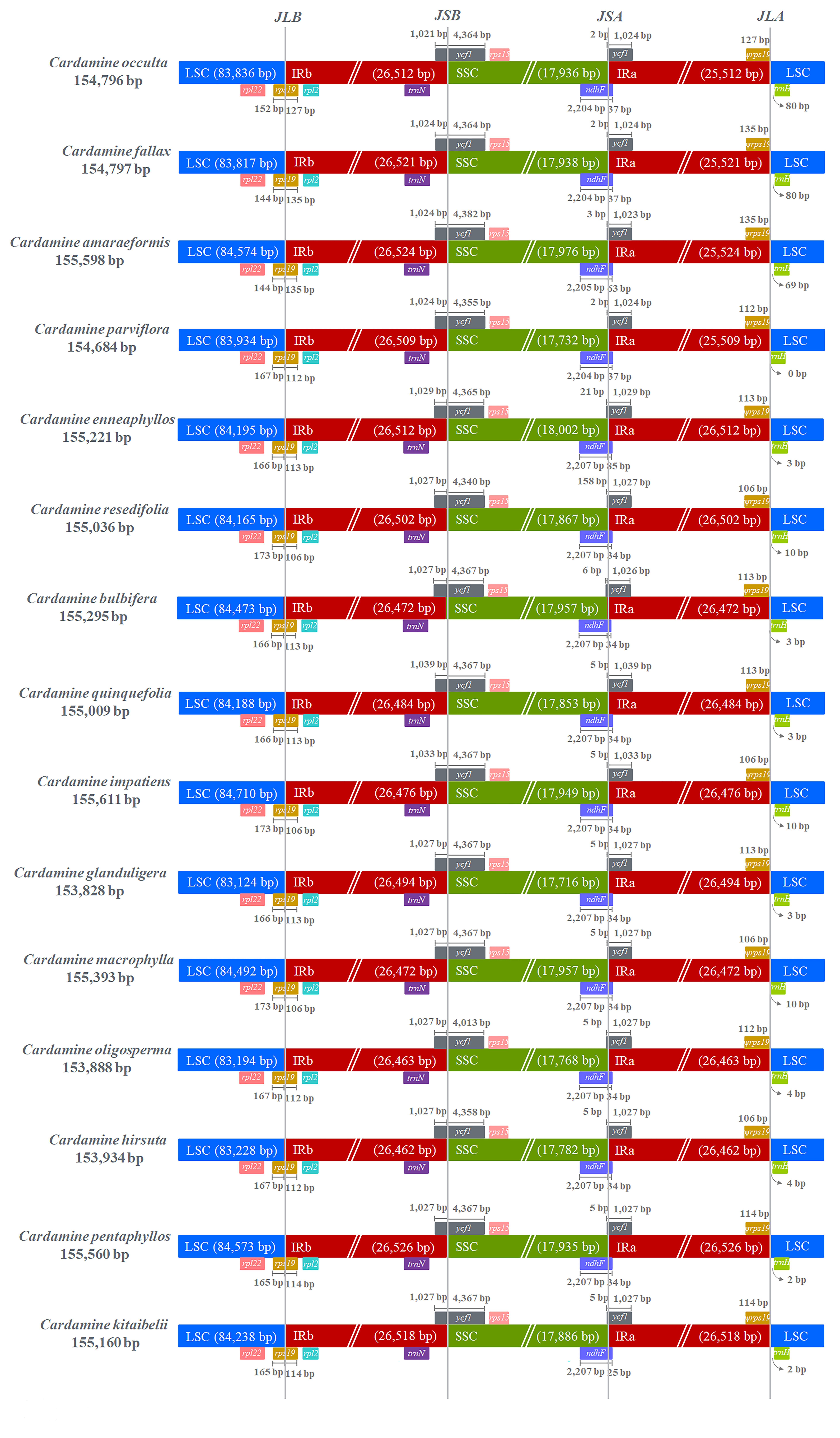 Fig. 2.
Fig. 2.Evaluation of the large single-copy (LSC), small single-copy
(SSC), and inverted repeat (IR) border regions of fifteen species of
Cardamine genera chloroplast genomes.
Genome-wide comparative analyses of the fifteen Cardamine cp genomes were achieved using mVISTA to estimate the level of sequence divergence. The cp genomes displayed strong sequence similarity, indicating that the plastomes are highly conserved (Fig. 3). Compared to the non-coding regions and single copy, the coding regions and IR were more conserved, with low variation among Cardamine.
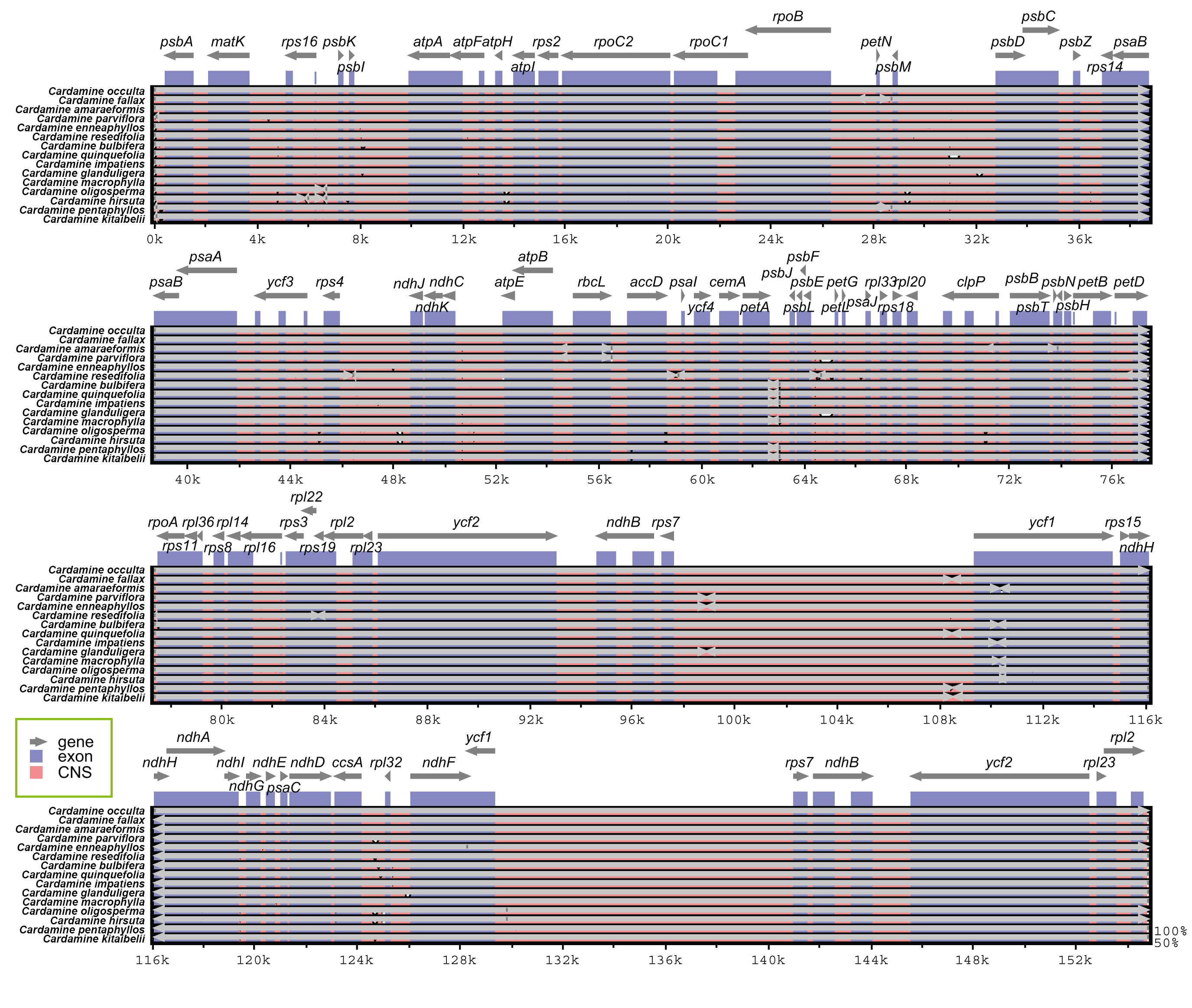 Fig. 3.
Fig. 3.Sequence alignment of fifteen species of Cardamine genera chloroplast genomes performed using the mVISTA program with Cardamine occulta as a reference. The top grey arrow shows genes in order (transcriptional direction) and the position of each gene. A 70% cut-off was used for the plots. The y-axis denotes a percent identity of between 50 and 100%, and the red and blue areas imply intergenic and genic regions, respectively.
The nucleotide diversity of 204 regions was evaluated using DnaSP software,
including 74 protein-coding genes and 128 intergenic and intron regions among
fifteen cp genomes of Cardamine genera. The results showed that the
maximum variable regions (
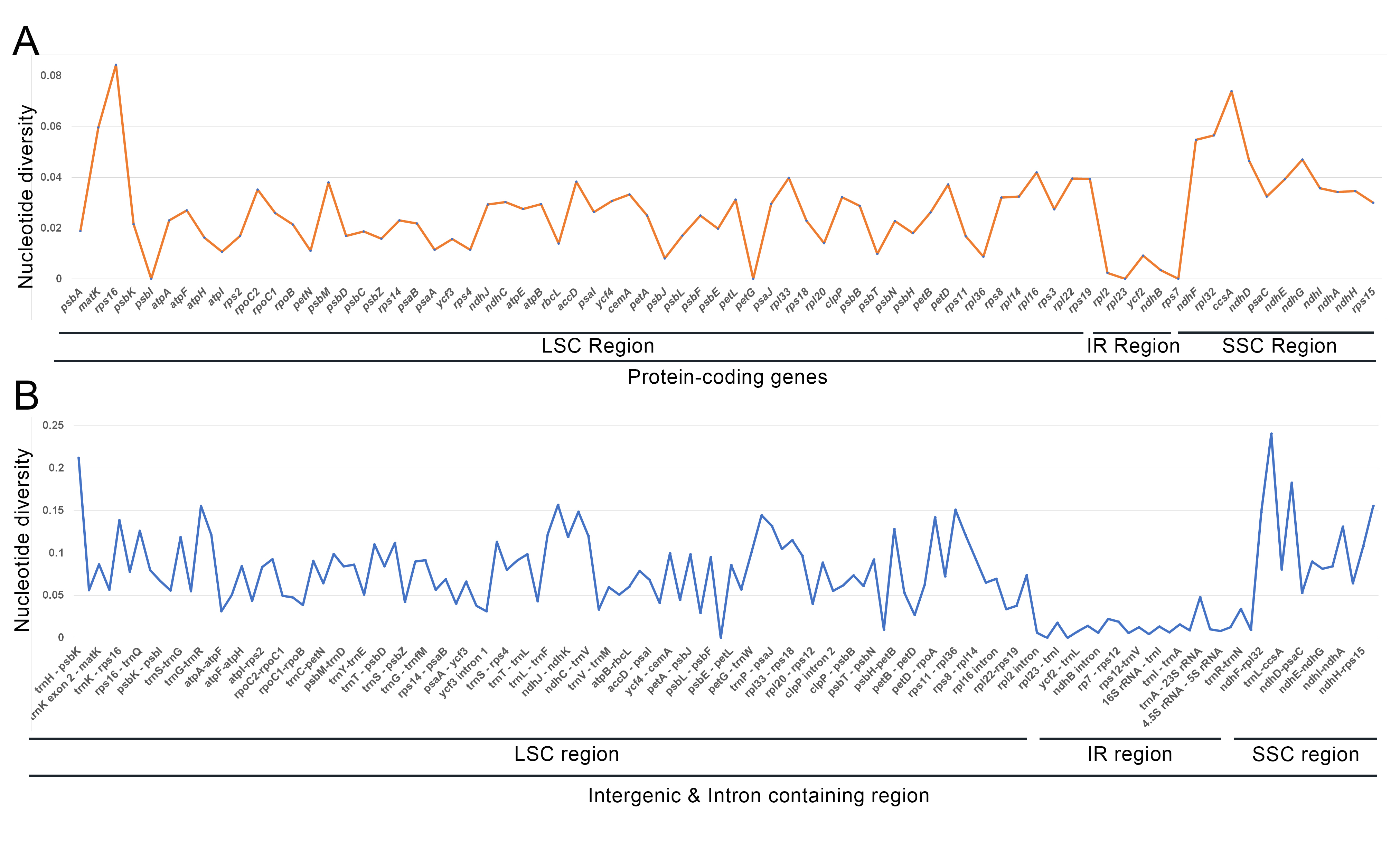 Fig. 4.
Fig. 4.Genetic diversity based on Kimura’s two-parameter model. (A) The P-distance value of protein-coding genes. (B) The P-distance value of intron and intergenic regions.
| Protein-coding regions | Nucleotide diversity (Pi) | Aligned length (bp) | No. of variable sites | IGS & Intron regions | Nucleotide diversity (Pi) | Aligned length (bp) | No. of variable sites |
| matK | 0.059642 | 1509 | 90 | trnH-psbK | 0.211765 | 170 | 36 |
| rps16 | 0.084388 | 237 | 20 | trnK-rps16 | 0.138756 | 418 | 58 |
| rpoC2 | 0.03514 | 4041 | 142 | rps16-trnQ | 0.125891 | 421 | 53 |
| psbM | 0.038095 | 105 | 4 | trnS-trnG | 0.118677 | 514 | 61 |
| ndhC | 0.030303 | 363 | 11 | trnG-trnR | 0.155405 | 148 | 23 |
| accD | 0.038298 | 1410 | 54 | trnR-atpA | 0.121107 | 289 | 35 |
| ycf4 | 0.030631 | 555 | 17 | trnE-trnT | 0.11 | 400 | 44 |
| cemA | 0.033333 | 690 | 23 | psbC-trnS | 0.111732 | 179 | 20 |
| petL | 0.03125 | 96 | 3 | ycf3-trnS | 0.112971 | 239 | 27 |
| rpl33 | 0.039801 | 201 | 8 | trnL-trnF | 0.121302 | 338 | 41 |
| clpP | 0.032149 | 591 | 19 | trnF-ndhJ | 0.156334 | 371 | 58 |
| petD | 0.037267 | 483 | 18 | ndhJ-ndhK | 0.118812 | 101 | 12 |
| rps8 | 0.032099 | 405 | 13 | ndhK-ndhC | 0.148148 | 54 | 8 |
| rpl14 | 0.03252 | 369 | 12 | ndhC-trnV | 0.12 | 800 | 96 |
| rpl16 | 0.041975 | 405 | 17 | petG-trnW | 0.100775 | 129 | 13 |
| rpl22 | 0.039583 | 480 | 19 | trnW-trnP | 0.144385 | 187 | 27 |
| rps19 | 0.039427 | 279 | 11 | trnP-psaJ | 0.131498 | 327 | 43 |
| ndhF | 0.054807 | 2226 | 122 | psaJ-rpl33 | 0.104326 | 393 | 41 |
| rpl32 | 0.056604 | 159 | 9 | rpl33-rps18 | 0.114833 | 209 | 24 |
| ccsA | 0.073961 | 987 | 73 | psbH-petB | 0.12782 | 133 | 17 |
| ndhD | 0.046481 | 1506 | 70 | petD-rpoA | 0.141935 | 155 | 22 |
| psaC | 0.03252 | 246 | 8 | rps11-rpl36 | 0.150943 | 106 | 16 |
| ndhE | 0.039216 | 306 | 12 | rpl36-rps8 | 0.121212 | 429 | 52 |
| ndhG | 0.047081 | 531 | 25 | ndhF-rpl32 | 0.147826 | 575 | 85 |
| ndhI | 0.035714 | 504 | 18 | rpl32-trnL | 0.240175 | 458 | 110 |
| ndhA | 0.034164 | 1083 | 37 | ccsA-ndhD | 0.182692 | 208 | 38 |
| ndhH | 0.034687 | 1182 | 41 | ndhI-ndhA | 0.130952 | 84 | 11 |
| ndhH-rps15 | 0.107843 | 102 | 11 | ||||
| rps15-ycf1 | 0.155063 | 316 | 49 |
Synonymous and non-synonymous substitution rates were calculated for 74
protein-coding genes of fifteen Cardamine genera cp genomes. The
K
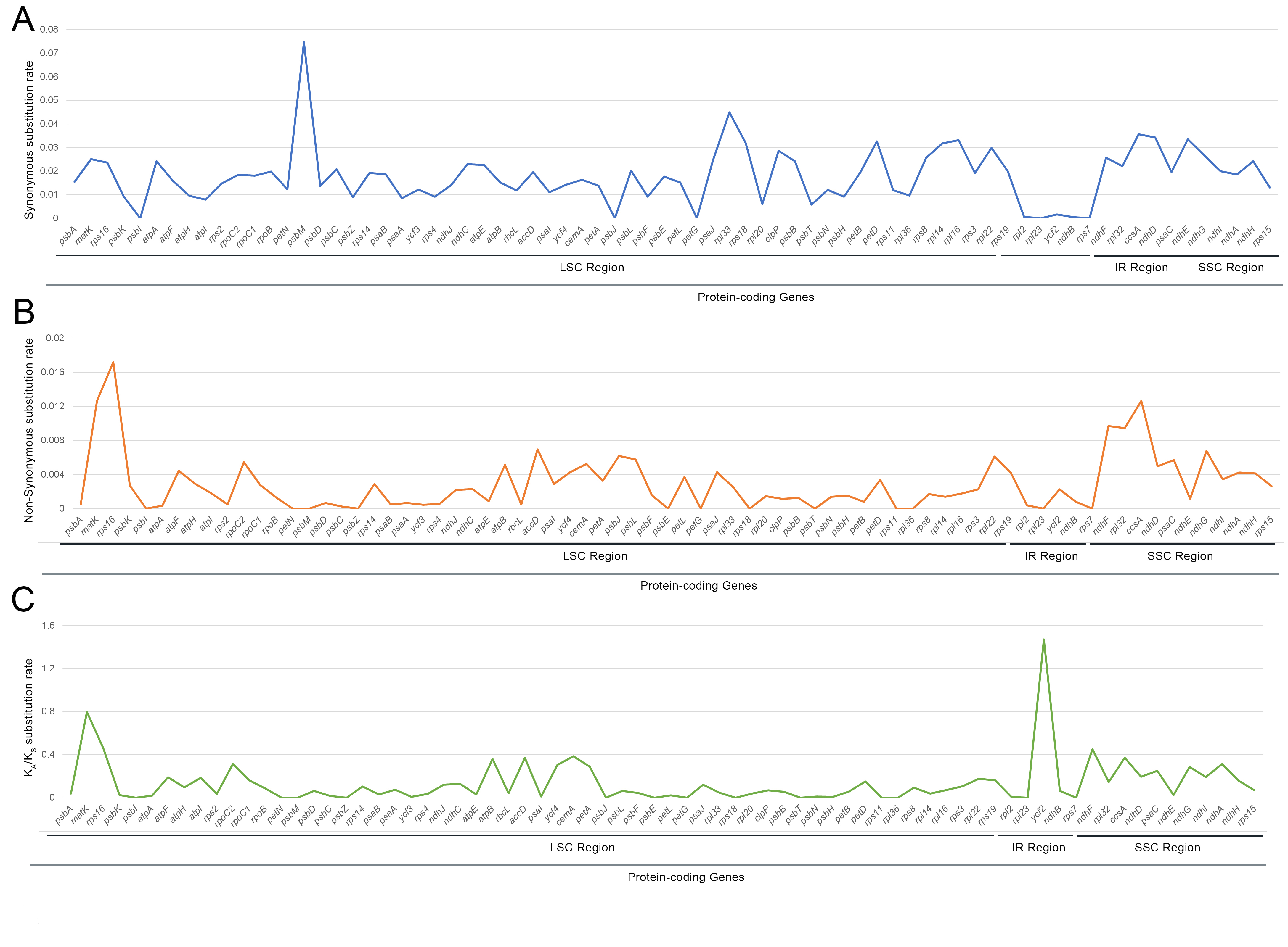 Fig. 5.
Fig. 5.Comparison of the ratio of non-synonymous (K
The selective pressure of thirteen protein-coding genes, such as four
NADH-dehydrogenase subunit genes (ndhA, ndhD, ndhG,
and ndhI), two subunits of cytochrome (petA and petD),
one ribosome small subunit genes (rps16), one subunit of ATP synthase
(atpB), and accD, ccsA, cemA, matK,
and ycf2 of fifteen species of Cardamine genera were analyzed
based on the substitution rate. If the substitution rate is
| Protein-coding genes | Comparison between models | 2ΔLnL | d.f. | p-value |
| atpB | M0 vs M3 | 3.883652 | 4 | 0.421980775 |
| M1 vs M2A | 0.472744 | 2 | 0.789486930 | |
| M7 vs M8 | 1.856066 | 2 | 0.395330561 | |
| M8a vs M8 | 0.473268 | 1 | 0.491487565 | |
| ccsA | M0 vs M3 | 99.613282 | 4 | 0 |
| M1 vs M2A | 73.543134 | 2 | 0 | |
| M7 vs M8 | 77.341918 | 2 | 0 | |
| M8a vs M8 | 73.073330 | 1 | 0 | |
| cemA | M0 vs M3 | 85.525976 | 4 | 0 |
| M1 vs M2A | 85.526090 | 2 | 0 | |
| M7 vs M8 | 85.531562 | 2 | 0 | |
| M8a vs M8 | 85.538130 | 1 | 0 | |
| matK | M0 vs M3 | 43.252730 | 4 | 0 |
| M1 vs M2A | 11.527882 | 2 | 0.003138718 | |
| M7 vs M8 | 7.7505199 | 2 | 0.020748942 | |
| M8a vs M8 | 7.3216859 | 1 | 0.006812747 | |
| ndhA | M0 vs M3 | 81.093012 | 4 | 0 |
| M1 vs M2A | 81.093202 | 2 | 0 | |
| M7 vs M8 | 81.104690 | 2 | 0 | |
| M8a vs M8 | 81.113370 | 1 | 0 | |
| ndhD | M0 vs M3 | 5.764528 | 4 | 0.217437134 |
| M1 vs M2A | 0 | 2 | 1.0 | |
| M7 vs M8 | 0.328019 | 2 | 0.848733535 | |
| M8a vs M8 | 0.037706 | 1 | 0.846034685 | |
| ndhF | M0 vs M3 | 42.00404 | 4 | 0.000000017 |
| M1 vs M2A | 14.27694 | 2 | 0.000793965 | |
| M7 vs M8 | 14.85864 | 2 | 0.000593592 | |
| M8a vs M8 | 14.24146 | 1 | 0.000160789 | |
| ndhG | M0 vs M3 | 71.71579 | 4 | 0 |
| M1 vs M2A | 64.05478 | 2 | 0 | |
| M7 vs M8 | 31.71765 | 2 | 0.000000130 | |
| M8a vs M8 | 29.72069 | 1 | 0.000000050 | |
| ndhI | M0 vs M3 | 696.4341 | 4 | 0 |
| M1 vs M2A | 619.6289 | 2 | 0 | |
| M7 vs M8 | 455.6721 | 2 | 0 | |
| M8a vs M8 | 455.7377 | 1 | 0 | |
| petA | M0 vs M3 | 93.69115 | 4 | 0 |
| M1 vs M2A | 93.73442 | 2 | 0 | |
| M7 vs M8 | 89.69042 | 2 | 0 | |
| M8a vs M8 | 88.736632 | 1 | 0 | |
| petD | M0 vs M3 | 42.008744 | 4 | 0.000000017 |
| M1 vs M2A | 18.072960 | 2 | 0.000118990 | |
| M7 vs M8 | 28.667442 | 2 | 0.000000596 | |
| M8a vs M8 | 18.07296 | 1 | 0.000021260 | |
| rps16 | M0 vs M3 | 2.750450 | 4 | 0.600415820 |
| M1 vs M2A | 1.880814 | 2 | 0.390468882 | |
| M7 vs M8 | 2.010804 | 2 | 0.365897514 | |
| M8a vs M8 | 1.879698 | 1 | 0.170368476 | |
| Ycf2 | M0 vs M3 | 25.882614 | 4 | 0.000033417 |
| M1 vs M2A | 20.461022 | 2 | 0.000036053 | |
| M7 vs M8 | 18.727454 | 2 | 0.000085780 | |
| M8a vs M8 | 17.756692 | 1 | 0.000025103 |
Repeat sequences were examined in the fifteen Cardamine plastomes. Six
hundred and ninety-one repeat sequences containing forward, reverse, complement,
and palindromic repeats, were observed among the fifteen Cardamineplastomes. Three hundred and twenty-two forward (46.6%) and 327 palindromic
repeats (47.3%) are relatively common among the detected repeats, whereas 21
(3.04%) of each reverse and complement repeats are comparatively rare (Fig. 6a). The complement repeats were absent in the species of C.
amaraeformis, C. enneaphyllos, C. hirsuta, and C.
parviflora. Similarly, the reverse repeats were absent in the C.
occulta cp genome. In addition, both reverse and complement repeats were absent
in the C. impatients and C. oligosperma species. In addition,
the length of the repeats (
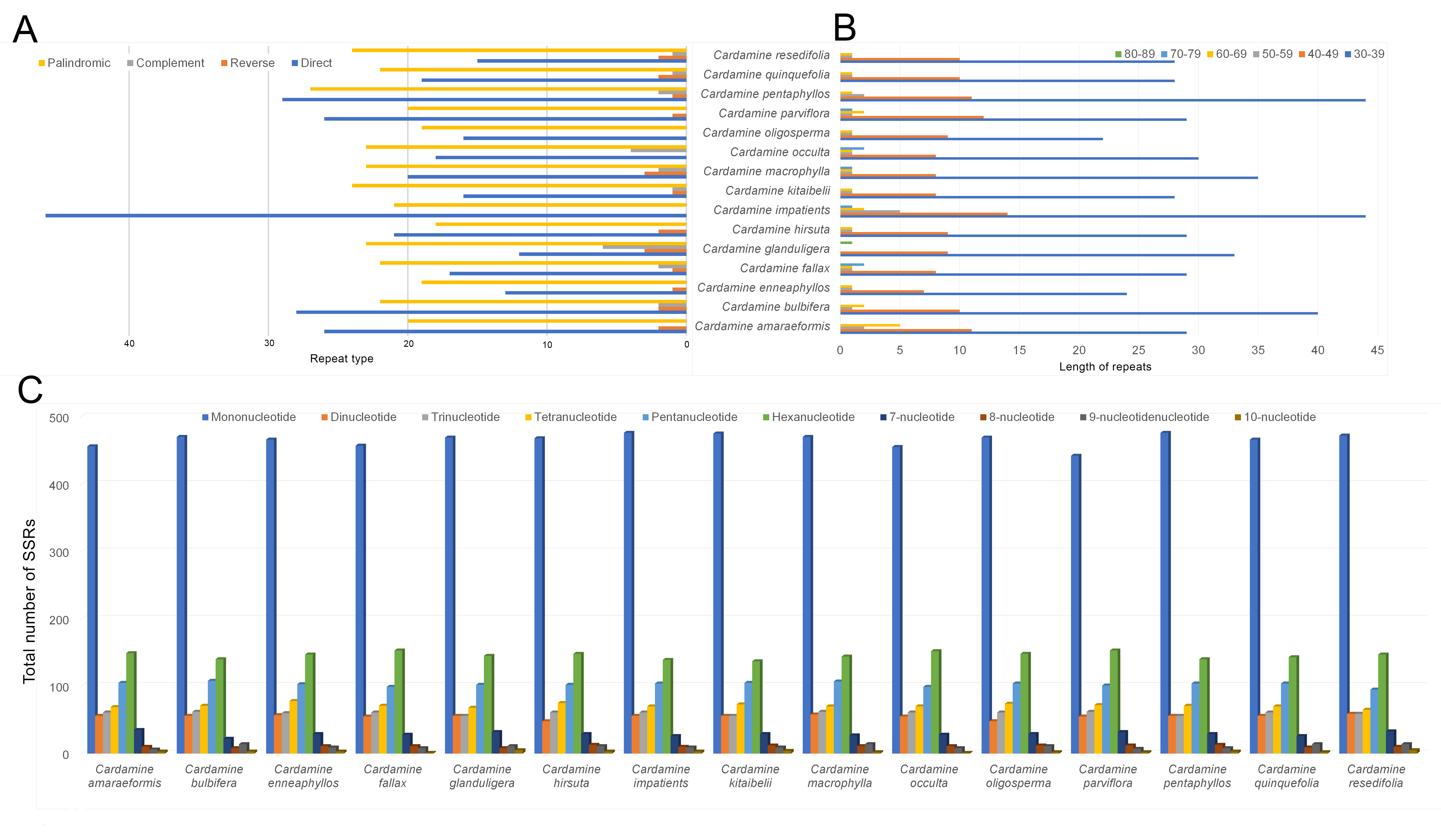 Fig. 6.
Fig. 6.Comparison of the distribution of different repeat types and SSRs in the fifteen species of Cardamine cp genomes. (A) The number of different types of repeats. F—forward repeats; R—Reverse repeats; P— palindromic repeats; C—complement repeats. (B) The length and the total number of repeat sequences present in their respective cp genomes. (C) Distribution of different types of SSRs.
A total of 14,298 simple sequence repeats (SSR) were identified in the fifteen species of Cardamine cp genomes with an average of 953 SSRs/genome. The SSRs detected ranged from 938 (C. parviflora) to 965 (C. macrophylla). The majority of the SSRs were mononucleotide repeats, which accounted for 48.87% of SSRs, followed by hexanucleotide repeats (~15.28%) and pentanucleotide repeats (~10.78%), tetranucleotide repeats (~7.46%), trinucleotide repeats (~6.29%) and dinucleotide repeats (~5.78%) and other repeat lengths from seven-nucleotide to 10-nucleotide repeats comprised 5.53% (Fig. 6c).
ML and MrBayes analyses were performed separately to determine the phylogenetic position and distance of C. occulta precisely. The five individual data sets of a combined total of 68 protein-coding genes, LSC, SSC, and IR regions and whole-genome of 40 cp genome sequences were used to imply the phylogenetic relationships between the closely related species of Brassicaceae. All five of both ML (Fig. 7; Supplementary Figs. 1–4) and Bayesian analyses (Supplementary Figs. 5–9) yielded similar trees. All the phylogenetic tree analyses showed that the species of Cardamine genera formed a monophyletic group. The topology of the phylogenetic tree showed that C. occulta has a close relationship with the species of C. fallax with a strong bootstrap value (100% for ML and 1.0 for MrBayes) (Fig. 7). Among the Cardamine clade, C. pentaphyllos and C. kitaibelii are the basal groups. The Cardamine clade was divided into two clades; C. bulbifera, C. quinquefolia, C. impatiens, C. glanduligera, C. macrophylla, C. oligosperma, and C. hirsuta formed one clade, and another clade consisted of C. occulata, C. fallax, C. amaraeformis, C. parviflora, C. enneaphyllos, and C. resedifolia with a 78% bootstrap value.
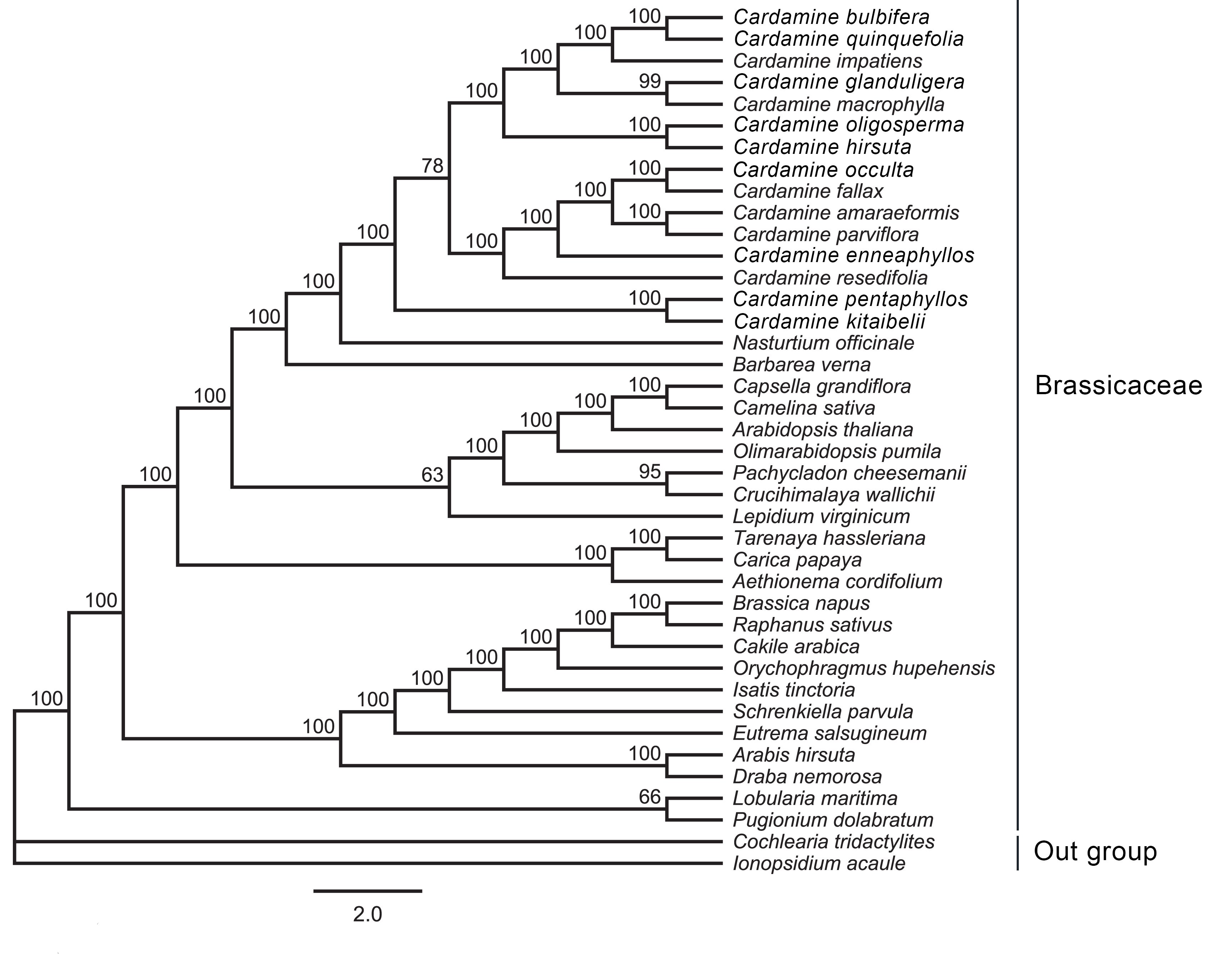 Fig. 7.
Fig. 7.Molecular phylogenetic tree based on 68 protein-coding genes of 40 Brassicales chloroplast genomes. Ionopsidium acaule and Cochlearia tridactylites were set as the outgroup. The tree was constructed by maximum likelihood analysis of the conserved regions using the RAxML program and the GTRGAMMA nucleotide model. The stability of each tree node was tested by bootstrap analysis with 1000 replicates. The bootstrap values are shown on the branches, and the branch length reflects the estimated number of substitutions per 1000 sites.
The species Cardamine occulta is distributed predominantly in Eastern Asia [9]. This species is quite similar to the European species, C. flexuosa, and is considered a single species [11] because these two species have not shown any morphological differences. Since 2006, these two species have been differentiated based on their ecological habitats [9, 10, 11, 13, 14]. Cardamine is a large genus in the Brassicaceae family of flowering plants that contains more than 200 species of annuals and perennials [9]. Thus far, fourteen chloroplast genomes have been sequenced and analyzed. On the other hand, no extensive and comparative studies of Cardamine genera have been carried out. Therefore, the present study sequenced the whole plastid genome of C. occulta using Illumina HiSeq 2500 platform and characterized the controversial species from South Korea. Comparative studies were carried out with fourteen other species of the Cardamine genera. The length of the complete chloroplast genome sequence of C. occulta is 154,796 bp and contains 131 individual genes, which is in the range of other species of Cardamine genera. The GC content of C. occulta is 36.3%, which is similar to all other species of Cardamine genera, suggesting that the distribution of the GC contents in the Cardamine cp genomes are consistent and highly conserved. Although the overall genomic structure, such as the gene order and gene number of the C. occulta, is identical to other Cardamine and Brassicaceae species except for the length of the atpB gene in the C. amaraeformis. The Cardamine plastomes were conserved, and no rearrangement events were found. All the species of Cardamine genera lost the protein-coding gene, initiation factor A (infA), in their cp genomes. Most of the angiosperms were lost independently from multiple angiosperm lineages, including other species within the Brassicaceae. This gene loss might have been due to an interruption of the nuclear-encoded DNA replication, recombination, and repair machinery that controls the cp genome and the evolution of the plant organelle genome [34].
The results of mVISTA analyses revealed high levels of similarity among the plastomes, indicating that the divergence of the C. occulta plastome is lower than that in other species of the Cardamine genera. Furthermore, lower sequence divergence in the IR region was detected compared to SC regions, which has been previously reported [8, 35, 36, 37]. One conceivable reason is that in the cp genome, which has multiple copies per cell, gene conversion with a slight bias in the contradiction of new mutations would reduce the mutation load in the two IR regions much more competently than in the single-copy regions because of the duplicative characteristics of the IRs [38, 39, 40, 41]. The expansion and contraction of the IR and single-copy convergence regions are considered the leading mechanism in driving the variation in the size of angiosperm plastomes, playing a vital role in their evolution [38, 42, 43, 44]. The present study did not identify any significant expansion and shrinkage in the IR/SC regions. Previous studies reported that the size of the whole cp genome does not always vary with the expansion or contraction of IRs [45, 46, 47, 48, 49, 50].
Comparative studies of the fifteen Cardamine cp genome
sequences revealed several regions of sequence polymorphisms. Among these
polymorphisms, most of the sequence variations were dispersed in the LSC and SSC
regions, whereas the IR regions displayed relatively lower sequence variations.
The lower sequence divergence of the IR region compared to the SC regions in Cardamine species and other plants may be due to a copy
correction among the IR sequences during gene conversion. Gene mutations and
rearrangements in the cp genome are not exhibited constantly throughout
the genome sequence. Instead, identifying the hypervariable regions in the
chloroplast genome is considered the hotspot region that serves as specific
molecular markers [51]. The present study identified 27 protein-coding and 29
intron and intergenic hypervariable regions. Among these, the maximum
hypervariable regions, such as the protein-coding genes (
This study analyzed the substitution rate in the fifteen species of the
Cardamine genera. C. occulta was used as a reference
genome in the present study and compared with other cp genomes. Initially, the
substitution rates of all the individual protein-coding genes of fifteen species
of Cardamine genera were averaged. The results showed that the ratio of
the K
The repeat units were distributed with high frequency and played a substantial role in the chloroplast genome evolution [59, 60, 61, 62]. The repeat types of the fifteen Cardamine plastomes comprised a variable number in their genomes. Liu et al. [60] reported that the variation in number and variety of repeats play a significant role in the plastome structural organization, but there was no correlation between these large repeat regions and rearrangement endpoints. In addition, microsatellite repeats are primarily present in the plastomes, which exhibit a high level of polymorphism and are used as a molecular marker in genetic studies [63, 64]. Simple sequence repeats (SSRs) play a major role during genome rearrangement and recombination [65]. The content of different SSRs and their distribution on various chloroplast regions were similar in their Cardamine species. The distribution of SSRs in the Cardamine plastome does not involve any genome rearrangement process. On the other hand, the existence of repeat sequences in the cp genome of Cardamine genera could be helpful for developing lineage-specific markers for genetic diversity and evolutionary studies.
The whole chloroplast genome of the plant offers a significant foundation to resolve the evolutionary, taxonomic, and phylogenetic studies [58, 66, 67, 68, 69, 70, 71]. The molecular phylogenetic analysis of both ML and Bayesian analyses in the current study showed that the species of Cardamine formed a monophyletic clade. The Cardamine clade is subdivided into two clades, and the species C. occulta is clustered with C. fallax with a substantial bootstrap value. On the other hand, the cytogenetic studies showed that the tetraploidy C. scutata (Diploid species C. amaraeformis and C. parviflora as the parents) and C. kokaiensis (Diploid species C. parviflora as the parent) are the parental for C. occulta [9, 11, 17]. In contrast, the diploid species C. amaraeformis and C. hirsuta are the parental species for C. flexuosa [9]. Moreover, the C. fallax was implied to be hexaploidy. Based on the DNA sequence data of C. fallax, it was postulated that this species or its diploid progenitors might have influenced the origin of C. occulta. Nevertheless, the cp genomes of C. flexuosa (European species), C. kokaiensis, and C. scutata need to be included to understand the phylogenetic position and their relationship with other Cardamine genera in future studies.
The complete chloroplast genome Cardamine occulta was sequenced, assembled, and analyzed in the present study. Valuable genomic resources were provided for Cardamine genera. Overall, the gene contents and arrangements were similar and highly conserved in the species of the Cardamine genera. Comparative analyses of the chloroplast genomes identified variable regions with potential application as species-specific DNA barcodes. Furthermore, thirteen protein-coding genes have diverged widely and under potentially positive selection, resulting from adaptation to the ecosystem. Finally, phylogenetic analyses of various cp data sets of both ML and Bayesian analyses show that C. occulta species has a closer genetic relationship to C. fallax. In conclusion, this study will facilitate future research, particularly resolving the controversial Cardamine clade. Nevertheless, in future studies, the cp genome of C. flexuosa (European species), C. kokaiensis, and C. scutata needs to be incorporated to understand the phylogenetic position and their relationship with C. occulta.
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov) under the accession number MZ043777. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA738458, SRR14833115, and SAMN19729360, respectively.
cp, chloroplast; LSC, large single-copy; SSC, small single-copy; IR,
inverted-repeats; tRNA, transfer RNA; rRNA, ribosomal RNA; K
GR and SJP designed the research study. GR performed the research, analyzed the data, and prepared a manuscript draft and figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was funded by grants from Scientific Research (KNA1-1-13, 14-1) of the Korea National Arboretum, Republic of Korea.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2704124.