
 , Chun Yang 2,*
, Chun Yang 2,*1 Department of Critical Care Medicine, The Third Affiliated Hospital of Soochow University, 213004 Changzhou, Jiangsu, China
2 Department of Anesthesiology and Perioperative Medicine, The First Affiliated Hospital of Nanjing Medical University, 210029 Nanjing, Jiangsu, China
3 Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
4 Department of Anesthesiology, The Second Affiliated Changzhou People’s Hospital of Nanjing Medical University, 213004 Changzhou, Jiangsu, China
Academic Editor: Graham Pawelec
Abstract
At present, there are seven known types of human coronaviruses (HCoVs), which can be further divided into two categories: low pathogenic and highly pathogenic. The low pathogenic HCoVs infect the upper respiratory tract, mainly causing mild, cold-like respiratory diseases. By contrast, highly pathogenic HCoVs mainly infect the lower respiratory tract and cause fatal types of pneumonia, which include severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), as well as the recent outbreak of coronavirus disease 2019 (COVID-19). Highly pathogenic HCoV infection has a high morbidity and mortality, which is usually related to the strong immune response induced by highly proinflammatory cytokines, which is also known as “cytokine storm”. Therefore, it is particularly important to explore the role of cytokine storm in the process of highly pathogenic HCoV infection. We review the epidemiological and clinical manifestations of highly pathogenic HCoV infection, and reveal the pathology of cytokine storm and its role in the process of highly pathogenic HCoV infection.
Keywords
- coronavirus
- cytokine storm
- SARS
- MERS
- COVID-19
- review
Coronavirus is an enveloped virus; with a single-strand, positive-sense RNA genome size of about 26–32 kb, it is the largest known RNA virus [1]. At present, there are seven known types of human coronaviruses (HCoVs): severe acute respiratory syndrome coronavirus (SARS-CoV); Middle East respiratory syndrome CoV (MERS-CoV); SARS-CoV2; and HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1, which cause only mild upper respiratory diseases [2, 3, 4, 5].
In recent years, novel coronaviruses have been detected periodically around the world. For example, SARS was caused by a SARS-CoV outbreak in Guangdong Province in South China from 2002 to 2003 [6]. A sustainable severe respiratory disease with MERS-CoV as the pathogen broke out in the Middle East in 2012 [7], and more recently, an outbreak of COVID-19 occurred in Wuhan, China, from the end of 2019 to the beginning of 2020 [5]. All three highly pathogenic HCoV infections caused severe pneumonia, which can further develop into fatal acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) with high morbidity and mortality [6, 8, 9, 10]. However, the specific mechanism of serious diseases caused by highly pathogenic HCoV infection remains unclear. It is noteworthy that excessive inflammatory response—that is, cytokine storm—is considered to play a vital role in fatal pneumonia caused by highly pathogenic HCoV infection [11]. Therefore, in this review, we summarize the epidemiology and clinical manifestations of highly pathogenic HCoV infections, and describe the pathology of cytokine storms and their role in the course of highly pathogenic HCoV infection.
SARS was first diagnosed in Guangdong, China, and quickly spread around the world in 2002–2003 [12, 13]. In March 2003, a new HCoV was identified as the pathogen of SARS and was promptly named SARS-CoV [14, 15]. By the end of the epidemic in July 2003, SARS-CoV had spread to 29 countries and regions; a total of 8098 patients were infected and 744 died (case fatality rate, 9.6%) [16]. At the beginning of the SARS epidemic, almost all diagnosed patients had been exposed to animals before the onset of the disease. After the SARS pathogen had been identified, SARS-CoV was found in palm civets on the market [17, 18]. However, new studies have shown that palm civets are only intermediate hosts, and the SARS-CoV found in them is actually transmitted by other animals [18, 19]. Moreover, multiple studies have reported the discovery of a novel coronavirus associated with human SARS-CoV in horseshoe bats [20, 21]. These lines of evidence suggest that horseshoe bats may be the natural hosts of SARS-CoV, whereas palm civets are the intermediate hosts.
SARS-CoV infection may exhibit a variety of clinical characteristics, ranging from asymptomatic or mild fever to acute respiratory tract infections, and even ARDS or death [12]. In addition to respiratory manifestations, such as fever and cough, SARS can also present nonpulmonary features such as myalgia and diarrhea [22]. In general, SARS-CoV replicates quickly after infecting the host. At this stage, patients with SARS will develop fever and cough, then develop high fever, hypoxemia, and pneumonia-like symptoms; eventually, about 20% of the patients will develop ARDS, and some of them may even die [11].
Since MERS-CoV was first isolated from the sputum of a 60-year-old man in Saudi Arabia in September 2012 [9], the number of MERS cases reported to the World Health Organization (WHO) has increased steadily. By December 2019, there have been a total of 2468 laboratory-confirmed cases of MERS-CoV infection, of whom 851 died, which translates to a mortality rate of about 34.5%. Of these, approximately 82% occurred in Saudi Arabia, and most of these cases resulted from outbreaks in hospitals and families [23]. In addition, similar to SARS-CoV, MERS-CoV spreads to humans across species. Increasing evidence has shown that the main intermediate hosts of MERS-CoV are camels, and its natural hosts are bats [24, 25, 26].
MERS-CoV can cause acute respiratory infection, but its severity ranges from asymptomatic to mild respiratory symptoms to life-threatening ARDS with high mortality [27]. The most common clinical manifestations are fever, sore throat, cough, myalgia, hard breathing, and other influenza-like symptoms, which rapidly develop into pneumonia [28]. In addition, MERS-CoV can also cause gastrointestinal symptoms, such as abdominal pain, vomiting, and diarrhea [29]. Most healthy individuals show mild to moderate respiratory diseases, but individuals with dysregulated immune response and severe respiratory diseases often develop ARDS [30].
In December 2019, a highly transmissible pneumonia, caused by a novel coronavirus SARS-CoV2, broke out in Wuhan, China [31, 32]. On February 11, 2020, the WHO named this novel coronavirus disease as COVID-19. Thus far, the virus has spread rapidly more than 200 countries and regions around the world. As of October 31, 2021, there have been a total of over 246 million confirmed cases of COVID-19, of whom nearly 5 million have died, and the number continues to rise [33]. Studies have shown that SARS-CoV2 can spread mainly from person-to-person through respiratory droplets or aerosol particles produced when infected people cough, sneeze, or talk [34]. Based on the analysis of the current epidemiological data, the basic reproductive number of SARS-CoV2 was estimated at 2.19 (95% confidence interval, 2.08 to 2.36), and median incubation period was estimated to be 5.1 days (95% confidence interval, 4.5 to 5.8 days), and it was confirmed that case and close contact isolation are effective in reducing transmission of SARS-CoV2 [35, 36]. It is worth noting that the genetic similarity between SARS-CoV2 and SARS-CoV is 79.6%, and the similarity with bat coronavirus is as high as 96% [37, 38]. Although the origin of SARS-CoV2 is unclear, current evidence points to bats as the likely natural host of SARS-CoV2 [39].
Available evidence suggests that the clinical setting of COVID-19 is complex. A series of studies have found that the most common symptoms of COVID-19 are fever, fatigue and dry cough [10, 40, 41, 42]. Meanwhile, the digestive symptoms, such as diarrhea, vomiting and abdominal pain, and neurological symptoms, such as olfactory and taste disorders, language disturbances, cerebellar ataxia and delirium, are also common in COVID-19 patients [43, 44, 45, 46]. In addition, ground-glass opacity and subpleural lesions were found to be common CT signs of SARS-COV2 infection [47]. Of note, a study has shown that more than half of the patients with severe COVID-19 have chronic diseases such as cardiovascular diseases, endocrine system disease and respiratory system disease, among whom 17% developed ARDS, and 11% deteriorated with multiple organ failure within a short period [40]. In a summary of a report of 72,314 cases from China, 81% were mild and 19% were severe and critical requiring mechanical ventilation [48]. Similar results were seen in a multicenter study in Germany, in which 17% of patients received mechanical ventilation and found that common comorbidities of COVID-19 were hypertension, diabetic, arrhythmias and kidney failure [49]. A cohort study conducted in the US showed that 20.3% of hospitalized COVID-19 patients died and 19.4% were admitted to ICU [50]. In addition, COVID-19 can produce a series of mental health challenges such as anxiety, depression, traumatic stress reactions and suicide [51]. Furthermore, a study has confirmed a significantly higher incidence of heart failure, stroke, chronic kidney disease stages 3–5 and chronic liver disease in COVID-19 patients after discharge from hospital than in the general population [52].
Here’s what we know, similar to influenza and respiratory syncytial virus (RSV), fever and cough are the most common symptoms of highly pathogenic HCoVs infection. In addition, studies have shown that the epidemiology of COVID-19 is related to temperature, radiation/sunlight and latitude [53, 54, 55]. Meanwhile, a recent study further confirmed that the COVID-19 incidence is negatively correlated with temperature and humidity [56]. These evidences demonstrated the seasonality of COVID-19.
However, highly pathogenic HCoVs are more likely to cause severe respiratory tract infection, ARDS and even multiple organ failure compared with low pathogenic HCoVs and seasonal influenza [57]. In addition, patients with COVID-19 frequently present with bilateral pneumonia and multiple ground-glass shadows on lung CT imaging, which provides a reliable detection method for early diagnosis of COVID-19 [58]. It is worth noting that some patients with COVID-19 are prone to fatal complications such as pulmonary embolism, septic shock or hemorrhagic stroke [59]. Furthermore, Idilman et al. [60] recruited 31 COVID-19 patients with suspected of having pulmonary thromboembolism who underwent pulmonary dual-energy computed tomography angiography and found that perfusion defects may be a sign of systemic microangiopathy with micro-thrombosis. These findings suggest that the clinical manifestations of patients infected with highly pathogenic HCoVs are not limited to severe respiratory symptoms, but also involve other organs and cause systemic damage. However, the specific mechanism is still unclear. It is important to note that some SARS patients with diminishing viral load still develop ARDS, suggesting that mechanisms other than viral virulence may mediate tissue damage [61]. Moreover, patients infected with highly pathogenic HCoVs showed significant changes in white blood cell count and immune biomarkers such as IL-6 [62]. Furthermore, a cohort study has found that hyperinflammatory syndrome, including secondary haemophagocytic lymphohistiocytosis, cytokine release syndrome, macrophage activation syndrome and macrophage activation-like syndrome of sepsis, was associated with the prognosis of COVID-19 [63]. Taken together, these studies substantiated the concept that immune mechanisms are involved in the occurrence and development of highly pathogenic HCoVs infection.
During virus infection, the innate immune response triggered by the host is the first line of defense against the virus. However, an overactive immune response may lead to immunopathology [64]. The innate immune system of the host recognizes viral pathogen-associated molecular patterns through pattern recognition receptors, thereby stimulating the expression of proinflammatory cytokines [65, 66]. These proinflammatory cytokines lead to the accumulation of inflammatory cells and acute inflammatory response, often leading to tissue or organ damage [67]. In most cases, the repair process after the beginning of the inflammatory response can fully restore the function of the tissue or organ, and some severe inflammatory reactions will lead to persistent tissue or organ dysfunction [68]. In some cases, excessive inflammatory cytokines such as interferons (IFNs), tumor necrosis factors (TNFs), interleukins (ILs), and chemokines (Table 1, Ref. [10, 55, 56, 57, 58, 59, 60, 66, 67, 68, 69, 70, 71]) are produced and accumulated, causing a cytokine storm and multiple organ dysfunction [68].
| HCoVs infection | Patient characteristics | Cytokines and chemokines | Ref. |
| SARS | SARS | IFN- |
Chien et al. [55] |
| Wong et al. [56] | |||
| Zhang et al. [57] | |||
| severe SARS | IFN- |
Cameron et al. [58] | |
| Theron et al. [59] | |||
| Cameron et al. [60] | |||
| MERS | MERS | IFN- |
Mahallawi et al. [66] |
| severe or moderate | IL-6, IL-1RA, IP10, MCP-1 | Shin et al. [67] | |
| MERS | |||
| severe MERS | IFN- |
Kim et al. [68] | |
| Min et al. [69] | |||
| COVID-19 | COVID-19 | IL-1, IL-6, IL-7, IL-8, IL-9, IL-10, IFN- |
Huang et al. [10] |
| severe COVID-19 | IL-2, IL-6, IL-7, IL-10, IP10, MCP-1, MIP1 |
Huang et al. [10] | |
| Chen et al. [70] | |||
| Liu et al. [71] | |||
| HCoVs, human coronavirus; SARS, severe acute respiratory syndrome; MERS, Middle East respiratory syndrome; COVID-19, coronavirus disease-19; IFN, interferons; IL, interleukin; IP10, interferon-gamma inducible protein 10; MCP-1, monocyte chemoattractant protein-1; TNF, tumor necrosis factor; RANTES, regulated upon activation normal T-cell expressed and secreted; MIP, macrophage inflammatory protein; G-CSF, granulocyte colony-stimulating factor. | |||
Innate immune response plays a key role in combating SARS-CoV infection. Studies
have shown that macrophages infected with SARS-CoV have low expression of
IFN-
It is noteworthy that many studies have shown that the levels
of proinflammatory cytokines (IFN-
MERS-CoV has a high morbidity and mortality. Although its pathological mechanism
remains unknown to date, accumulating evidence indicates that immune response
plays an important role [78]. Studies have shown that macrophages infected with
MERS-CoV highly express IL-12, IFN-
Recent studies have shown that there is a significant proinflammatory cytokine
response in the acute phase of human infection with MERS-CoV, and the serum
concentrations of IFN-
Many patients with MERS-CoV infection, therefore, exhibit excessive proinflammatory cytokines and chemokines, which leads to a cytokine storm and plays an important role in the pathogenesis of MERS-CoV.
COVID-19, now considered a worldwide pandemic disease with high morbidity and mortality, is caused by SARS-CoV2. In the latest study, tissue biopsy samples obtained from an autopsy of a dead patient with COVID-19 showed that the pathological features of COVID-19 were very similar to those of SARS-CoV and MERS-CoV infection [86, 87, 88]. Studies have shown that lymphocytes, particularly CD4+ T and CD8+ T cells, are significantly reduced in almost all patients with COVID-19 [89, 90]. In addition, C-reactive protein, neutrophil count and the neutrophil-to-lymphocyte ratio in the peripheral blood of patients with COVID-19 are increased, and that the neutrophil-to-CD8+ T cell ratio in patients with severe COVID-19 was significantly higher than that in patients with mild diseases [91]. Therefore, understanding the complex immune dysregulation is essential to the diagnosis and treatment of COVID-19.
The study found that plasma concentrations of IL-1, IL-7, IL-8, IL-9, IL-10,
IFN-
Cytokine storms play an important role in highly pathogenic HCoV infection.
Current studies have found that the main elevated cytokines in the plasma of
critically ill patients with SARS, MERS, or COVID-19 are IFN-
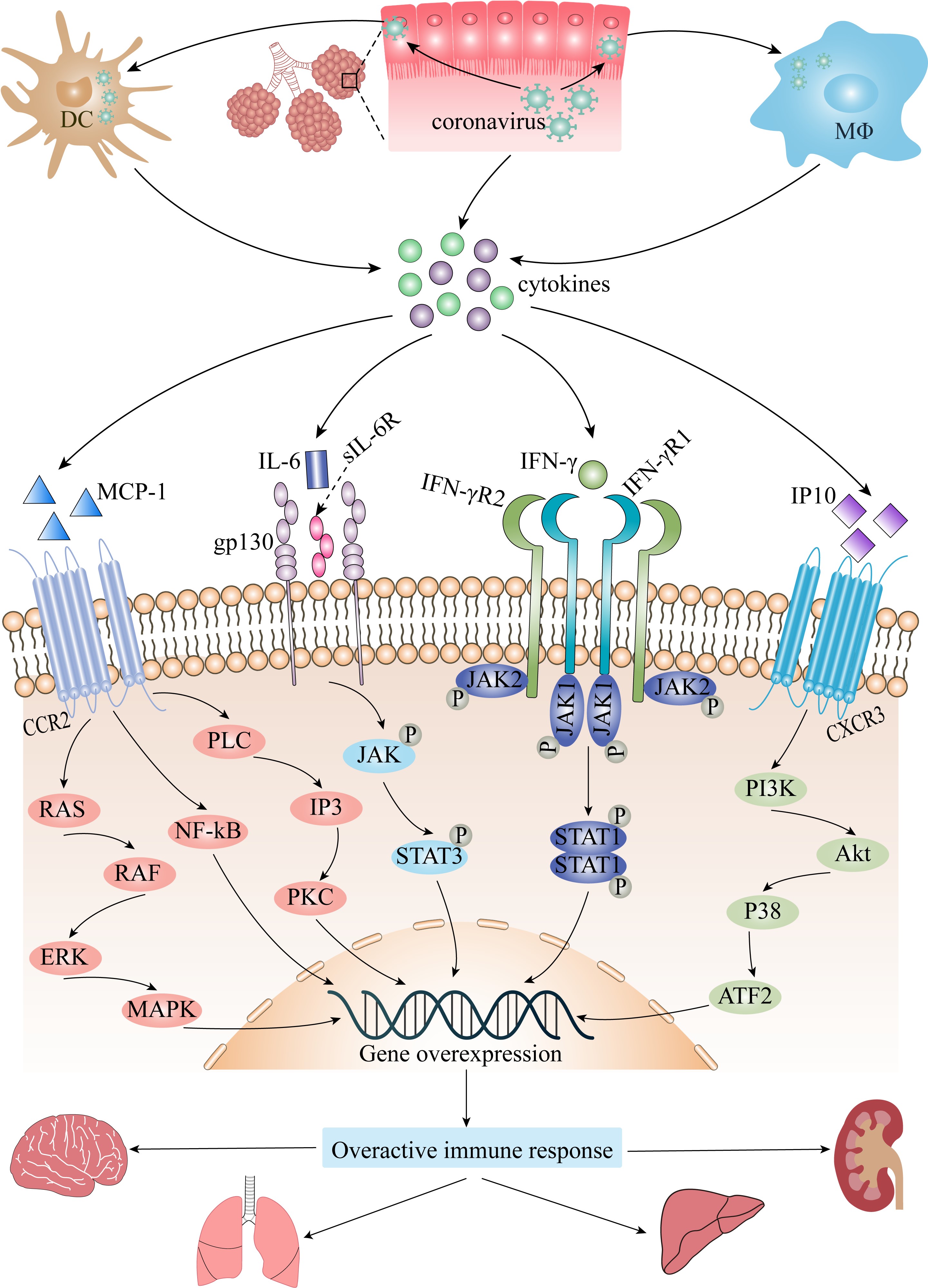
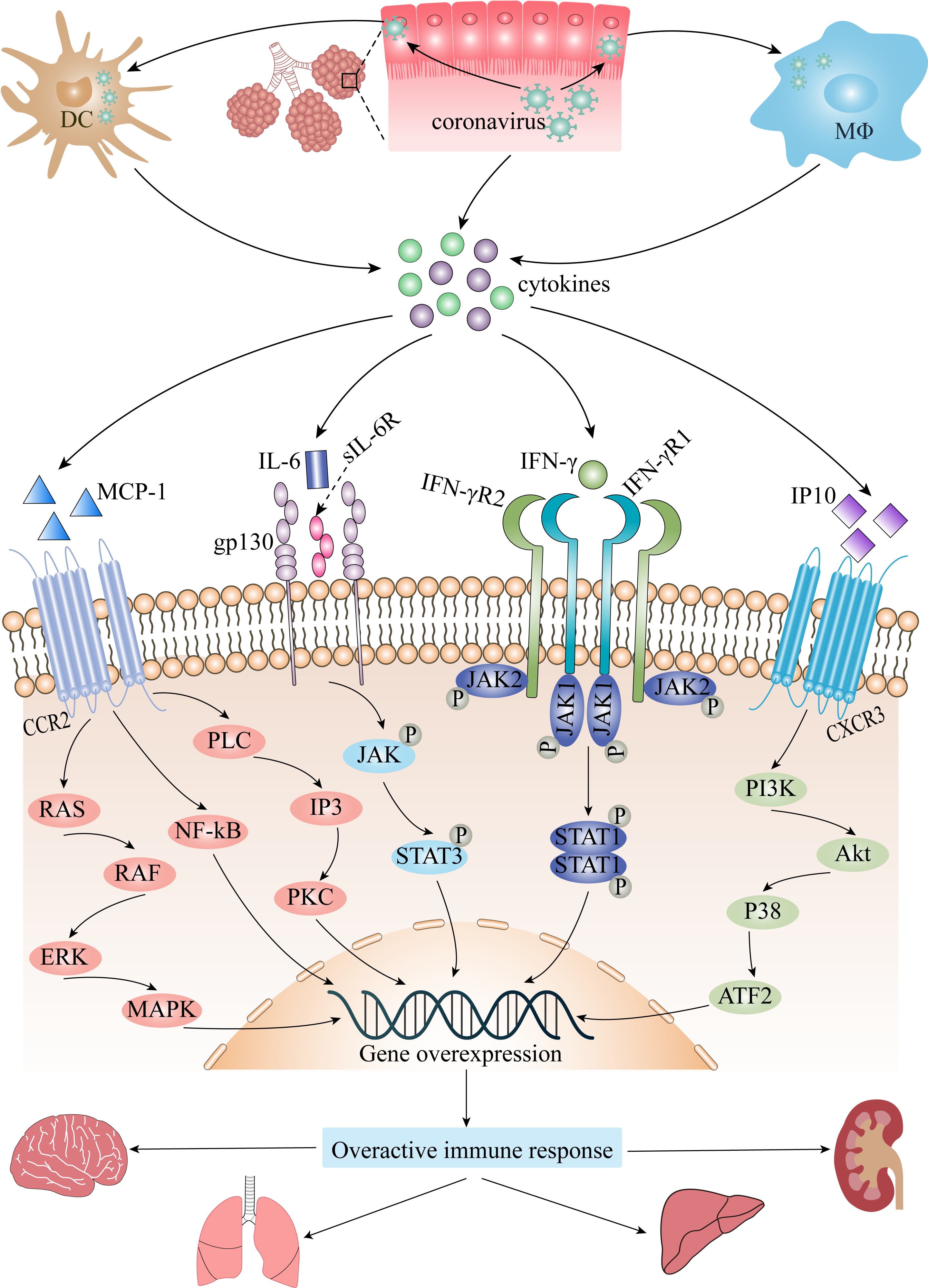 Fig. 1.
Fig. 1.The role of cytokine storm in coronavirus infection. Akt,
protein kinase B; ATF2, activating transcription factor 2; CCR2, CC chemokine
receptor 2; CXCR3, CXC chemokine receptor 3; DC, dendritic cell; ERK,
extracellular signal regulated kinase; gp130, interleukin-6 receptor
subunit-beta; IFN-
IFNs are a class of cytokines that play an important role in the innate immune
response induced by viral infection [96]. According to the specificity of the
receptor, they can be divided into three types—type I, type II, and type III.
IFN-
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon
genes (STING) signaling pathway is important for the production of IFN. cGAS, a
pattern recognition receptor of cytoplasmic double-stranded DNA (dsDNA),
catalyzes the reaction between GTP and ATP and generates the small molecule
cyclic GMP-AMP (cGAMP) after sensing the dsDNA derived from pathogens or damage
of host tissues, and then activates STING to induce the phosphorylation of
TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), which
ultimately induces IFN expression and secretion [100, 101]. Previous reports have
indicated that both SARS-CoV and SARS-CoV2 enter cells via binding with
angiotensin converting enzyme 2 (ACE2) receptor to cause tissue and organ damage
[62]. In addition, self-DNA release due to tissue damage can over-activate the
cGAS-STING pathway and cause lung injury [102]. Meanwhile, as mentioned
previously, IFN-
IL-6 is an important cytokine that participates in the regulation of immune
response [103]. Its receptors include IL-6 receptor-
Although IL-6 is significantly elevated in patients with severe COVID-19 and MERS, as previously noted, and the cytokine storm is strongly associated with patient mortality. However, it should be noted that the cytokine response of patients with severe COVID-19 is distinct from that of patients with bacterial sepsis, in particular, the elevated IL-6 levels are significantly lower [107, 108]. Very recently, a study has confirmed that routine use of tocilizumab in hospitalized COVID-19 patients is not supported [109]. Moreover, studies have shown that anti-IL-6 receptor treatments fail to benefit the survival of hospitalized patients with COVID-19, and even increase the secondary infections in severe patients [110, 111]. These results suggest that IL-6 plays a complex role in the occurrence and development of COVID-19, and the specific mechanism remains unclear. Studies have indicated that plasma plasminogen activator inhibitor-1 (PAI-1) in patients with severe COVID-19 is positively related to IL-6 levels, and the production of PAI-1 can be reduced by inhibiting IL-6 signaling pathway [112]. PAI-1 is a factor associated with vascular endothelial injury, and its increase indicates vascular endothelial lesions and coagulation dysfunction. Interestingly, thromboembolic events be more frequent in COVID-19 associated cytokine storm compared to other cytokine storms [113]. Therefore, the abnormally elevated IL-6 in patients with highly pathogenic HCoV may be involved in the occurrence of thromboembolic events in severe patients by promoting the production of PAI-1. In addition, low 25 (OH) vitamin D levels were associated with disease severity in patients hospitalized with COVID-19 [114]. Furthermore, studies have found that vitamin D3 is associated with the inflammatory response of virus infected patients, especially can reduce the level of IL-6 in patients with RSV infection [115]. Does this imply that the pathogenic mechanism of IL-6 in the occurrence and development of coronavirus infection is regulated by vitamin D3. In conclusion, current studies have shown that IL-6 plays a crucial role in the process of highly pathogenic HCoV infection, and more studies are needed to further clarify the specific mechanism in the future.
IP10, a chemokine with a molecular weight of 10 kDa, is induced by
IFN-
It is worth pointing out that IP10 was significantly elevated in severe SARS, MERS and COVID-19 patients, but the specific role of IP10 remains unclear. Study has shown that IP10 can inhibit vascular endothelial repair, which means IP10 can promote thrombosis [124]. Interestingly, autopsy pathological evidences have found that thrombosis is a common clinical manifestation in patients with severe COVID-19 [125]. In addition, a previous study suggested that IP10 has neurotoxic potential [126]. Meanwhile, some scholars believe that IP10 plays a key role in olfactory dysfunction in COVID-19 patients [127]. These lines of evidence suggest that IP10 may be involved in the development of highly pathogenic HCoV infection by damaging vascular endothelia and exerting neurotoxicity. However, the specific mechanisms that cause these pathological processes are still being explored. It is known that macrophages expressing high levels of CXCR3 can be recruited by IP10 to produce large amounts of IL-6. In addition, high level of IL-6 can induce the high expression of IP10 in the cerebrospinal fluid of patients with subarachnoid hemorrhage [128]. Thus, whether IP10 and IL-6 form a positive feedback loop in the occurrence and development of the disease and eventually cause cytokine storm. It has been found that a high concentration of IP10 in the serum of patients with H1N1 can mediate the formation of ALI by activating the phosphatidylinositol 3-kinases (PI3K)-protein kinase B (Akt)-p38-activating transcription factor 2 (ATF2) signaling pathway [129]. Based on this, we speculate that excessive IP10 in the serum of critically ill patients with highly pathogenic HCoV may also mediate tissue or organ injury by activating the PI3K-Akt-p38-ATF2 signaling pathway.
MCP-1 is a chemokine with a molecular weight of 13 kDa that belongs to the CC
subfamily [130]. It can be expressed by a variety of cells, including monocytes,
macrophages, endothelial cells, smooth muscle cells, fibroblasts, epithelial
cells, mesangial cells, astrocytes and microglial cells [131, 132]. Its specific
receptor, CCR2, is a G protein coupled receptor that crosses the membrane seven
times [131]. After binding to CCR2, MCP-1 activates a series of signaling
pathways, regulates the activation and chemotactic migration of target cells, and
then participates in the regulation of immune response [133]. The specific
signaling pathways include phospholipase C (PLC)-inositol
1,4,5-trisphosphate (IP3)-rotein kinase C (PKC), nuclear factor-kappa B
(NF-
Highly pathogenic HCoV infections can cause respiratory diseases with varying
severity, with high morbidity and mortality. Increased levels of proinflammatory
cytokines and chemokines were found in the plasma of patients with SARS, MERS, or
COVID-19, and this increase was related to the severity of the disease.
Therefore, it is considered that cytokine storm is associated with the pathogenic
mechanism of highly pathogenic HCoV infection. In addition, the increased
cytokines in the plasma of critically ill patients with SARS, of MERS and
COVID-19 were mainly IFN-
ACE2, angiotensin converting enzyme 2; Akt, protein kinase B; ALI, acute lung
injury; ARDS, acute respiratory distress syndrome; ATF2, activating transcription
factor 2; CCR2, CC chemokine receptor 2; cGAMP, cyclic GMP-AMP; cGAS, cyclic
GMP-AMP synthase; COVID-19, coronavirus disease-19; CXCR3, CXC chemokine receptor
3; DC, dendritic cell; dsDNA, double-stranded DNA; ERK, extracellular signal
regulated kinase; GAF, IFN-
CY and BZ designed the work. RX, CL, XX, YH, BZ and CY collected and reviewed the references. RX wrote the first draft. CY and BZ wrote and reviewed the final version of the manuscript. All authors discussed and contributed to the manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Chun Yang is serving as one of the Editorial Board members of this journal. We declare that Chun Yang had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.