





1 Department of Dermatology, Traditional Chinese and Western Medicine Hospital of Wuhan, Tongji Medical college, Huazhong University of Science and Technology, 430022 Wuhan, Hubei, China
†These authors contributed equally.
Academic Editor: Giuseppe Ingravallo
Abstract
The skin is the largest barrier organ of the human body and serves to protect the internal structure of the body from the harmful environment. The epidermis forms the outermost layer and is exposed to the environment. Keratinocytes are important constituent cells of the epidermis and alter their morphology and structural integrity through a highly complex differentiation process referred to as cornification. Abnormalities in the process of epidermal cornification can lead to skin barrier dysfunction. The epidermal differentiation complex (EDC) is a gene cluster located within a 2 Mb region of human chromosome 1q21. EDC is responsible for epithelial tissue development and for properties of the stratum corneum. One of the most important features of psoriasis is the abnormal terminal differentiation of keratinocytes. However, the relationship between EDC and the occurrence of psoriasis is still unclear. In this review, we summarize current knowledge regarding the physiological functions of EDC and discuss its possible contributions to the pathogenesis of psoriasis.
Keywords
- psoriasis
- epidermal differentiation complex
- filaggrin
- late-cornified envelope
- involucrin
- loricrin
- S100
The outermost layer of the skin, or epidermis, provides a tight barrier for the human body. The cellular composition of the epidermis consists mainly of keratinocytes, melanocytes, Langerhans cells and Merkel cells. Upon leaving the basal layer, keratinocytes begin a complex mechanism of terminal differentiation that culminates in formation of the stratum corneum. This process is known as epidermal differentiation. The epidermal differentiation complex (EDC) is a cassette of genes present in a 2 Mb region of human chromosome 1q21. It is comprised of 62 coding genes present within four gene families, namely: filaggrin (FLG) and FLG-like, late cornified envelope genes (LCEs), small proline-rich regions (SPRRs), and S100 genes (Fig. 1). EDC encodes structural and functional proteins that have a profound effect on terminal differentiation of the human epidermis [1, 2, 3, 4, 5].
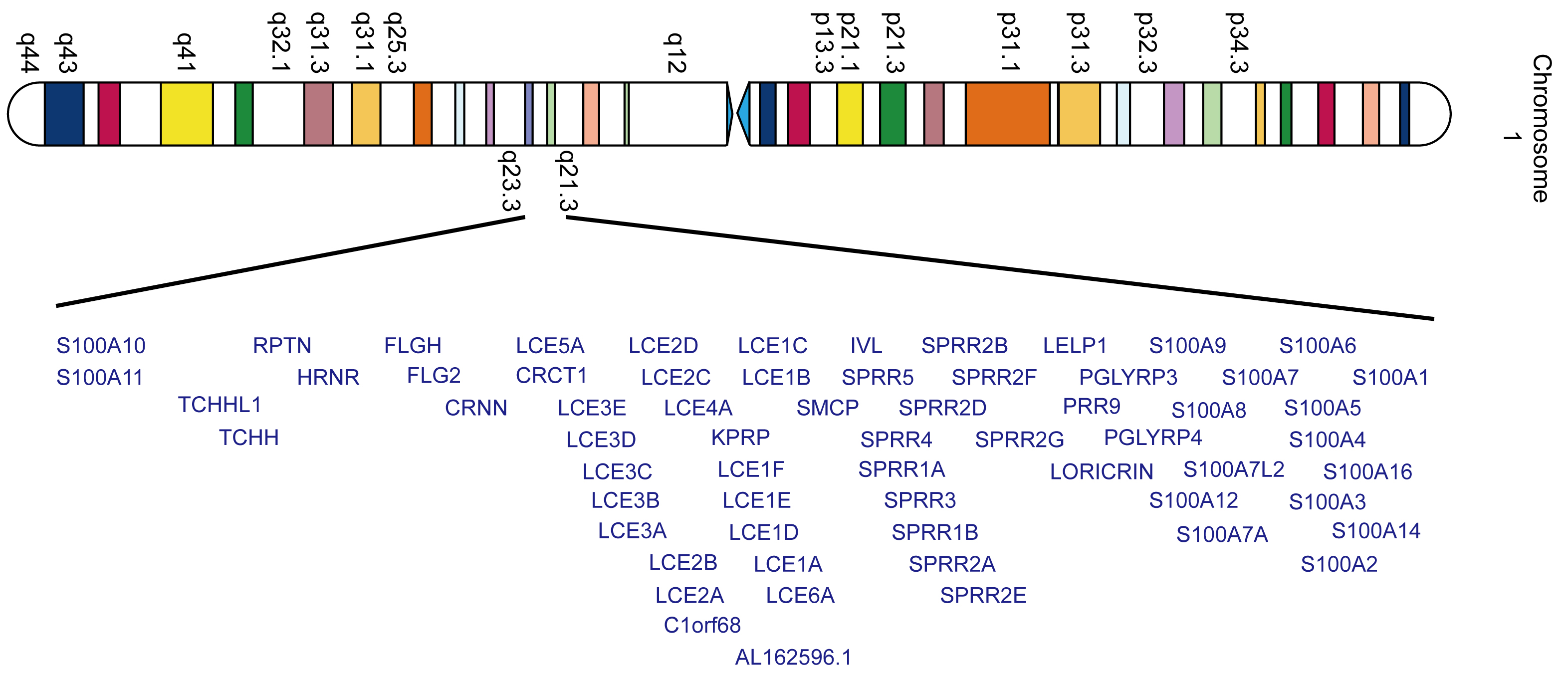 Fig. 1.
Fig. 1.Schematic representation of human EDC genes on chromosome 1q21.
Epidermal cells undergo a set of programmed proliferation /differentiation events and express specific proteins during an ordered and sequential process (Fig. 2a). Following formation of the spinous layer, the cells acquire keratohyaline granules containing mostly profilaggrin. Profilaggrin is cleaved into filaggrin monomers of approximately 37 kDa which can cause keratin filaments to aggregate into tight bundles. Other structural proteins including loricrin (LOR), involucrin (IVL), small proline-rich proteins (SPRRs), and late cornified envelope proteins (LCEs), are expressed later in the process. Subsequently, the epidermal proteins are cross-linked by transglutaminases to form a cornified envelope. However, abnormalities of this process may lead to barrier dysfunction, resulting in skin disorders such as ichthyosis vulgaris, atopic dermatitis (AD), psoriasis, and skin-related neoplasia [6, 7, 8, 9, 10]. The turnover time of normal skin cells is approximately one month, whereas in psoriasis the epidermal cells are replaced in just 4 days [11]. The keratinocyte is the main building block of the epidermis and is the target cell for the major cytokines involved in the psoriatic inflammatory process. The onset and development of the psoriatic phenotype occurs because of increased proliferation and impaired differentiation of keratinocytes (Fig. 2b) [12, 13].
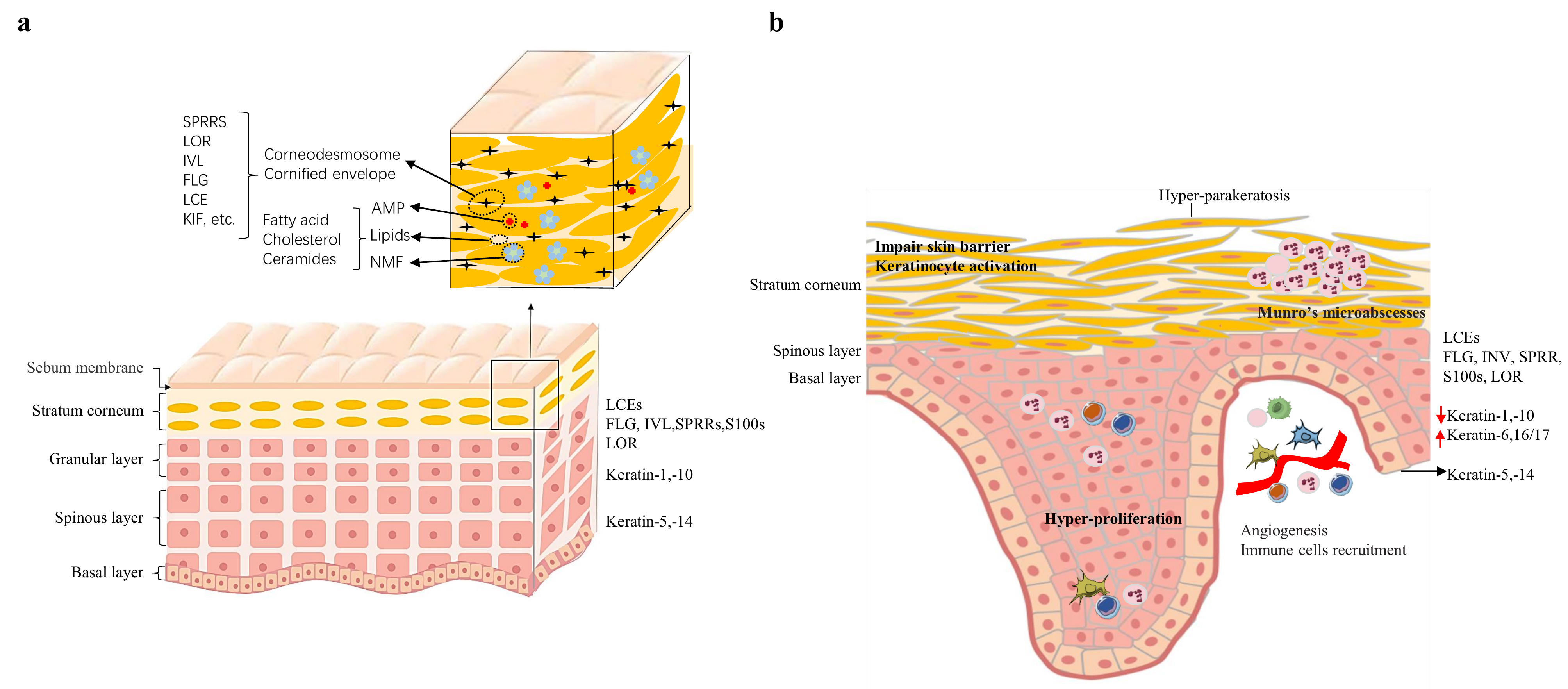 Fig. 2.
Fig. 2.The normal and psoriatic epidermic structure. (a) Epidermal structure and establishment of the epidermal barrier. During the different stages of epidermal differentiation, epidermal cells express different proteins. (b) Altered epidemic structure present in psoriasis. AMP, antimicrobial peptides; NMF, natural moisturing factor; KIF, keratin intermediate filament; IL, interleukin.
Human filaggrin (FLG) is a filament aggregation protein derived from profilaggrin and containing more than 10 filaggrin-repeat units. FLG plays a critical role in the formation of the skin barrier [14]. Based on its cDNA sequence, human profilaggrin contains 25% serine, 15% glycine, 12% histidine, and 10% arginine residues. The profilaggrin precursor mRNA is transcribed as the keratinocytes differentiate into cornecytes [15]. The protein is then synthesized and phosphorylated in the epidermal granular layer. It is subsequently dephosphorylated and cleaved into monomeric filaggrin by a complex group of proteases during the keratinocyte differentiation process from granules to cornified cells [16]. Filaggrin monomers bind to keratin and aggregate the intermediate filaments, thus causing cytoplasmic collapse and the flattening of keratinocytes. Filaggrin is then degraded into free amino acids as the corneocytes move outward through the inner layers of the cornified layer. Although filaggrin exists only transiently, it maintains hydration of the stratum corneum and provides a solid barrier for the skin [17].
Filaggrin expression in psoriatic lesions is significantly reduced compared with
normal skin [18]. In addition, several studies have shown that inflammatory
axis-related cytokines, such as IL-17a, TNF-
A number of studies have been performed to determine whether FLG deficiency is involved in the pathogenesis of psoriasis (Table 1, Ref. [18, 25, 26, 27]). Hüffmeier and colleagues reported that the expression of FLG genes was downregulated in psoriatic skin. However, mutations linked to filaggrin deficiency showed no obvious associations with psoriasis vulgaris or psoriatic arthritis in a genetic analysis of German populations [18]. A case-control study described a significant difference in FLG P478S (rs11584340) genotype frequencies between psoriasis patients and controls in Taiwan, suggesting this polymorphism plays an important role in genetic susceptibility to psoriasis [25]. A novel nonsense mutation in FLG, p.K4022X, was reported in a Chinese family with psoriasis coexisting with ichthyosis vulgaris, as well as a significant association with the occurrence of psoriasis in the Chinese population [26]. However, parallel studies conducted by other researchers reported inconsistent results for this mutation. A case-control study conducted to investigate the R510X and 2282del4 mutations in profilaggrin found no significant association with psoriasis in the Irish and UK populations [27]. Similar results were obtained from a general population study and from a meta-analysis [28], indicating these profilaggrin null mutations are not associated with the occurrence of psoriasis. Moreover, loss-of-function mutations in FLG do not appear to play a major role in childhood psoriasis [29].
| Population | Variant | Study group | Result | Reference number |
|---|---|---|---|---|
| Ireland and UK | R501X (rs61816761) | Ps (n = 691) | no significant association (p = 0.075) | [27] |
| Controls (n = 2117) | ||||
| 2282del4 (rs558269137) | Ps (n = 691) | no significant association (p = 0.932) | ||
| Controls (n = 2117) | ||||
| Germany | R501X (rs61816761) | PsV (n = 737) | no significant association (p = 0.398) | [18] |
| Controls (n = 721) | ||||
| PsA (n = 720) | no significant association (p = 0.675) | |||
| Controls (n = 721) | ||||
| 2282del4 (rs558269137) | PsV (n = 702) | no significant association (p = 0.386) | ||
| Controls (n = 704) | ||||
| PsA (n = 703) | no significant association (p = 0.291) | |||
| Controls (n = 704) | ||||
| China (Taiwan) | P478S (rs11584340) | Ps (n = 314) | a significant association (p = 0.020) | [25] |
| Controls (n = 611) | ||||
| China (Mainland) | p.K4022X (rs146466242) | Ps (n = 414) | a significant association (p = 0.011) | [26] |
| Controls (n = 500) | ||||
| Ps means psoriasis, PsA means Psoriatic arthritis, PsV means Psoriasis vulgaris. | ||||
Genetic mutations in FLG and changes in gene expression may therefore be involved in the susceptibility to psoriasis, but these associations have yet to be fully elucidated. More research is thus required to better understand the role of FLG mutations in the pathogenesis of psoriasis.
Since 2008, the LCE gene cluster on chromosome 1q21 has been recognized as a predisposing site for psoriasis (Table 2, Ref. [30, 31, 32, 33, 34, 35]) [30, 31]. This cluster is further divided into six groups, LCE1-LCE6, based on their relevant amino acid sequence, genomic organization and expression pattern [36]. Most LCE genes are expressed in granular keratinocytes during keratinocyte differentiation and are amongst the final cornified cell envelope (CE) components to be cross-linked to this structure. The expression of LCE2 is upregulated by calcium, while that of LCE1 and LCE2 is induced by UV light [37]. Differential regulation of LCE gene expression occurs in the epidermis of psoriatic skin. Quantitative PCR showed that the expression level of all members of LCE3 was too low to be detectable in normal skin, but was significantly upregulated in psoriatic lesions. In contrast to LCE3, expression levels for the LCE-1, -2, -5 and -6 groups were significantly downregulated in psoriasis [38]. Similar results were observed for the expression of LCE genes after tape stripping [39]. Using immunohistochemistry, LCE3 proteins were detected in the stratum spinosum (SS) and stratum granulosum (SG), but not in the stratum corneum (SC) [40]. Interestingly, Climbazole can also induce the expression of LCE2 and LCE3 genes in keratinocytes when used to treat dandruff and where the skin barrier is thought to be dysfunctional [41, 42].
| Population | Variant | Study group | Result | Reference number |
|---|---|---|---|---|
| Europe/America | LCE1C (rs6701216) | Ps (n = 233) | a significant association (p = 6.2 |
[32] |
| Controls (n = 519) | ||||
| Mongolia | LCE1C (rs6701216) | a significant association (p |
[31] | |
| LCE1B (rs12023196) | ||||
| LCE3A (rs4845454) | PsV (n = 305) | |||
| LCE3D (rs512208) | Controls (n = 383) | |||
| LCE3D (rs4112788) | ||||
| LCE3D (rs4085613) | no significant association (p | |||
| LCE3A (rs1886734) | ||||
| China | LCE3A (rs4845454) | a significant association (p |
[30] | |
| LCE3A (rs1886734) | ||||
| LCE3D (rs4112788) | Ps (n = 1139) | |||
| LCE3D (rs4085613) | Controls (n = 1132) | |||
| LCE1B (rs12023196) | ||||
| LCE3C_LCE3B-del | ||||
| LCE3D (rs512208) | [35] | |||
| Germany | LCE3C_LCE3B-del | PsA (n = 650) | no significant association (p = 0.088) | [33] |
| Controls (n = 937) | ||||
| Italy | LCE3C_LCE3B-del | PsA (n = 424) | a significant association (p = 0.03) | [34] |
| Controls (n = 450) | ||||
| Ps means psoriasis, PsA means Psoriatic arthritis, PsV means Psoriasis vulgaris. | ||||
Real-time quantitative polymerase chain reaction (qPCR) analysis has shown that human LCE1 and LCE2 genes, especially LCE1C, LCE2A and LCE2B, are mainly expressed in the epidermis [37]. The LCE1 group can be trans-activated by p53 and is thought to have tumor suppressor functions by regulating the activity of PRMT5 [25]. A GWAS analysis of European and American populations published in 2008 reported that LCE1C (rs6701216) was a potential susceptibility site for psoriasis [32]. The LCE1C (rs6701216) and LCE1B (rs12023196) genes were later also found to have a strong association with psoriasis vulgaris in a population from Inner Mongolia [31].
The LCE3 gene cluster can be further divided into five subgroups
(LCE3A, LCE3B, LCE3C, LCE3D and LCE3E) with different
structures and functions. Their expression is barely detectable in normal skin
and in non-psoriatic lesions [37]. However, LCE3 expression is
induced in the epidermal layer of psoriasis lesions and after superficial skin
injury. It has been speculated that LCE3 may play an important role in
repair of the skin barrier after superficial injury, whereas other LCE
members may play significant roles in the maintenance of normal skin barrier
function [2, 38]. Several cytokines associated with psoriasis, including
TNF-
GWAS analysis has revealed that LCE3A gene mutations (rs4845454 and rs1886734) in the Chinese Han population were associated with the occurrence of psoriasis, suggesting this gene may be a predisposing factor for psoriasis [30]. Both LCE3B and LCE3C have been associated with the development of psoriasis in different ethnicities [33, 43]. In several different populations, the frequency of LCE3C_LCE3B-del is much higher in psoriasis patients compared to controls [44, 45]. Analysis of 2,831 samples from several European countries demonstrated an association between the LCE3C_LCE3B-del variant and psoriasis, consistent with a large family-based study published in 2009 [33]. Meanwhile, it was also confirmed that LCE3C_LCE3B-del is associated with psoriasis in the Chinese population [30]. A multicenter meta-analysis has confirmed that LCE3C-LCE3B-del is a susceptibility site for psoriasis in multiple European and Asian populations [45].
A possible association between LCE3C_LCE3B-del and arthritic psoriasis (PsA) remains uncertain due to insufficient data. No significant associations were observed between LCE3C_LCE3B-del frequency and PsA in German and Tunisian populations [33, 44, 46]. However, other studies have reported that LCE3C_LCE3B-del was associated with PsA in Italian and Spanish populations [47, 34]. Although LCE3C_LCE3B-del is a well-established risk factor for psoriasis, it is not associated with the Koebner Phenomenon in psoriasis [48]. Other studies have shown that LCE3C and LCE3B are associated with certain immune diseases, such as rheumatoid arthritis and systemic lupus erythematosus [49, 50].
With regards to LCE3D, the LCE gene cluster on 1q21 (rs4112788 and rs4085613) was found to have a close association with psoriasis in a large GWAS study of the Chinese population [30]. Based on the analysis of clinically relevant psoriasis vulgaris subtypes, significant associations were observed between the severity of cutaneous manifestations and LCE3D variants [51]. A novel missense variant in LCE3D (rs512208) was reported in the Chinese Han population through a large-scale sequencing study of psoriasis patients [35]. A missense variant in the LCE3D locus (rs512208) was also reported to be the most important risk factor for psoriasis patients from Inner Mongolia [31]. Interestingly, proteins from the LCE3 family show broad-spectrum antimicrobial activity, with LCE3A being the most potent [52, 53].
The expression of LCE4A and LCE5A is barely detectable in human tissues, whereas LCE6A genes are upregulated during keratinocyte differentiation [37]. However, there is still a lack of evidence to link these three genes with the pathogenesis of psoriasis.
At least 24 different S100 gene members have been identified to date,
most of which are part of the EDC located on human chromosome 1q21 [54, 55].
These proteins belong to the Ca
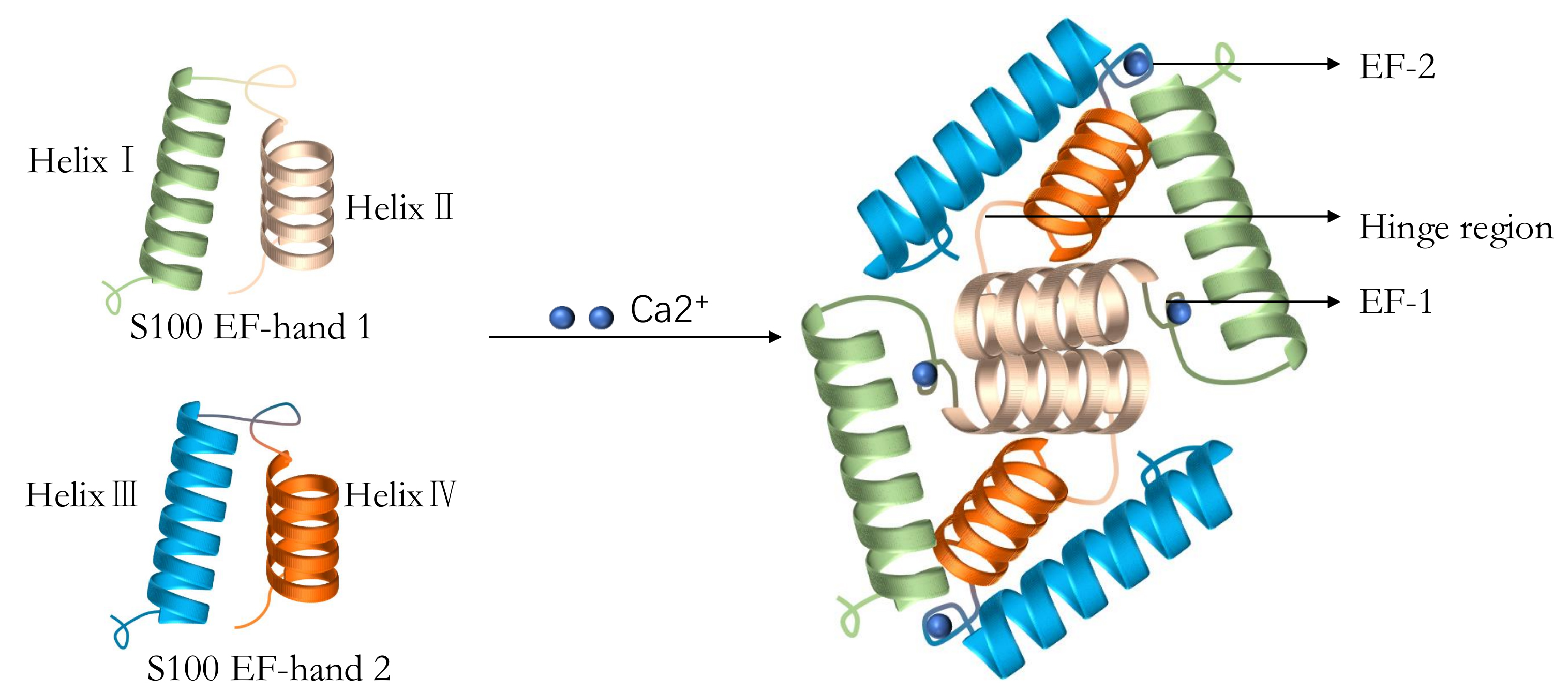 Fig. 3.
Fig. 3.
The typical structure of S100 proteins including two EF-hands,
four
Epigenetic reprogramming is known to be an important factor in the development
of psoriasis, with epigenetic regulation of S100 in particular having received a
lot of attention. Recent results indicate that S100 protein expression may be
correlated with the extent of DNA methylation in S100 gene regulatory
regions, generally presenting as a negative correlation. Differentially
methylated CpGs in S100 were reproducibly identified between psoriatic and normal
skin tissues, including S100A5, S100A9 and S100A12. Several studies have also
reported that the abnormal methylation of S100 protein returns to a normal level
after anti-TNF-
No consensus has been reached on the relationship between S100B and psoriasis. S100B protein is overexpressed in psoriatic patients and shows a significant association with disease severity according to the Psoriasis Area and Severity Index (PASI) score [63]. However, another study found no significant association between S100B protein levels in the serum and the PASI score of psoriatic patients [64].
S100 protein A2 is expressed at high levels in psoriasis and has a protective effect against UV light [65, 66]. The S100 proteins A4 and A6 are expressed in both the hair follicle bulge and during hair follicle germination. They are significantly associated with the activation of hair follicle stem cells, suggesting they play a crucial role in epidermal renewal [67].
The S100 proteins A7, A12 and A15 are highly expressed in psoriatic skin and blood. Serum levels of S100A7 and S100A12 are significantly associated with psoriasis activity [68]. There are some inconsistent results, however. For example, Borsky and colleagues did not find any association between S100A7/A12 levels and disease severity [69]. Furthermore, S100A7 protein can increase the levels of stress-induced, psoriasis-related angiogenic factors, which in turn act on dermal endothelial cells to promote angiogenesis [70]. The interaction between overexpressed S100A7 and Jab1 may contribute to p27Kip1-dependent proliferative dysfunction in psoriasis [71]. The S100A7 serum level is also correlated with the occurrence of subclinical atherosclerosis in psoriasis [72]. The S100 A15 protein is highly homologous with S100A7. Th17 cytokines play an important role in the pathogenesis of psoriasis by inducing the expression of pro-inflammatory S100A15 through the IL-17AR in keratinocytes. This process can be suppressed by the vitamin D analog calcipotriol, or by narrow-band UVB. Therefore, S100 A15 protein may be a promising marker for the treatment response in psoriasis [73, 74].
Several studies have investigated the association between the S100 proteins
A8/A9 and psoriasis. Both S100A8 and S100A9 are expressed and released by
keratinocytes and activated leukocytes during skin inflammation and wound
healing, including psoriatic lesions [75, 76, 77]. The tetramerization of S100A8/S100A9 induced by calcium can inhibit the pro-inflammatory function of
S100A8/S100A9 dimers [78]. The expression of S100A8 and S100A9 is increased in
imiquimod-induced, psoriasis-like skin inflammation and can be stimulated by
several of the psoriasis-associated cytokines or chemokines, in particular IL-17A
and IL-1
In conclusion, several studies have suggested that S100 proteins play an important role in psoriasis because of their contribution to the severity and progression of disease, although the mechanisms remain mostly unclear.
Loricrin (LOR) and Involucrin (IVL) are structurally similar and highly homologous proteins. They have unique internal domains that are cross-linked by glutamine transaminase to bind different glutamine, proline, and serine-rich repeats [3]. There is evidence that LOR and IVL have important roles in terminal epidermal differentiation, while also being involved in the maintenance of epidermal homeostasis.
LOR is the major structural protein of the epidermal keratinocyte envelope (CE) and is mainly expressed in the granular layers and superior spinous layer [3]. Patients with LOR mutations share some common features, including diffuse palmoplantar hyperkeratosis [78, 80, 81]. However, Loricrin knockout (LKO) mice show only mild and transient erythema in the neonatal period, which then improves in adult mice. This may be explained by compensatory mechanisms that upregulate the expression of other barrier proteins such as SPRRP2D, SPRRP2H, and Lce1 to make up for the loss of LOR expression [82, 83]. Psoriasis-associated cytokines such as IL17A and IL22 are known to downregulate the expression of LOR, which then damages the skin barrier function in psoriasis [84, 85]. Currently however, there is still a lack of evidence regarding the possible mechanism of action of LOR [86].
Another important CE protein, IVL, is only expressed in the granular and
supra-spinous layers in normal epidermitis. IVL is one of the main components of
CE and is a marker of the early differentiation of keratinocytes [87]. Several
studies have suggested a trend for increased IVL expression in patients with
psoriasis [88]. Moreover, it is known that some cytokines related to psoriasis
(IL-1, IFN-
In brief, LOR and IVL may have important roles in maintaining skin barrier function and homeostasis. It is reasonable to speculate these proteins may also have different roles in the initial and maintenance phases of psoriasis. More research is needed to reveal the mechanisms by which LOR and IVL contribute to psoriasis through their effects on terminal differentiation of the epidermis.
Although past research has mostly focused on genes that code for proteins, it is now clear that non-coding RNAs play an important part in the regulation of many biological processes. This has led to several new research fields, and in particular the roles of microRNAs (miRNA) and lncRNAs. RNA-seq technology has identified many differentially expressed lncRNAs between normal and psoriatic skin. Interestingly, the EDC is one of the highest lncrna density regions and was significantly enriched for at least 28 lncRNAs [93, 94, 95]. These results suggest not only a distinct role for lncRNAs in the development of psoriasis, but also provide further evidence for an important role of the EDC in the pathogenesis of psoriasis. Although several studies have found that miRNAs can regulate the proliferation and differentiation of keratinocytes, regulation of gene expression in the EDC complex of psoriasis patients requires further exploration [96]. Due to their specific expression and function, further research on both miRNA and lncRNA could lead to the discovery of new biomarkers for the diagnosis, prognosis and monitoring of therapeutic effects in psoriasis.
The EDC is composed of four gene families: filaggrin/FLG-like, LCE genes, S100 genes, and small proline-rich region (SPRRs, including LOR and IVL). EDC genes encode structural and functional proteins that have a profound impact on terminal differentiation in the human epidermis leading to the formation of a solid physical and chemical barrier of skin. Psoriasis is a common chronic inflammatory skin disease that can be triggered by multiple risk factors. This disease involves a number of processes including antigen presentation, transcriptional regulation, immune cell activation, inflammatory cytokine networks, and cell signaling. Abnormalities in the proliferation and differentiation of keratinocytes are the main pathophysiological manifestations of psoriasis. Traditionally, psoriasis has been considered as a Th1/17 cell-mediated and IL-23/IL-17 inflammatory axis-dependent systemic disease that is based on a complex genetic disorder and modulated by environmental factors.
Over the past few decades, considerable progress has been made in our understanding of the role of the EDC in the pathogenesis of psoriasis. However, much more remains to be learned about the pathogenesis of this disease.
Several studies have shown that mutations in EDC genes can not only trigger the development of psoriasis, but are also associated with the progression and severity of disease. Although some genetic risk loci for psoriasis have been identified by GWAS analyses, further studies are needed to determine the mechanisms by which EDC gene products affect keratinocyte differentiation and proliferation. In theory, a single or combination of EDC gene variants could affect differentiation of the epidermidis and its integrity. Once the keratinocyte differentiation process from granules to cornified cells is interrupted, the cornified envelope loses its normal physical function. As a consequence, skin barrier damage will be exacerbated in the presence of external mechanical stimulation, resulting in disruption of skin microbiota homeostasis, invasion of pathogens, activation of innate or adaptive immune responses, infiltration of inflammatory cells into the epidermis and dermis, and abnormal proliferation of keratinocytes. Meanwhile, destruction of the skin barrier may also initiate the repair process to upregulate the expression of other EDC genes such as LCE3 and IVL [37, 89]. Increased expression of S100-related genes is correlated with the severity of psoriasis [76]. Overexpression of the EDC family of genes may promote the secretion of more inflammatory factors by keratinocytes, rather than inducing the denuclearization process. Nevertheless, several EDC genes are downregulated, including FLG. Both innate and adaptive immunity lead to the activation of keratinocytes, resulting in the production of Th1/17-related cytokines which further influences the expression of EDC-related genes and damages the epidermal barrier. This process can lead to a cascading inflammatory response loop and to abnormal proliferation of keratinocytes (Fig. 4).
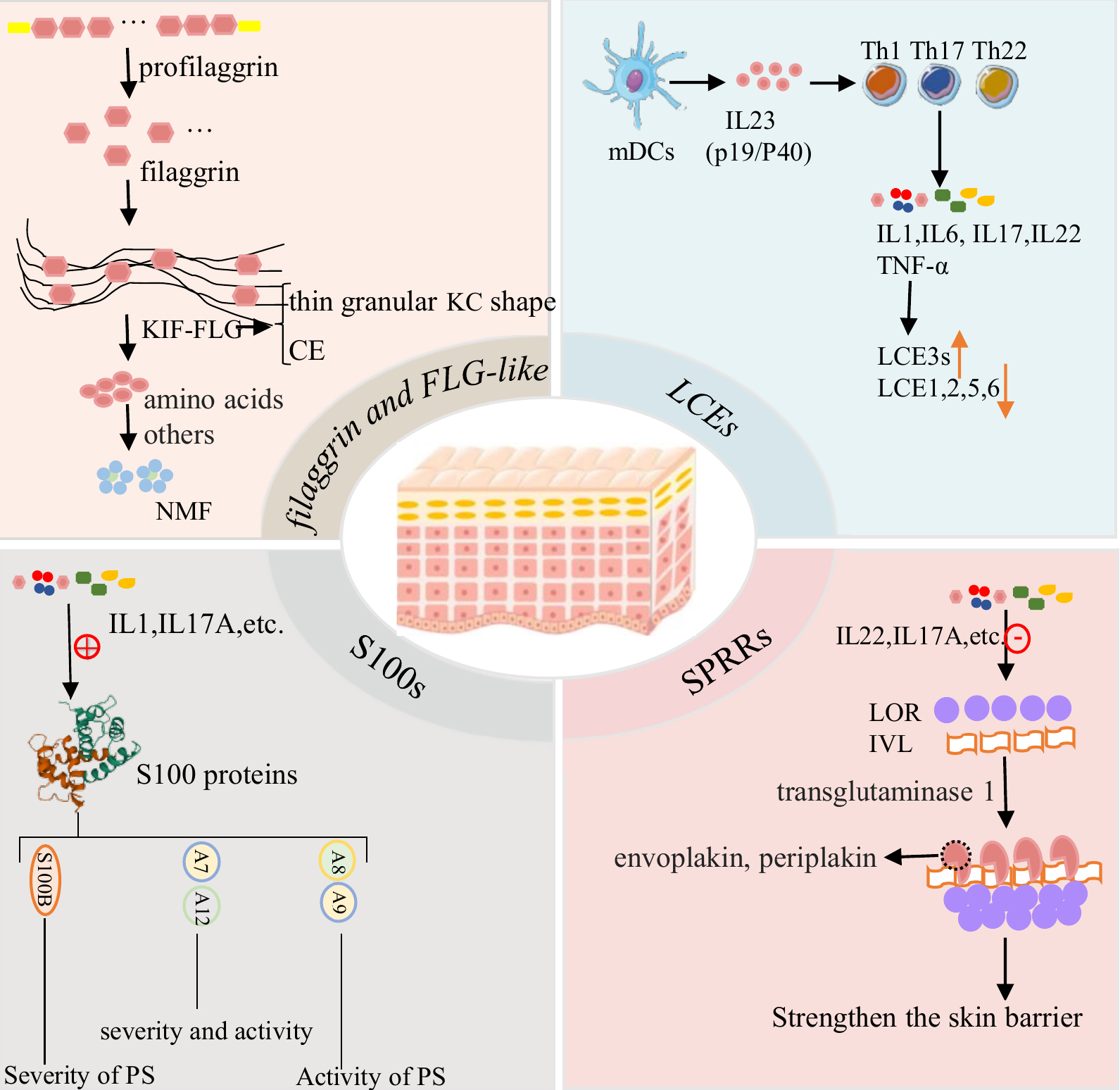 Fig. 4.
Fig. 4.The possible mechanisms of the expression of EDC genes in the occurrence and development of psoriasis.
There is currently a lack of suitable animal models that accurately reflect the clinical features of human psoriasis. These could serve as a useful preclinical research tool for testing new candidate therapeutics and for exploring the pathogenesis of psoriasis. What occurs when a specific EDC gene is silenced or overexpressed in mice? In particular, do keratinocyte abnormalities caused by the absence or overexpression of EDC genes contribute to the occurrence and development of psoriatic-like inflammation in mice? Large cohort studies of EDC gene variants in patients with different stages of psoriasis and from various world-wide populations may reveal additional genetic risk factors for psoriasis. The development of suitable animal models might also lead to the discovery of novel candidate drugs for the future treatment of psoriasis.
DQ and LM prepared the initial draft of the review. LQ reviewed and ensured that the descriptions are accurate. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.