

1 School of Dentistry and Medical Sciences, Charles Sturt University, Wagga Wagga, NSW 2678, Australia
2 ACRF Department of Cancer Biology and Therapeutics, The John Curtin School of Medical Research, Acton, ACT 2601, Australia
Academic Editors: Giuseppe Ingravallo and Graham Pawelec
Abstract
The title usage of Latin Quo vadis ‘where are you going’ extends the question Unde venisti from where ‘did you come?’ posed in the accompanying paper and extends consideration of how ancient eukaryotic and eumetazoan functions of progesterone receptor membrane component (PGRMC) proteins (PGRMC1 and PGRMC2 in mammals) could influence modern human health and disease. This paper attempts to extrapolate to modern biology in terms of extensions of hypothetical ancestral functional states from early eukaryotes and the last eumetazoan common ancestor (LEUMCA), to relativize human metabolic physiology and disease. As novel cell types and functional specializations appeared in bilaterian animals, PGRMC functions are hypothesized to have continued to be part of the toolkit used to develop new cell types and manage increasingly complex tasks such as nerve-gut-microbiome neuronal and hormonal communication. A critical role of PGRMC (as one component of a new eumetazoan genetic machinery) is proposed in LEUMCA endocrinology, neurogenesis, and nerve-gut communication with possible involvement in circadian nicotinamide adenine dinucleotide synthesis. This model would explain the contribution of PGRMC to metabolic and differentiation/behavioral changes observed in age-related diseases like diabetes, cancer and perhaps aging itself. Consistent with proposed key regulation of neurogenesis in the LEUMCA, it is argued that Alzheimer’s disease is the modern pathology that most closely reflects the suite of functions related to PGRMC biology, with the ‘usual suspect’ pathologies possibly being downstream of PGRMC1. Hopefully, these thoughts help to signpost directions for future research.
Graphical Abstract
Keywords
- steroid biology
- eukaryogenesis
- membrane-associated progesterone receptor
- neurogenesis: neurobiology
- synapse
- cyP51A1
- heme
- redox
- metabolism
- translational control
- eumetazoa
- eumetazoans
- LEUMCA
- gastrulation
- gastrulation organizer
- blastoporal axial organizer
- pluripotent stem cells
- TMEM97
- sigma-2 receptor
- cell motility
- sleep
- epigenetics
- aging
- aging clock
- tyrosine phosphorylation
- epithelial-mesenchymal transition
- EMT
- cancer
- diabetes
- intestinal microbiome
This paper is meant to be read after the accompanying paper [1]. Hopefully the reader has been guided to a vantage point perspective that encompasses early eukaryotic roles for membrane-associated progesterone receptor (MAPR) proteins in heme homeostasis, mitochondrial regulation, steroid biology and oxygen responses, as well as recognition of the last eumetazoan common ancestor (LEUMCA) as the evolutionary platform from which our bilaterian body plan evolved, and the extrapolated possible roles of progesterone receptor membrane component (PGRMC) proteins in modern human biology. Most vertebrates have two pgrmc genes, pgrmc1 and pgrmc2, following a gene duplication in the early chordate lineage [2, 3]. Following the conventions of the accompanying paper, this paper will refer to PGRMC1 or PGRMC2 proteins for mammals, or to PGRMC proteins for non-chordates. For common reference, all amino acid numbering refers to the cognate residues of human PGRMC1.
This evolutionary PGRMC1 vantage point provides a conceptual framework from which to assess the role of PGRMC phosphorylation (then and now), where the field currently stands, and where its future trajectory should be oriented: Quo vadis? While this question might have been more accurately asked of the LEUMCA, perhaps from a modern medical perspective we should consider it to incorporate: Unde venis pervenit? (‘Where have you arrived’? But it is meant to mean: “Now that we know this about PGRMC, where should medical science go?”).
The interconnected systems of a modern bilaterian body (e.g., central nervous system mediated coordination of behavior based upon sensory inputs, hormonal and nervous system control of body functions, etc.) all evolved in small sequential steps from the LEUMCA, an organism with a body complexity presumably somewhat similar to that of cnidarians [1]. If there were processes that continuously required PGRMC tyrosine phosphorylation events (at some stage of the life cycle) during that evolutionary pathway (as conservation of Y139 and Y180 among eumetazoans would suggest), then we can expect those processes to be very important in our own biology.
Take, for example, neural coordination of muscle contraction, or of secretory glands. In the LEUMCA, as in cnidarians [1], these cells were adjacent. As more complex bilaterian body plans developed then the basic existing LEUMCA functions had to be adapted to be effective over longer distance via circuitous routes (e.g., nerve axons and hormones). There is a strong case that PGRMC involvement in processes like vasculogenesis, hormonal secretion into the blood, or axon guidance and synaptic function (etc.), represent bilaterian adaptations of pre-existing LEUMCA PGRMC functions. From this vantage point, which is why the narrative of the accompanying paper [1] has circuitously brought us here, stopping to appreciate the lookouts along the way, we can proceed to consider PGRMC functions in modern human biology, as well as to reflectively interpret human biology in terms of PGRMC function.
The gastrulation organizer produces the symbiotic microbiota cavity, or gut. The LEUMCA was the first organism in the eumetazoan lineage to possess a specialized gut (excluding ctenophores, which apparently evolved nerves and gut independently: accompanying paper [1]), and so this topic presents an apt starting point to consider human health.
The intestinal microbiota is intimately associated with human life from early fetal stages to adult [4]. Bacteria have possibly been involved in the evolution of animals from the time of the LEUMCA, or before. All animal epithelia are colonized by bacteria. Microbial communities that colonize the gut of cnidarians differ from those of the ectoderm, suggesting gut-specific roles, although their functions are poorly studied (reviewed by [5]). In triploblastic bilaterians the gut forms a microbe-filled tube, where the gut flora ferments digested food into products that can be used by the host animal under anoxic conditions, e.g., from worms [6] to mammals [7]. In nematodes, bacterial metabolites regulate host muscle mitochondria to attenuate age-related mitochondrial fragmentation, which increases worm lifespan [8]. As discussed in the accompanying publication [1], sleep arose in the LEUMCA, with the evolution of a nervous system. In the diploblastic body plan of the LEUMCA, the physical distance between gut bacteria and neurons was small.
The author is unaware of any evidence that the cnidarian sleep-like state [9] involves bacteria at all. However, a historically early and at the time perplexing discovery is highly interesting in this context. Although it has received scant recognition, in the author’s opinion, this is a classic reminder of how international research progress can be retarded due to the sometimes speciously misguided failure of grant review panels to recognize important research directions.
In the late 1980s and early 1990s a team at the University of Newcastle in Australia were working on slow wave sleep and sleep disturbances, where they had generated a series of solid publications [10, 11, 12]. By the early 1990’s they had biochemically fractionated blood to identify a bioactive peptide. It was discovered (and duly ignored for years) that human sleep is regulated by bacterial peptides [13, 14]. The very observation that none of the key involved authors (Brown, Price, King or Husband) was able to generate follow-up publications in that area is informative that medical funding grant panels scorned and rejected the seismically paradigm-stirring hypothesis of what must have been their next research project funding applications. Scientific meetings and the top journals of the day featured talks on transcription factors (Myc, Jun, Fos, AP1, cyclic AMP-response element binding protein (CREB), p53, etc.), transcriptional regulators such as Rb and P300, or an excitingly (or repetitively) growing string of kinases. Fecal bacteria influencing sleep were not on the radar of the tolerable!
The author personally attended a visiting scientific seminar given by Dr. Brown in the early 1990s, while undertaking PhD studies in Hannover, Germany, and so can recall this case well. It is ironic that years later PGRMC own research brings the author back to the same system, to stumble across this work while investigating that sleep originated in the first organism to develop a gut with presumably specialized gut flora. Having experienced the grant funding system in the same country for a dozen years, the author comments on the retardation of knowledge acquisition by 1990s grant panels by providing quotes from two recent reviews which acknowledge that Brown and colleagues were monumentally correct.
(1) “It appears that a crucial role in the production of reactive oxygen species can be attributed to gut microbiota, due to their ability to shape our behavior and neurodevelopment through their maintenance of the central nervous system” [15].
(2) “Gut microbial metabolites influence central and hepatic clock gene expression and sleep duration in the host and regulate body composition through circadian transcription factors” [16].
The gut microbiota may well also influence human ageing [17]. In terms of PGRMC biology, consider this as another potential system where original LEUMCA functional foundations laid down by the gastrulation organizer may have been co-opted and adapted during evolution of the vertebrate body plan and its physiology.
Glycolytic biology is another potential (hypothetical) area where PGRMC gut cell functions may have been inherited from the LEUMCA. In mammals, colonic epithelial cells (colonocytes) consume oxygen to promote gut lumen hypoxia associated with obligate anaerobic healthy gut microbiota, as related in the accompanying paper [1]. The degree of hypoxia is important to the health of the host mammal. Failure to maintain hypoxic levels permits the expansion of facultative anaerobes, leading to dysbiosis that is associated with several pathologies [7].
In the gut epithelium, stem cells buried in hypoxic crypts divide to produce progeny of increasing degrees of differentiation which are continually pushed up out of the crypts and along the surface of tower-like villi. Mature colonocytes on the villi surface absorb nutrients from the gut lumen, with which they are in direct contact. In this process, stem cells in intestinal crypts perform Warburg glycolytic metabolism. Cells change to aerobic metabolism as differentiated progeny cells move from crypts to the epithelium of the intestinal villi. The differentiation process involves activation of fatty acid catabolism and oxidative phosphorylation by mature colonocytes, consuming oxygen to promote hypoxia of the gut lumen. Thereby, colonocyte aerobic metabolism is central in maintaining a healthy gut microbial population [7].
In light of recent recognition that PGRMC1 modulates Warburg/glycolytic metabolism [18, 19, 20], it is likely that PGRMC1 tyrosine phosphorylation (1) was involved with the origin of the gut, as the first structure formed after/by gastrulation, and (2) is associated with the manipulation of oxygen levels associated with Warburg versus oxidative phosphorylation metabolism that affects microbiota composition that has been associated with multiple pathologies, including inflammatory bowel disease [21], cardiovascular disease [22], neurodegenerative disorders [23], and ageing [8, 24], among other diseases. The general field has been reviewed [25], as has the role of gut microbiota in disease [26]. Whereas a direct role for PGRMC1 in most of these processes remains speculative, evidence for its role in Warburg metabolism and the embryogenic origin of animal tissues via the organizer is strong.
The intestinal tract is the site of insulin secretion because the pancreas is topologically connected to the intestinal epithelium via the common bile duct. It has been suggested that altered PGRMC protein activity could be strongly associated with diabetes [27, 28]. Because of the insulin/glucagon-like effects induced, a role of PGRMC1 phosphorylation in diabetes is very likely (e.g., insulin receptor activation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling and induced vesicle fusions to plasma membrane in myocytes [27], as well as insulin-like effects of PGRMC1 over-expression on metabolic enzymes [19]). Indeed, Craven’s group has demonstrated PGRMC1 regulation of the sub-cellular translocation of both the insulin receptor and glucose transporters to the plasma membrane [27], which are accompanied by increased glycolysis. Further to the ‘pan-metabolic’ role of PGRMC1, Sabbir et al. [29] also reported physical association between PGRMC1 and hexokinase, the first glycolytic enzyme. Atif et al. [30] reported that high levels of progesterone (P4) reduce both cytoplasmic glycolysis and mitochondrial oxidative phosphorylation in glioblastoma cells. They did not provide data on the mechanism of mitochondrial regulation, but discussed possible PGRMC1 involvement, reminiscent of the P4-induced and PGRMC1-associated Warburg effect observed in gestational diabetes [31].
Bearing in mind this ‘pan-metabolic’ biology, the Korean group of Lee et al. [32], using PGRMC1 knockout cells and mice, demonstrated in cultured hepatocytes that PGRMC1 is involved in the regulation of phosphoenolpyruvate carboxy kinase (PEPCK), one of the key enzymes of gluconeogenesis (the mutually exclusive inverse pathway to glycolysis). The mechanism involved the PGRMC1-mediated activation of cAMP synthesis by adenyl cyclase, followed by protein kinase A (PKA)-mediated phosphorylation and activation of the nuclear transcription factor cAMP-response element binding protein (CREB) to induce the gene for the gluconeogenic enzyme PEPCK. The induction of PEPCK required PGRMC1 since it was impaired in PGRMC1 knockout cells.
Interestingly, the inhibitor AG-205 promoted hepatocyte PEPCK expression. Because AG-205 was designed to occupy the heme-binding site of MAPR proteins [33], its binding to PGRMC1 (or other MAPR proteins) probably interferes with heme-binding (although it also has PGRMC-independent effects as discussed in the accompanying paper [1]). Recall that immediately adjacent to the heme-binding cleft is the MAPR interhelical insertion region (MIHIR) motif (see [1]) that putatively interacts with the actin cytoskeleton via a coiled-coil motif, and which acquired a tyrosine at one of the coiled-coil (CC) heptad repeat residues in the LEUMCA [34] where tyrosine phosphorylation would prevent CC-dependent protein interactions [35] (See Y139 in Fig. 10 of the accompanying paper [1]). PGRMC1 does interact with actin cytoskeletal and mitochondrial proteins [36], and actin cytoskeletal protein complexes with PGRMC1 are perturbed by AG-205 [37].
The results of Lee et al. [32] imply that non-heme bound apo-PGRMC1 can lead to activation of cAMP production. Adenylyl cyclase is commonly activated by G-protein-coupled receptors (GPCRs) and leads to PKA activation which is the major effector of the glucagon (anti-insulin) response. Whether cAMP production involved a trimeric G protein was not assayed, however the response occurred in the absence of glucagon, and so was not driven by the glucagon receptor (a GPCR).
PEPCK induction was stimulated by P4 in culture but not in living mouse hepatocytes. In terms of eumetazoan evolution this presents a fascinating apparent development. In cultured hepatocytes, P4 led to increased glucose production. However, in normal healthy mice, P4 suppressed glucose production following insulin induction. Yet, under conditions of insulin deficiency or impaired insulin response, P4 stimulated hepatic gluconeogenesis in mice, similar to the response of cultured hepatocytes [32]. Therefore, hormonal input by insulin apparently overrides a cellular level effect of P4. This possibly reflects an ancestral PGRMC function that has been modulated by the insulin/glucagon system during the evolution of the LEUMCA to deuterostome lineage (and the insulin/glucagon system). If so, then the activity of insulin-induced protein phosphatase 1 may alter the response of PGRMC1 that is induced by P4 (which was not assayed), implying that P4-induced PGRMC1 phosphorylation status regulates gluconeogenesis.
Sabbir [18] showed that P4 can induce changes in PGRMC1 phosphorylation, ubiquitination and sumoylation, which are coupled to altered glycolytic biology and nuclear translocation. Similar effects are also observed by treatment with mifepristone (also known as RU-486), which has conventionally been considered as a specific inhibitor of the classical nuclear progesterone receptor (PGR) as a pregnancy abortion treatment. Rahman and colleagues showed that mifepristone also influences PGRMC1 signaling. For ovarian [38] and testicular [39] cancer models mifepristone leads to PGRMC1 translocation to the nucleus which is associated with altered gene expression, increased proliferation, migration, and invasiveness in mouse xenograft tumor models. PGRMC1 post-translational modifications were not examined, but Sabbir’s findings would predict that mifepristone affects particularly PGRMC1’s phosphorylation and sumoylation states.
Ubiquitinated and sumoylated PGRMC1 run as higher molecular weight complexes in sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS PAGE), as visualized by Western blot. It is unclear from the data published in the Lee et al. [32] study whether higher molecular weight PGRMC1 species were involved in PEPCK induction. They referred to ‘monomeric’ PGRMC1, by which they apparently meant the 25 kDa species in Western blot. All Western blots presented showed only the 25 kDa band of PGRMC1 (the higher molecular weight gel regions are not shown). If heme-mediated dimers [40] were present, their subunits would also have resolved as 25 kDa ‘monomers’ in these gels, and the Kabe et al. [40] paper describing dimeric PGRMC1 was not cited. As such, Lee et al. [32] seemed to omit consideration of higher molecular species (sumoylated, ubiquitinated, other?), and the meaning of their use of ‘monomeric’ PGRMC1 remains unclear. Ignoring the concept of ‘monomeric PGRMC1’, the study did show that PGRMC1 protein levels were involved in PEPCK induction.
The results presented by Lee et al. [32] also imply that the functions of heme-bound holo-PGRMC1, e.g., cytochrome P450 (CYP450) regulation, are separable from those of heme-free apo-PGRMC1, consistent with the model of the accompanying paper’s Fig. 6A [1]. This is important when we consider the scenario that the affinity of heme chelating MAPR tyrosinate residues for heme depends upon the oxidation state of the iron atom. If we additionally consider that MIHIR coiled-coil protein interactions could (1) be inhibited by heme, and (2) be regulated by tyrosine phosphorylation [34, 35], then this framework may contribute towards separating and functionally stratifying the various multiple functions of PGRMC1.
The reader is encouraged to contemplate these issues in terms of the combination of ancient and new functions during eukaryogenesis, upon which were superimposed at least new regulatory modes (if not more probably new functions) that seem to have enabled the descendants of the LEUMCA to develop complex body plans with multiple tissue types, which underlies many aspects of human biology. Establishing communication between these cell types requires not only cell migration during embryology, but also metabolic regulation of and communication between adult cells.
The insulin/glucagon system that regulates blood glucose levels developed in
response to this selective pressure. The same Korean mouse knockout group have
shown that PGRMC1 regulates fatty acid synthesis in hepatocytes [41]. Similar
effects are seen in cancer cells [42], and PGRMC1 phosphorylation site mutants
inversely affect the abundance of fatty acid synthesis and
In summary, following the main hypothesis of this paper that PGRMC1 function was ancestrally related to metabolic control which it can now manifest in a variety of manners, it seems apparent that such perturbations of metabolic flux could easily be associated with diabetes. However, until the various functional attributes of PGRMC proteins are better identified, allowing them to be individually pharmacologically addressed, this acknowledgement does not immediately suggest therapeutic avenues. This situation reflects the failure of grant agencies to recognize the importance of the PGRMC signaling system.
It may be helpful to reconsider neurobiology from the vantage point proffered to us by the observation that PGRMC tyrosines appeared coincidentally with gastrulation and neurons in the LEUMCA, and that PGRMC (at least according to the model proposed here) is ancestrally related to redox sensing via steroidogenesis, heme synthesis, and metabolic regulation functions in early eukaryotes. The extant LEUMCA descendants with most primitive body plans are the cnidarians. These possess nerve nets that coordinate sensory information and motility to satiate hunger [44, 45, 46].
The accompanying paper [1] details how alterations between glycolytic and oxidative metabolism are important for both neural and gut endothelial biology in animals. It also proposed that the origin of both tissues may have been in response to hypoxic stress during the Sturtian glaciation, which hypothetically involved tyrosine phosphorylation of PGRMC, a key ancestrally overarching regulator of mitochondria and metabolic flux. Perhaps this provides a prism through which to view neural metabolism. Active and inactive neurons may be foundationally hardwired to switch between metabolic states because of their evolutionary history. i.e., the mechanism of being a neuron may rely on functions which were useful in LEUMCA neurons, and which modern neurons are compelled to reiterate because of their evolutionary history. This hypothesis deserves investigation.
There is circumstantial evidence that the original neural-gut circuitry may still exist in mammals. As recounted in the accompanying paper [1], the major animal groups may have independently evolved central nerve cords [47], so that we would not expect to observe conservation of a gut-brain neural anatomy between chordates and insects, nematodes, or mollusks.
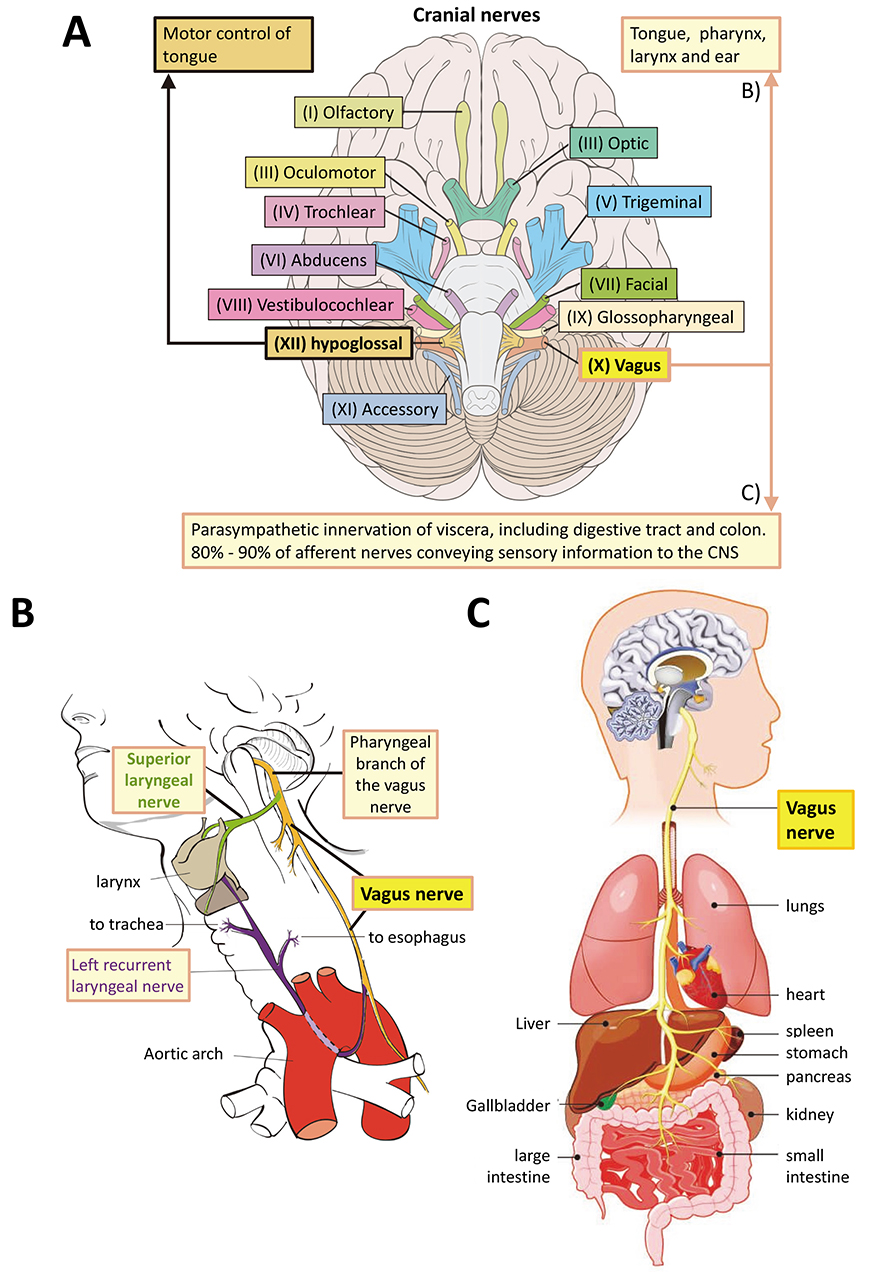
However, the hindbrain structures of the mammalian brain evolved first [48, 49]. The most basal of the twelve central nervous system (CNS) cranial nerves are XII (hypoglossal nerve, innervates the tongue), XI (Accessory nerve, innervates muscles of neck), and X (vagus nerve, main parasympathetic nerve that innervates the thorax including gut) (Fig. 1A, Ref. [50, 51]). Of these, the accessory nerve is a relatively late evolutionary development, having appeared during chordate evolution. The cell bodies of the both the hypoglossal and vagus nerve neurons are initially indistinguishable amongst the neuroblasts in the ventral hindbrain. They grow axons ventrally and dorsally respectively from mouse embryonic day e9.0 [52].
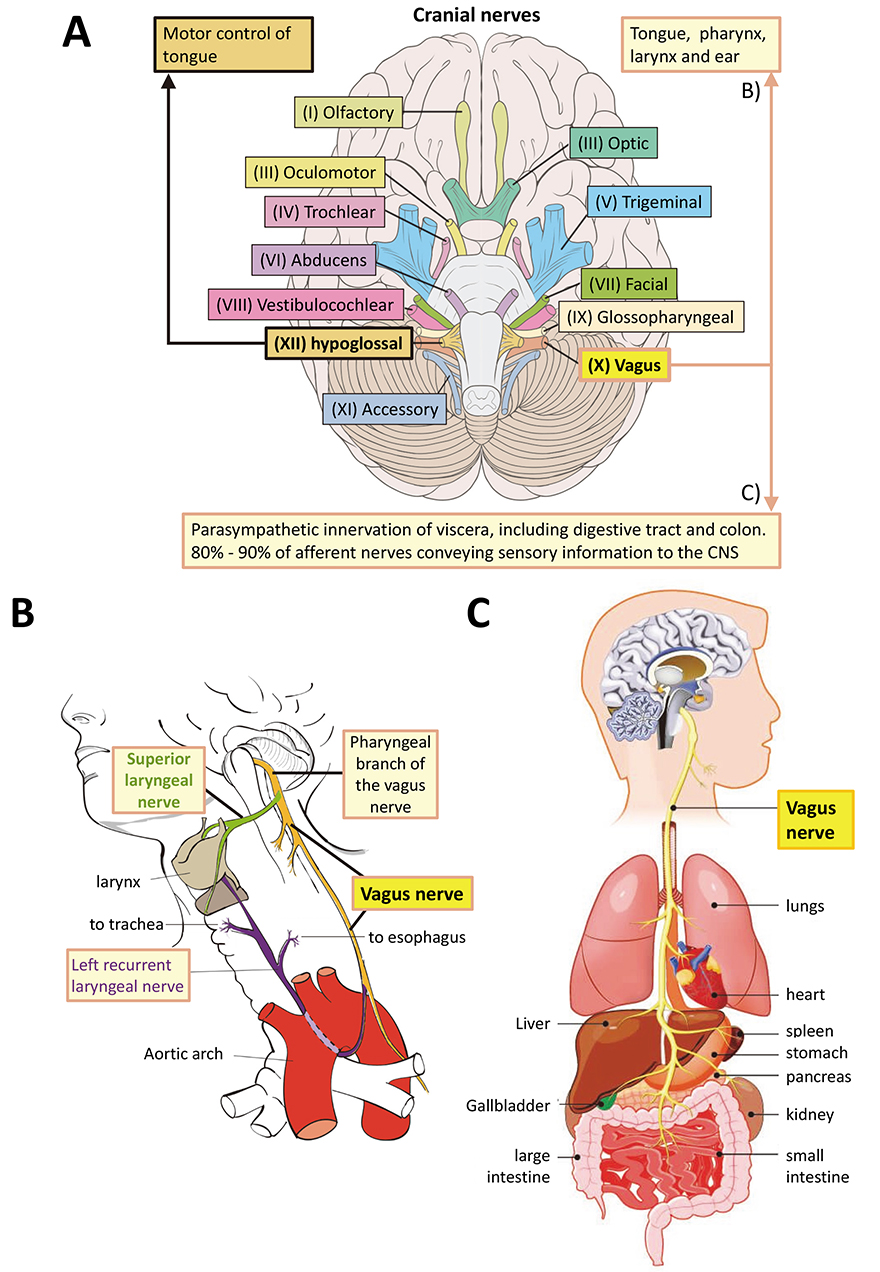 Fig. 1.
Fig. 1.The vagus nerve. (A) The twelve cranial nerves, highlighting the positions of the vagus (X) and hypoglossal (XII) nerves in the human CNS, as a representative of vertebrates. For annotated detail see [50, 51]. The original image by Patrick J. Lynch was taken from https://commons.wikimedia.org/w/index.php?curid=15108118 under a CC BY 2.5 Creative Commons license and was altered by adding labels and annotation. It is provided under the terms of the same license. (B) Innervation by the pharyngeal branch of the vagus nerve. Original image by Wikimedia author Jkwchui. Reproduced under a Creative Commons CC BY-SA 3.0 license from https://commons.wikimedia.org/wiki/File:Recurrent_laryngeal_nerve.svg. Changes to labelling were made. The image is free to reproduce under a CC BY-SA 3.0 license. (C) Organs innervated by the vagus nerve. Note that several tissues innervated by the vagus nerve in B and C reflect gut and mouth relationships that may have been inherited from the LEUMCA. Mesodermal tissues arose first in bilaterians. Reproduced with permission from Biology Dictionary, https://biologydictionary.net/vagus-nerve/.
For our purposes these represent the evolutionarily oldest part of the vertebrate hindbrain, which is the oldest part of the entire brain [48, 53], and therefore the one most likely to share ancestral features with the LEUMCA nervous system. The hypoglossal nerve exerts motor control over the tongue, while the vagus nerve innervates tongue, pharynx, larynx as well as the entire viscera extending to the colon [50, 51] (Fig. 2, Ref. [19, 54, 55, 56, 57, 58, 59, 60]). Therefore, the vagus nerve innervates the entire gastro-intestinal tract from mouth to anus.
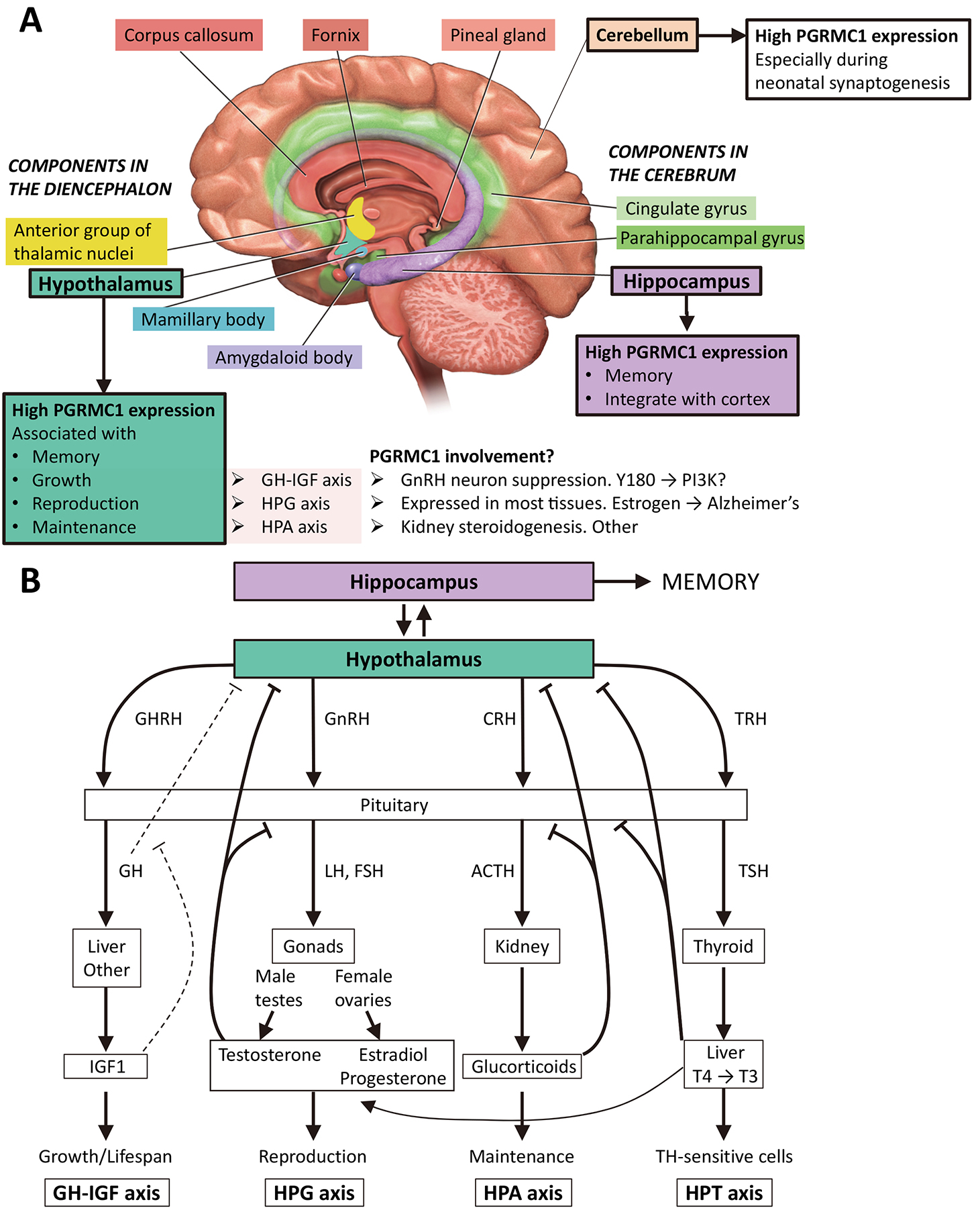
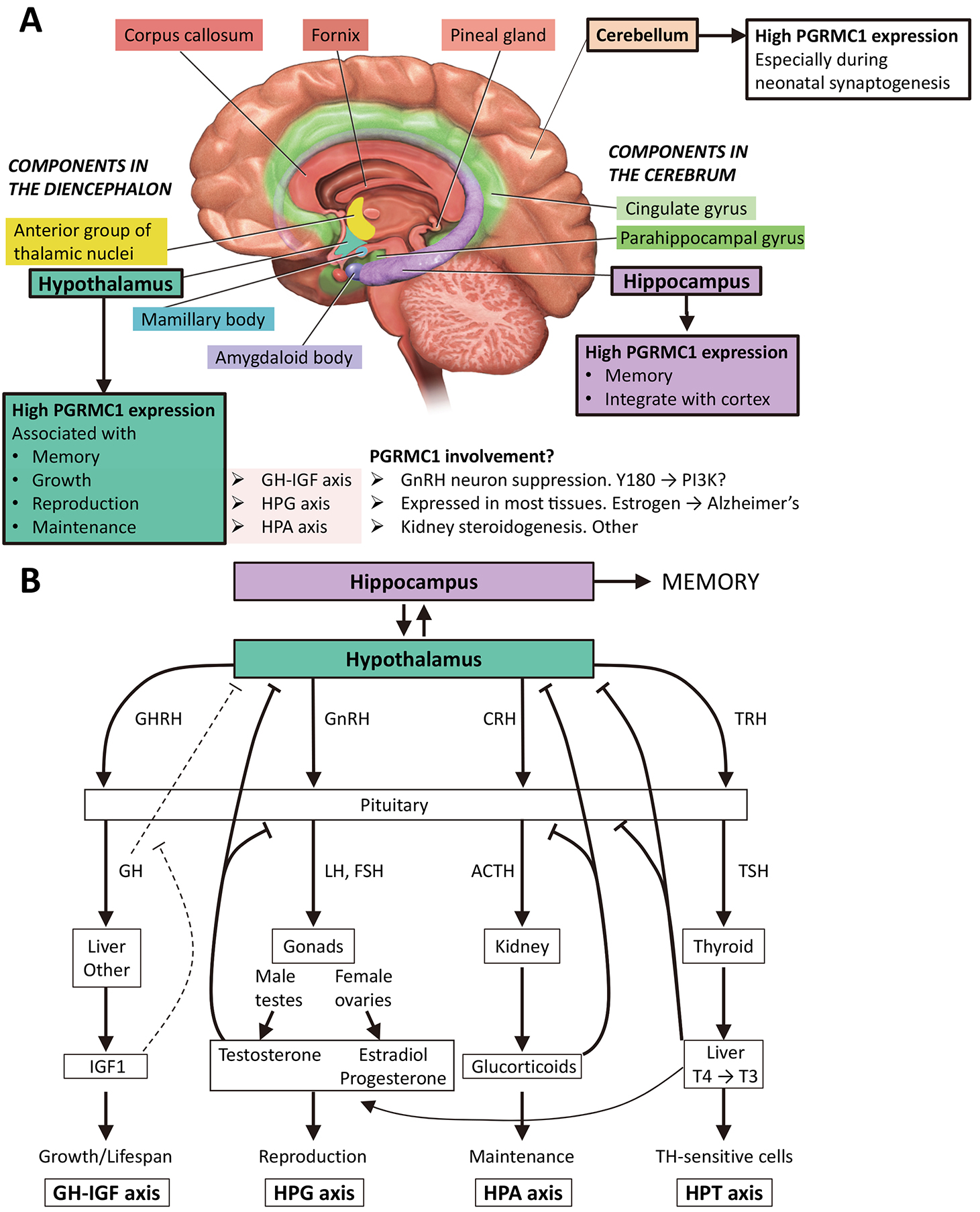 Fig. 2.
Fig. 2.The hippocampus and hypothalamus in body metabolism and memory. (A) The image shows the limbic system, which deals with emotions and memory, and where PGRMC1 expression is reportedly highest in the adult mouse [54, 55]. The hypothalamus is depicted as associated with memory [56], as well as growth, reproduction and maintenance, which depend upon the growth hormone–insulin-like growth factor (GH-IGF) axis, the hypothalamic-pituitary-gonadal (HPG) axis, and the hypothalamic-pituitary-adrenal (HPA) axis, respectively [57]. The PI3K/Akt pathway is a powerful regulator of GH-IGF, and PGRMC1 regulation of PI3K through Y180 [19] could influence this. The HPG axis is heavily involved in learning, involving sex steroids and other mechanisms [58]. As key regulator of sterol availability PGRMC may be involved in the HPA axis, however no clear role has been identified. The limbic system image is modified from [59]. (B) Major whole-body axes of the hypothalamus. The diagram is based loosely upon [57], with addition of the hypothalamic–pituitary–thyroid (HPT) axis, which influences juvenile growth and adult fertility [60]. Note that all these systems have evolved since the last common bilaterian ancestor that was descended from the LEUMCA which acquired PGRMC1 tyrosine phosphorylated Y139 and Y180. CRH, corticotropin-releasing hormone; GH, growth hormone; GHRH, growth hormone-releasing hormone; GnRH, gonadotropin-releasing hormone; IGF1, insulin-like growth factor 1; T3, triiodothyronine; T4, thyroxine; TRH, thyrotropin releasing hormone; TSH, thyroid-stimulating hormone. T4 and T3 (numbers refer to iodines) can both be made in the thyroid. T4 can be also converted to T3 in other organs, such as the liver (shown) [60].
As such, the gut/brain neural connections of the vagus nerve, the most ancient of the vertebrate cranial nerves that primarily regulates involuntary actions via the parasympathetic nervous system, are potentially the evolutionary products of innervation that was proposed to have existed between two neural centers (apical and blastoporal) since the LEUMCA [61]. That system may have innervated the original gut and mouth of the first bilaterian and would originally have resembled the nervous arrangement of the diploblastic LEUMCA. The parasympathetic nervous system is a part of the autonomic nervous system that controls the activity of the smooth and cardiac muscles and glands, functions which must be very similar to what the urbilaterian nervous system inherited from the LEUMCA.
Perhaps that is related to the observed prominent involvement of the vagus nerve in the axis between gut microbiota and the brain [62] and its dramatic influences over CNS function [63, 64], if those neural connections have been conserved since the first neurons mediated communication between gut and sensory nerve centers in the LEUMCA. If PGRMC was ancestrally related to neural function, we expect it to feature prominently in vagus neurons, including during embryogenesis. It is fully conceivable that communication between neurons and gut epithelium from the LEUMCA provided a platform that was built upon and reformed, rather than replaced, in complex bilaterians. If so, then PGRMC1 involvement in synaptic membrane trafficking, LDLR internalization, and involvement in insulin/glucagon regulation of metabolic regulation may represent vestiges of that ancient system. PGRMC biology may form an indispensable part of the fabric of eumetazoan body architecture. To the author’s knowledge, this has not been explored. Note that this is a deductive process. Strong experimental evidence does not exist to support this hypothesis.
The nematode ventral midline-1 (Vem-1) homolog of PGRMC1 is involved in the guidance of some axons during the establishment of the ventral nerve cord. A single neuron called AVG (anterior ventral neuron G) pioneers the right ventral cord axon tract, depositing signals that will subsequently be used by other axons. Vem-1 expression was detected at gastrulation and later in early anterior head neurons, including neurons of the nerve ring and the AVG that extend axons into the ventral nerve cord. The AVG cell nucleus is near the posterior base of the brain (analogous to the origin of the vagus nerve), and its axon migrates towards the posterior, secreting the ligand netrin/UNC-6 (uncoordinated-6: uncoordinated due to faulty nervous system) which is part of an evolutionary conserved guidance system involving the receptor deleted in colorectal carcinoma (DCC)/uncoordinated-40 (UNC-40) (reviewed by [65, 66]). Subsequent axon guidance uses the secreted netrin/UNC-6 of pioneer nerves like AVG as guidance during formation of the ventral nerve cord. Vem-1 interacts physically and genetically with UNC-40, the C. elegans homolog of the netrin receptor DCC, and vem-1 gene deletion results in failure of the AVG neuron but not others to faithfully extend axons along the correct pathways. As we saw in the accompanying paper [1], the Netrin/DCC system first appeared in the LEUMCA [67], and one of the circumstances under which PGRMC1 was first identified in mammals was under the synonym of ventral midline antigen (VEMA), involved in axon migration of the embryonic mouse central nerve cord [68, 69].
Interestingly, a comparative holistic model has been recently proposed by Wang et al. [57] for coordinated cell metabolism and immune functions coordinated through the hypothalamus. An extension of that model is proposed here, where primal PGRMC biology directs profound deep-level changes to cell gene expression and metabolism, related in part to its ancient role in mitochondrial regulation.
PGRMC1 is expressed abundantly in the hypothalamus, where it is thought to exert local immunomodulatory functions [54, 55]. Wang et al. [57] discuss the hypothalamus as associated with three major whole organism feedback axes concerned with growth (growth hormone–insulin-like growth factor (GH-IGF) axis), reproduction (hypothalamic–pituitary–gonadal (HPG axis), and homeostasis (hypothalamic-pituitary-adrenal (HPA) axis). Another major axis is the hypothalamic–pituitary–thyroid (HPT), which is involved in juvenile growth and adult fertility [60] (Fig. 2B). Neurosteroidogenesis of P4, and associated reorganization of neuronal actin cytoskeleton feature prominently in the development and differentiated function of these axes [70, 71, 72, 73].
The HPA axis activity can affect memory, e.g., via stress signaling to the hippocampus [74] which involves glucocorticoid steroid hormones. PGRMC1 was originally identified under the synonym ‘inner zone antigen’ and a cytochrome P450-regulating protein associated with renal glucocorticoid steroidogenesis [75, 76], and PGRMC1 is also located in gonads [77, 78], pituitary [79], cortex, hypothalmus including hypothalamic nuclei involved in female reproduction, as well as in the hippocampus in neonatal and adult mice [55, 80]. The latter is recognized as a central component in memory formation [81]. So PGRMC1 is expressed in all the major cell types involved in the HPA axis, as we might expect if the system had evolved from a LEUMCA precursor that resembled modern cnidarians. It also inhibits gonadotropin-releasing hormone receptor (GnRH) neurons of the hypothalamus in response to P4 [82], and is expressed in the thyroid [83] and liver [41]. Remarkably, PGRMC1 is expressed in many of the tissues involved in the major whole organism feedback loops of the hypothalamus, and there is evidence of its direct involvement in many of the cell responses involved in those pathways (Fig. 2). Once more, this thought process is hypothetically deductive. The model requires experimental verification.
Another organism-wide regulatory system involves glucose homeostasis via the insulin-glucagon system. If we view the insulin response as a vestige of communication between gut, neurons, and muscle of the ancestral LEUMCA, via insulogenic secretory cells, then its manifestions in the central nervous system could be highly informative. These include (but are not limited to) glucose homeostasis [84, 85], dietary intake [86, 87, 88], neuroprotection [89], neuron growth and differentiation [90], synaptic activity [91], and memory formation [92], and could validly be added as a fifth axis to Fig. 2B. Insulin resistance leads to hippocampal dysfunction: impaired neuroplasticity and decreased cognitive function, as well as increased risk of Alzheimer’s disease (AD) [93, 94]. The relationship between insulin biology, neuronal health, and memory has indeed prompted the concept that Alzheimer’s disease can usefully be considered as type 3 diabetes [95]. The author would extend this to propose that the functioning central nervous system can be usefully considered to be dependent on ancestral PGRMC functions, and that this extends to AD and quite probably many other neuropathologies. We will pursue this below.
The involvement of PGRMC1 in steroidogenesis and P4 responses in the nervous
system and cells associated with the female reproductive system is established
and has been extensively reviewed previously [75, 76, 78, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114]. It will not be
discussed further here beyond noting that the competing reaction to that
catalyzed by lanosterol-14-demethylase (CYP51A1) and PGRMC1 involves the
24-dehydrocholesterol reductase-mediated reduction of lanosterol to
dihydrolanosterol, which requires NADPH but not oxygen (see Fig. 3 in the
accompanying manuscript [1]). Interestingly, this enzyme activity (under the
synonym seladin-1) protects against amyloid beta (A
The S2R is a pharmacological activity with possible therapeutic relevance to cancer [116, 117, 118, 119] and neurological disorders [120, 121, 122]. For reviews on the history of Sigma-1 receptor and S2R field see [118, 123, 124, 125]. A relatively detailed review of recent development in S2R biology is provided here to provide adequate background for non-versed readers for the section on Alzheimer’s disease.
S2R activity was initially localized to an 18–22 kDa [126] or 21.5 kDa [127] membrane protein. The finding that PGRMC1, with predicted molecular weight of 21.67 kDa, was cross-linked to a photoactivable S2R ligand [128] led to an initial degree of confusion as to whether PGRMC1 itself was the S2R, rather than a member of a protein complex containing S2R. It soon became apparent that whereas S2R activity was decreased by reducing PGRMC1 levels in some cell systems [118, 128, 129], in others this was not the case [130, 131, 132].
S2R-ligand-dependent affinity purification and mass spectrometry identification of associated proteins revealed that the small integral membrane protein transmembrane protein 97 (TMEM97) was crucial for S2R activity [133]. TMEM97 was previously known as meningioma-associated protein 30 (MAC30) [134]. It binds ligands within the hydrophobic interior of the lipid bilayer [135, 136]. Initial commentary on the identity of TMEM97 with S2R took the position that its molecular cloning completed the unambiguous characterization of this receptor class [137], and resolved its “identity crisis” with PGRMC1 [138]. This sentiment has become so well rooted that both NCBI (NP_055388.2) and UniProt (Q5BJF2) have now currently reannotated the TMEM97 protein as “sigma intracellular receptor 2”. In both cases the gene name is TMEM97. This altered nomenclature was adopted despite having been originally characterized with nomenclatural priority as MAC30 (meningioma is the most common type of head cancer), and the fact that S2R/TMEM97 is indeed overexpressed in a variety of other cancers [123, 139, 140].
TMEM97 is one of a suite of genes induced by sterol regulatory element-binding protein (SREBP)-2 under low cholesterol levels (the system regulated by PGRMC [41, 141, 142]), that is involved in the endosomal lysosomal compartment where it is associated with LDL cholesterol transport-regulator Niemann-Pick C1 (NPC1) [143]. It is also a member of a group of related proteins that by similarity are likely to possess sterol isomerase activity [144]. In fact, although the two human sigma receptors (S1R and S2R) are unrelated to each other, they are both related to other families with sterol isomerase domains [144], and so seem connected under the broader umbrella of sterol biology. Their overlapping ligand affinities are probably the result of convergent evolution (see [123]).
Sterol biochemistry is central to PGRMC function, whether as a member of the Insig-1/SCAP complex that senses sterol levels and regulates activation of the mevalonate pathway by sterol regulatory element binding proteins [41, 141, 145], involvement with CYP51A1 (the most conserved eukaryotic CYP450) in the 14-demethylation of lanosterol (the very first sterol modification, and therefore the earliest to evolve) [142, 146, 147], or its conferral of responsiveness to progestogens, including P4 (reviewed in [97, 147] or to other steroids such as estrogen [148, 149, 150, 151, 152]). And of course the finding that the enzymes of the entire mevalonate pathway, lanosterol cyclase, and the CYP51A/PGRMC reaction, that 14-demethylates the first sterol lanosterol, all came from bacterial rather than archaeal genes [153]. PGRMC may justifiably be thought of as a godfather of steroid biology, exerting effects that appeared at least a billion years before the appearance of nuclear steroid receptors in early animals.
It is then perhaps no surprise (in hindsight) that PGRMC1 was found to form a complex with the LDLR and TMEM97/S2R [43], which obviously provides the solution of a lipid supply problem that appeared after a vascular circulatory system and specialized tissue functions in chordates, where most steroid synthesis occurs in the liver but is consumed in the periphery. The author had highlighted the involvement of TMEM97 in sterol transport [28], and was fortunate enough to have predicted such a PGRMC1::LDLR interaction [97], which promoted Mach and colleagues [133] to investigate the potential interaction of PGRMC1, TMEM97 and LDLR. The PGRMC1/TMEM97/LDLR complex was responsible for a pathway that elevated rates of LDL internalization over and above the background constitutive levels. Since actively growing cells require more cholesterol, this was consistent with observations that S2R activity was elevated in growing cells [139]. Removal of TMEM97 from this system totally removed the binding of their S2R ligand, RHM-4 [43].
As discussed in the accompanying paper [1], the author’s laboratory has generated preliminary data that tryptophan-rich sensory protein (TSPO) may form an obligate part of a TMEM97-containing S2R ligand-binding complex, where both TSPO and TMEM97 are both required for S2R activity. TSPO and PGRMC1 both interacted with TMEM97 in proximity ligation assay, but not with each other [154]. TSPO binds heme and cholesterol [155]. That PGRMC1 and TSPO may be functionally related is further strengthened by several MAPR-related candidate phyla radiation (CPR) bacterial genes for cytb5MY proteins being found in operons that include a bacterial TSPO gene, in a phylogenetic distribution suggesting that the ancestral cytb5MY-encoding operon contained TSPO [153]. The endogenous 20(S)-hydroxycholesterol ligand of TMEM97 [136] is formed by CYP11A1 as the cholesterol steroid skeleton is transferred from the outer to inner mitochondrial membrane during the synthesis of pregnenolone, the first animal steroid hormone [156]. Deductively reconstructing, one could assume that the TMEM97 ligand system arose after the origin of animals. It remains possible that a 20-hydroxylase activity could have acted on one of the steroids from the metabolic pathways of lanosterol synthesis [1] before the evolutionary appearance of cholesterol, and later adopted cholesterol as its substrate.
That the ‘identity crisis’ of S2R may not yet be fully solved by its identity with TMEM97 is further supported, once more by publications from the Mach laboratory. Having demonstrated that TMEM97, PGRMC1 and LDLR form a complex, and that S2R ligand binding was abrogated by TMEM97 depletion [43], they also showed that S2R-induced cytotoxicity was unaffected by TMEM97 or PGRMC1 depletion. Even depleting both together had no effect on S2R-mediated cytotoxicity [139, 157]. Furthermore, while the double knockout reduced the rate of fluorescent S2R ligand SW120 uptake, the level of internalized SW120 of knockout cells eventually reached the same levels as control cells [157]. Sereti et al. [158] also concluded that S2R-ligand-mediated cytotoxicity did not correlate with levels of S2R/PGRMC1 protein abundances, and hence that the cytotoxic mode of action does not involve a TMEM97/PGRMC1 complex. One possibility is that another as-yet unidentified S2R activity such as TSPO is responsible for S2R-mediated cytotoxicity, and hence that TMEM97 represents just one S2R receptor. If so, then the identity of the further binding activity could provide new insights to address the pharmacology of pathology which TMEM97 does not exert. Another possibility is that the PGRMC2 protein could partially substitute for absent PGRMC1 function, however a perplexity of possibilities exists. For instance, S2R ligands could conceivably bind to non-protein membrane components.
As an aside, the COVID-19 disease-causing severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) protein Orf9c is present in a protein complex with
TMEM97 [159]. It is interesting to speculate whether TMEM97 may be involved in
the endocytic entry of virus to the cell. There is no association between PGRMC1
and SARS-CoV-2, however PGRMC1 does exert a negative influence over influenza A
virus infection [160]. There, infection leads to downregulation of PGRMC1, which
is associated with reduced interferon-
Finally, a closing note to the discussion of a S2R::PGRMC1 complex, PGRMC1
involvement in the Warburg effect aligns with a report that a sigma-2 receptor
ligand increases aerobic glycolysis, elevating hypoxia-inducible factor 1 alpha
(Hif-1
PGRMC proteins may underlie many of these root-level gastrulation organizer-initiated processes in eumetazoans, including humans. Furthermore, and this may well be wrong, but AD appears to the author personally to be the disease that is most directly related to perturbations of the PGRMC signal system described here [162]. A such, its consideration can surely be broadly revealing.
The author has had only a tangential association with neuroscience research,
precipitated by his development of proteomics technologies and possession of
molecular cell biology signal transduction expertise in a company where others
excellently performed neural cell culture [163, 164]. Nevertheless, based on
having PGRMC1 expertise the author was recruited in 2013 by Cognition
Therapeutics Inc. (CogRx, Pittsburgh, PA, USA) as scientific advisory board
member to their AD therapeutics program (see conflict of interest statement).
They had developed an anti-AD S2R ligand series which displaced synaptic
oligomeric amyloid beta (A
PGRMC1 is involved in a membrane trafficking pathway which is required for the
mechanism of action of synaptorestorative drug CT1812 (Note that CT1812 binds
S2R, whereas A
A
It was mistakenly thought that PGRMC1 was the S2R, as explained above, which is why the author was recruited by CogRx. The narrative will expound below why this may have been serendipitous because otherwise the author would not have thought about AD in depth, and how the grand-scale biology of PGRMC, as outlined here and in the accompanying paper [1], may be central to the pathobiology of AD.
AD is an area of critical and burgeoning importance. Worldwide, the 33 million
sufferers in 2018 already carried a social cost exceeding US$1 trillion. In
2020, an estimated 700,000 people in the United States aged 65 and older had
Alzheimer’s when they died [173, 174], compared with 627,039 deaths in the same
country for the COVID-19 pandemic from February 2020 until July 27th 2021 [175].
Unless improved interventions are developed, the cost is expected to double by
2030, ballooning to more than 150 million cases by 2050 [176]. Governments
recognize this and funding bodies in several nations have been prioritizing AD
research. Large pharmaceutical companies spent more than US$75 billion on R&D
for AD in 2016 and sponsored
Despite the growing huge market and unmet social need, the large pharmaceutical
companies - the only bodies capable of bringing treatments to market - have been
withdrawing from AD research (e.g., Pfizer in Jan 2018, Biogen in March 2019)
after three decades of unproductive research pursuing the traditional “amyloid
cascade hypothesis” [26, 168, 181, 182]. The amyloid cascade paradigm proposed
that errors in the production, processing, and aggregation of A
Much grant agency support continues to flow to project proposals based upon the Amyloid Cascade Hypothesis historical model, despite mice with plaques developing no memory impairment and three major beta-site amyloid precursor protein (APP) cleaving enzyme (BACE) inhibitor anti-plaque strategy drug failures in late 2018 [26, 192] (for detailed discussion see [190]). In a field where the historical success rate for discovery of new treatments has been exceedingly low and requires new impetus [26, 176, 193], the growing and tragic social burden is exacerbated by those treatments which have been approved being unable to prevent progression, but merely temporarily ameliorating some symptoms [26, 168, 178, 194]. New insights and alternative mechanisms for drug development are desperately needed [26, 194], and here, perhaps, is an example.
CogRx [122, 169] pursued an alternative version of the amyloid hypothesis
[195, 196, 197, 198], neglected by most studies at the time and all drug developments that
had until then entered clinical trial. It predicted that the key neurotoxic
A
CogRx recently demonstrated [208] that the naturally occurring human A
Interestingly, Biogen recently resurrected their previously abandoned Aducanumab
program: a monoclonal antibody that recognizes both A
In October 2019 Biogen announced they would be filing for FDA approval, which
was met with a mix of cautious optimism and skepticism by analysts at the time
[168, 212, 213, 214], and after FDA approval was granted in 2021 (but only for
participation in clinical trials) [178, 180, 210, 211, 215, 216]. The Biogen
presumed mechanism of action for Aducanumab involves specific targeting of
aggregated A
It is fully possible/probable that marginally beneficial effects are due to
A
In proposing that PGRMC1 may be central to AD [162], the author feels obliged to provide the community with a deeper rationale. As a non-neuroscientist, this may seem presumptuous, if not audacious. For a non-technical overview of the complexity over which the author has only limited understanding, see Herrup [190]. In the following, we walk through some AD- or neuron-related biology and make connections with the story that has been developing for PGRMC function in this and the accompanying paper [1], and never assuming that addressing PGRMC1 biology might hold all answers to AD therapy.
If PGRMC-dependent processes were foundational to the evolution of the nervous system, as inherited from the LEUMCA, we could reasonably expect to see the involvement of neural innervation of mesodermal and gut structures by the vagus nerve (as discussed above), and hormonal regulation of these systems that evolved with vasculature systems. Glucose-sensing neurons are at multiple locations in peripheral and central nervous systems. The most extensively studied brain region is the hypothalamus, but the various regions are thought to be synaptically highly interconnected.
Brain glucose-sensing neurons make sympathetic and parasympathetic connections
to target organs that modulate metabolism, such as liver, pancreas and visceral
adipose [218], all of which are innervated by the vagus nerve. It has long been
recognized that vagal stimulation can enhance pancreatic insulin release [219, 220], which the author is hypothetically relating to PGRMC function. Part of the
reduced pancreatic
The vagus nerve plays a clear role in pancreatic activity, and also in the regulation of signals from brain regions that influence hedonic components of feeding behaviors [220]. Here, glucose homeostatic neural mechanisms are largely controlled by nutrient and hormone effects on the hypothalamus and brainstem ganglia. Conversely, hedonic drive is controlled by the mesolimbic dopaminergic (DA) system. Hormones and nutrients can directly regulate the DA system, or indirectly regulate via the hypothalamus or brain stem neurons. Hormones include satiety signals such as leptin (from adipocytes), insulin (pancreas), or glucagon-like peptide-1 (GLP-1) (from the gut, or from GLP-1-expressing neurons), or opposing hunger signals such as ghrelin (from the stomach), orexins (mostly from hypothalamus), or neuropeptide Y (sympathetic neurons), as reviewed [220].
We are here considering the modern mammalian derivative of the ancestral
communication between gut, nerves and behavior that was initially associated with
the evolution of the LEUMCA organizer, and the first eumetazoan nervous system.
To assess the potential involvement of PGRMC in satiety control (the
insulin/glucagon system), consider that PGRMC1 is involved in the regulated
subcellular translocation of the following proteins to the plasma membrane: the
insulin receptor and glucose transporters GLUT-4 and GLUT-1 [27], and the GLP-1
receptor in pancreatic
The insulin/glucagon system regulates not only glycolytic activity, but also the
system of fatty acid storage of excess energy from glycolysis. By regulation of
glycogen synthase kinase 3 (GSK-3
Lipid synthesis is regulated by glucose. High glucose levels increase sterol and fatty acid synthesis via SREBP1 and SREBP2 [225]. SREBP1 is N-glycosylated in the presence of high glucose, which regulates its activity [226]. Some non-cholesterol sterol (lathosterol, cholesteronol, desmosterol) levels are good predictors of hyperglycemia and the development of type 2 diabetes [227]. PGRMC1 forms a complex with SREBP1 and SREBP cleavage activating protein (SCAP) [145]. PGRMC1 knockdown promotes dysregulation of this system leading to hepatic steatosis [41], and PGRMC1 modulates lipid homeostasis in adipose [228] and cancer breast cells [42], and in cardiac muscle cells which are highly dependent upon fatty acid oxidation [229].
Thus, excessive energy available as glucose seems to be converted to fat under the watchful eye of the PGRMC/SREBP1 system [230]. This reflects the eukaryogenic role proposed in the accompanying paper [1]. Accordingly, perturbed regulation of the PGRMC/SREBP system is thought to be related to increased fatty acid and cholesterol synthesis by antipsychotic drugs [141], perturbed lipid homeostasis and oncogenic progression in breast cancer [42], and a deletion of PGRMC2 is associated with fatty acid variations in the milk of dairy cattle [231].
As well as regulating fatty acid synthesis, SREBP activates the pentose phosphate pathway [232], the major source of reducing power to counteract oxidative stress [233] (notable in a hypothesis associating PGRMC with oxidative biology and eukaryogenesis [1]). SREBP1 is one of the major metabolic regulators induced by mammalian target of rapamycin c (mTORc) in response to low energy levels, along with Myc and Hif-1 [232] (also related to the biology of aging, which is discussed below). As we saw in the accompanying paper [1], PGRMC phospho-tyrosines and Hif-1 both entered the genome during the Sturtian glaciation in response to presumed altered metabolic requirements, coincidentally with gastrulation and the organizer [1], and this is likely to be part of the organism-wide deep biological associations of PGRMC.
In a section linking SREBPs and their regulation by PGRMC, and noting the interaction of PGRMC with membrane trafficking and components of the actin cytoskeleton, it would be remiss to omit that from Drosophila to humans SREBP1 is also regulated by mechanical stress propagated from the extracellular matrix via geranylgeranyl pyrophosphate, a key mevalonate pathway (MVP) intermediate, where RhoA-dependent actomyosin contractions inhibit the activation SREBP1 [234]. Mevalonate pathway activity and regulation is important in normal and pathological activity of many body functions, including the cardiovascular system, cancer and neurobiology [235, 236, 237, 238]. Please remember that these arguments are presented as part of a non-confirmed hypothesis. Further studies would be necessary to validate the relationships between the above features.
If neurobiology exploits some aspect of PGRMC biology inherited from the origin of eukaryotes, as adapted by the eumetazoan organizer systematics [1], it may be related to redox and/or metabolic switches and/or membrane trafficking actin cytoskeletal regulation hypothesized above to have been involved in eukaryogenesis. Membrane remodeling is one function of PGRMC1 that is active at synapses [165]. Perhaps neurogenesis requires the evocation of a PGRMC-directed cell state with cytoskeletal organization inherited from the first truly eukaryotic cell. If so, this probably involves altered mitochondrial function and glycolysis. The author stresses that this is a purely conjectural hypothesis which at this stage lacks experimental evidence.
P4 clearly exerts PGRMC1-dependent effects on neurons. It induces a neuroprotective signal in primary rodent neural cultures, involving the secretion of brain-derived neurotrophic factor (BDNF) from glia cells which promotes neural survival and synaptogenic conditions. This involves PGRMC1-dependent activation of extracellular signal-regulated kinase (ERK)5 [239, 240], which can be negatively regulated by the microRNA (miRNA) let-7i that targets the PGRMC1 mRNA for destruction [241].
In a similar primary culture model, a series of publications from Hou and
colleagues [242] at Shijiazhuang in China’s Hebei Province have shown that
P4-mediated neuroprotection from A
In human embryonic kidney derived HEK293 cells, Sabbir [18] noted alterations in phosphorylation, ubiquitination and sumoylation of PGRMC1 following P4 treatment, and these were associated with altered glucose metabolism. The activation of the Ras pathway in neuronal survival [243] is suggestive of the involvement of tyrosine phosphorylation in this process. Although HEK293 cells are far removed from neurons, it is possible that similar processes occur in P4-treated neurons. Indeed, Hou and colleagues [20] demonstrated the involvement of PGRMC1 in elevated glycolysis in response to P4 treatment in animal and cell culture AD models. Therefore, PGRMC1 biology in this instance is closely associated with the disease state of AD. We could extrapolate from arguments developed above that Ras activation might imply PGRMC1 tyrosine phosphorylation, and that this in turn would affect PGRMC1’s membrane trafficking functions. Once more, this is conjecture.
As stated above, PGRMC1 is part of the S2R complex that is targeted by CogRx
ligands such as CT1812 which allosterically alters the affinity of S2R/TMEM97 for
soluble A
The limbic system houses a series of separate functions associated with such central human functions as behavior, motivation, olfaction, long term memory and emotion [246]. This region of the brain consists partly of diencephalon (part of the midbrain) and of cerebrum (part of the forebrain) (Fig. 2) and is therefore a much later evolutionary development than the hindbrain. PGRMC1 is also widely expressed in the cerebellum where it is thought to be involved in synapse formation [114]. Such abundant expression levels of PGRMC1 may mean that important PGRMC neural functions inherited from primitive neurons have been retained into the more complex structures of the higher vertebrate brain.
AD symptoms of A
Glasgow et al. [249] recently showed that LTP requires presynaptic
netrin signaling to post-synaptic DCC (the same ligand-receptor pair involved in
PGRMC-related axon guidance [66, 69], which appeared in the LEUMCA [1]). This
induces Ca
P4 induces such synaptic actin cytoskeleton remodeling, and PGRMC1 has been
implicated in the response (although whether its role was neurosteroidogenic
and/or P4 binding was not clear) [73, 250]. The simplest connection between the
LTP-promoting effects of CogRx molecule CT1812 and the mechanism of Glasgow
et al. [249] is that a PGRMC1-DCC dependency is perturbed by A
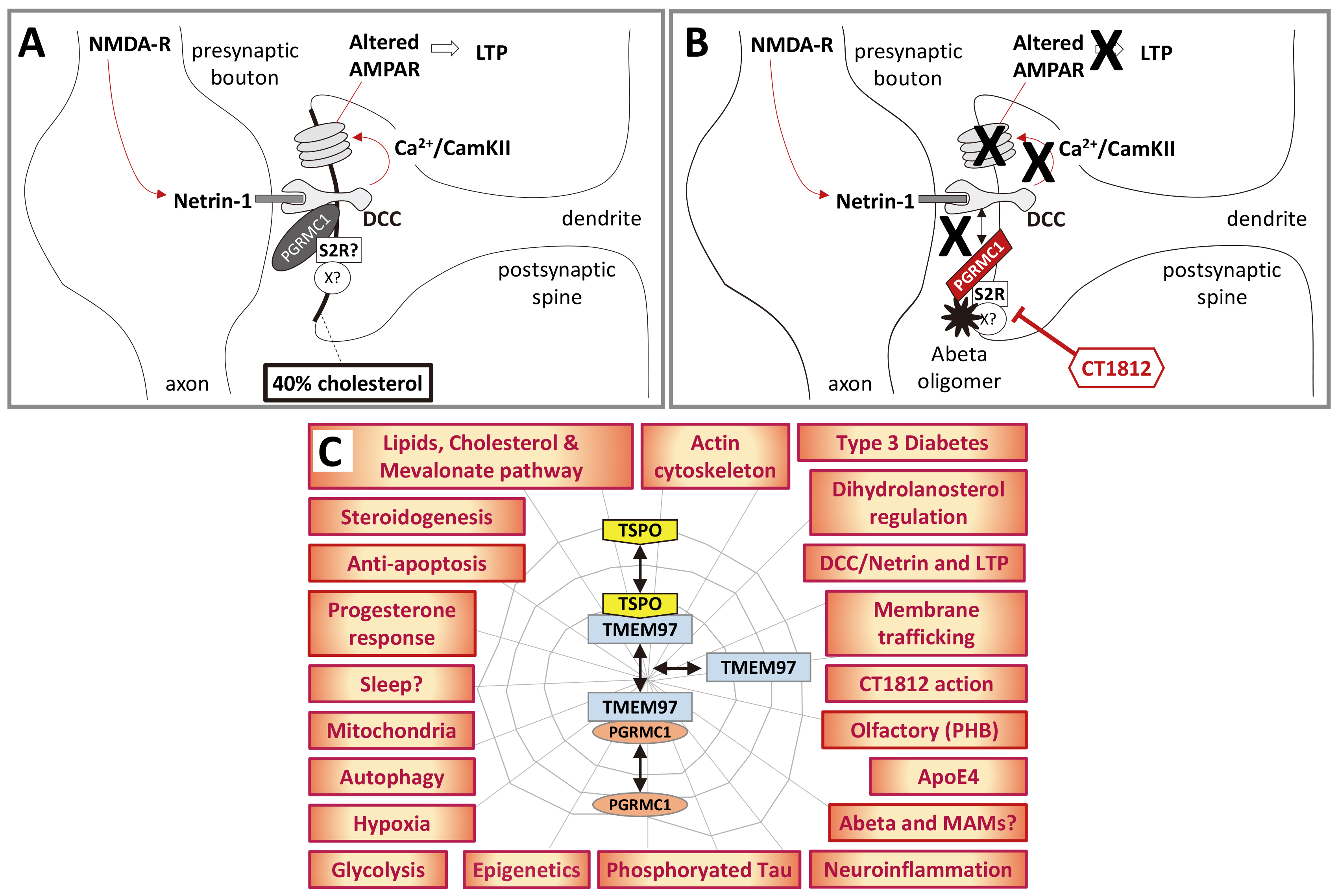
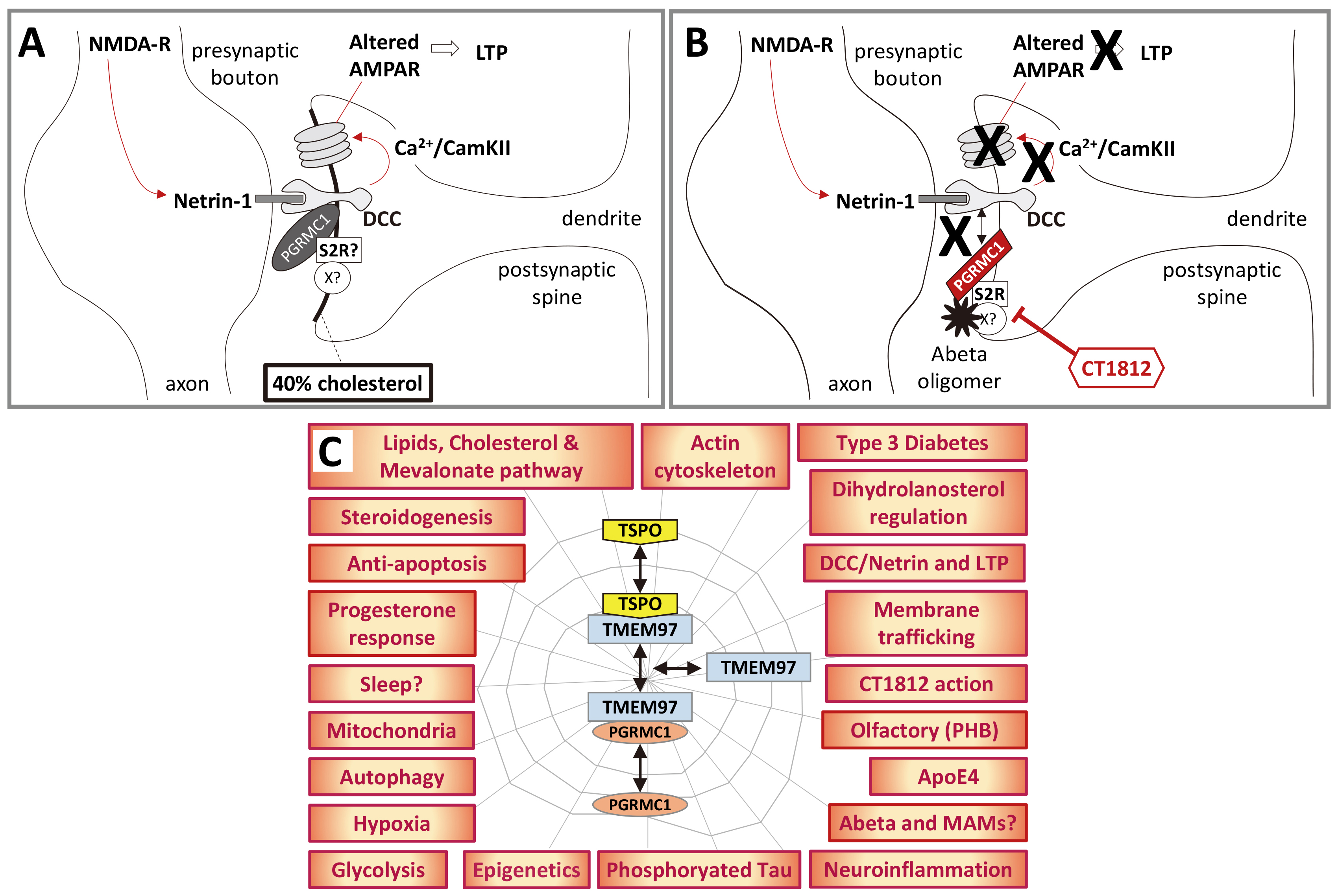 Fig. 3.
Fig. 3.A hypothetical model of PGRMC1 involvement in AD. (A)
This crude schematic proposes that PGRMC1 is required for DCC function by
incorporating the mechanism of action of LTP-promoting synaptorestorative
Cognition Therapeutics drug CT1812 [121, 122, 165] with the mechanism of LTP
described by Glasgow et al. [249], acknowledging that PrP [251] or other
proteins in a PGRMC1/S2R complex could be the major A
If the last seemingly audacious statements are not fully misguided, then we
predict that manifestations of AD pathology should reflect PGRMC1 biology. These
would include mitochondrial effects [19, 252], PGRMC1 membrane-trafficking [28, 147], sterol biology [1, 43, 97], Warburg/glycolysis [18, 19, 20], autophagy
[253, 254, 255, 256], diabetes [252], hypoxia, and epigenetics [257]. The biology should
also include the known mechanism of LTP, as described above, whereby presynaptic
netrin activates post-synaptic DCC, initiating Ca
We also must expect the PGRMC1-centric model of AD to account for the ‘usual
suspect’ key AD biomarkers apolipoprotein E4 (ApoE4), A
Mitochondrial dysfunction is among the earliest pathogenic alterations found with AD, which is manifest long before the accumulation of amyloid plaques [168, 252, 260], and has even been proposed to be the driving force behind AD and worthy of drug development [261, 262, 263]. Mitochondria are central to metabolic processes and to apoptosis, and are involved in P4-dependent mechanisms of neuroprotection [264], which involve PGRMC1 [103, 114]. Lower P4 levels are associated with AD symptoms in rodent models [265] and humans [266].
We have recently shown that PGRMC1 is associated with altered metabolic changes in cancer cell culture and drives mitochondrial functional changes [19]. Wu et al. [20] have shown that so-called PGRMC1 inhibitor AG-205 blocks P4-induced neuronal glucose uptake that is required for learning. Recall that assumptions of AG-205 being PGRMC1-specific are untrue (as discussed in the accompanying paper [1]), and its effects could easily be due to other MAPR proteins, or even non-MAPR proteins. However, AG-205 does seem able to antagonize certain PGRMC1 functions [267].
Dysfunctional mitochondrial association at mitochondrial endoplasmic reticulum-associated membranes (MAMs) is associated with several neurodegenerative diseases including AD [268, 269]. The fungal cognates of MAMs are called ER-mitochondria encounter structure (ERMES) which have been shown by genetic and biochemical means to facilitate phospholipid and calcium exchange. The resemblance of eukaryotic lipids to those of bacteria, rather than the archaeal proto-eukaryotic symbiotic host cell, is related to the transfer of mitochondrial membrane lipids from mitochondria at ERMES [270, 271]. Adding to the burgeoning list of AD-phenotypes associated with PGRMC1 biology, Sabbir et al. [29] have shown that PGRMC1 knockdown disrupts mitochondria-MAM interactions in HEPG2 but not HEK293 cells. That is in striking alignment with the overarching hypothesis pursued in the accompanying paper [1], that during eukaryogenesis the original eukaryotic PGRMC function was ancestrally related to mitochondrial regulation of metabolism.
Mitochondrial dysfunction is one of a suite of overlapping neurodegenerative mechanisms common to AD and diabetes [272, 273], as well of course as cancer. The cell biology of diabetes and AD are closely related [274]. The association of PGRMC1 biology with that of diabetes is described above.
As a provider of non-glucose carbon skeletons for energy production, autophagy can be considered briefly here. The involvement of PGRMC1 in autophagy was described in the accompanying paper [1]. Suffice to reiterate that autophagy levels are reduced in AD, and that increasing the level of mitophagy (a subtype of autophagy) can improve cognition in AD models [256, 275].
Kim et al. [276], from the Korean mouse knockout collaboration, also showed that PGRMC1 is required for the growth of ductules during mammary gland development in response to P4. This may seem unrelated to neurology yet may provide useful information on the potential role of neuronal PGRMC1. As discussed elsewhere in this paper but reiterated here, the nematode VEM-1 homologue of PGRMC1 interacts with UNC-40 (one of the uncoordinated genes names due to impaired neurogenesis when they are mutated) to direct embryonic axon guidance of neurons of the central nerve cord. This function is conserved from nematodes to mammals [66, 68, 69]. Mammals have two orthologues of UNC-40. One is DCC that is involved in axon guidance [66] and long term potentiation [249], and the other is Neogenin [277, 278]. Both UNC-40 orthologs interact with their mammalian ligand Netrin.
Neogenin is also involved in Netrin-mediated axon guidance, migration of T-cells across the blood/brain barrier, inflammation, and angiogenesis [277]. It has long been known that the extending growth bud of the mammary end bud during the growth of mammary ductules involves interactions between Netrin and Neogenin [278]. With the recent discovery that this process depends upon PGRMC1 [276], we can extrapolate to hypothetically propose that this Netrin/DCC/Neogenin migration function reflects an ancient eumetazoan function related to establishing specialized communication modules between different cell types as the primitive LEUMCA body grade was stretched and elaborated by subsequent evolution. It probably involves PGRMC1 tyrosine phosphorylation acquired by the LEUMCA, and actin cytoskeletal rearrangements to mediate a migrating cell front (be it axonal extension or cell migration). Note that this is an undemonstrated hypothesis. PGRMC1/2 also promotes luteal vascularization [77], however there is no indication that this depends upon DCC. Indeed, PGRMC1 regulates VEGF expression [150, 279], which may be responsible for all PGRMC1-mediated vascularization.
Considering functions related to PGRMC1 biology, it has long been recognized
that AD-associated insulin resistance and metabolic dysregulation is intertwined
with A
In striking concordance with a main hypothesis of this paper, that perturbations
in AD reflect manifestations of PGRMC1 biology retained since eukaryotic origins
where the ancestral archaeal host cell consumed amino acids to feed the
endosymbiotic proto-mitochondrion [1, 284], substantial changes in branched chain
amino acid metabolism accompany AD. Although effects are observed in samples from
human cerebrospinal fluid and animal models, the underlying basis remains poorly
characterized [285]. Branched chain amino acid perturbations were among those
predicted by pathways analysis of cells expressing different PGRMC1
phosphorylation mutants [19]. It is unclear whether AD-specific branched chain
amino acid metabolism is related to the
Neuropathologies like AD have been hypothesized to be essentially brain metabolic diseases related to mitochondrial function and metabolic control, which can be addressed by dietary modification and ketone body production to drive metabolism from glycolysis towards mitochondrial respiration [287]. That would be in accord with a model where ancestral PGRMC1-mediated regulation of cell metabolism was perturbed in AD. Accordingly, a 12-week modified ketogenic diet randomized crossover trial improved patient daily function and quality of life scores, and resulted in a non-significant trend towards improved cognition [288].
The PGRMC1 relevance of this concept is further strengthened by the finding that cholesterol oxidation is linked to altered glucose uptake, and cholesterol oxidation has indeed been proposed to be a main mechanistic cause of AD, leading for instance to impaired insulin-dependent glucose uptake [289]. The involvement of PGRMC1 in the mechanism of action of a drug that reverses AD symptoms [122, 165], the role of PGRMC1 in DCC interactions [66], where DCC signaling is also required for the mechanism of LTP [249], and the role of PGRMC1 in regulating glucose [18, 19, 20] and sterol [43, 97] metabolism, seem to juxtapose too many coincidences for there not to be a causal relationship between PGRMC1 and AD, as developed further below.
All brain cells are always metabolically active. Resting oxygen consumption relies on oxidative phosphorylation. When neurons are activated their rate of glycolysis increases [290] (reviewed by [291]). This increase in glycolysis has been interpreted in terms of elevated requirements for energy associated with synaptic activity, and requirement to synthesize neurotransmitters [168, 291, 292].
However, pre-existing pools of neurotransmitters exist in synaptic vesicles ready to be fused to the presynaptic plasma membrane upon activation. If increased energy yield alone were the driving force, then the substantially higher yield of oxidative phosphorylation should be favored unless energy must be generated quickly. Glycolysis generates ATP approximately 100 times faster than oxidative phosphorylation and has been suggested to be the favored metabolic mode for rapidly dividing cells, such as cancer cells [293]. Indeed, glycolysis is required for transition through the G1/S cell cycle checkpoint, where glycolytic intermediates provide the precursors for metabolites such as nucleotides that are required prior to entering S-Phase [294]. A complex of PGRMC1 and PGRMC2 controls the G1/S checkpoint [295]. However, neural glycolysis does not easily fit these models.
While metabolic modelling and empirical measurements can accurately explain the metabolic flux of neural glucose consumption once we assume that glycolysis is involved [290, 291, 292, 296], it does not adequately explain why glycolysis is required for active neurons in the first place. The usual explanation would be that energy is required quickly, or that carbon skeletons are required for the synthesis of neurotransmitters [291], however the requirement for glycolysis does not seem to have been adequately explained. For instance, neuronal glycolysis is induced by increasing 6-phosphofructo-2-kinase (PFK2) activity, which activates the rate limiting glycolytic reaction [297]. But that only redirects the question to why does PFK2 need to be upregulated.
Perhaps the answer lies in an unexpected area to which we have been blinded by our preconceptions of the specialized nervous system, glycolysis, and energy metabolism. What if neuronal biology requires a PGRMC1 functional status (or mitochondrial state) that is proglycolytic, but glycolysis per se is not the key driving output? Rather, what if glycolytic biology permits an epigenetic cell state that is permissive for the expression of neuronal genes because of our evolutionary history and associated constraints. Once more, this is a hypothesis.
Once we accept the presence of a PGRMC-accessible switch between metabolic states in the LEUMCA, and that the LEUMCA had already developed a system of cell-type differentiation to produce specialized cells [1], it is fully conceivable that the neuronal differentiation pathway was able to evolve in the LEUMCA by employing gene combinations whose activity was originally epigenetically linked to the glycolytic state, but which were themselves not directly involved in glycolysis. This is a novel if highly speculative contrivance on the author’s part, whereby the very identity of the neuronal state could depend upon the epigenetic maintenance of glycolytic metabolism, and the connected co-expression of epigenetically linked neuronal identity-specifying genes. This hypothesis would be consistent with the ability of perturbed PGRMC1 activity to confer loss of neuronal differentiation in AD concomitant with impaired glycolysis. Over evolutionary time, the non-glycolytic enzymes would presumably have acquired allosteric metabolic regulation which now links their activity tightly to the metabolic state.
This could all be regulated by the flux of glycolytic carbons through
mitochondria: the metabolic process which best fits PGRMC’s proposed ancestral
role. The carbons of glucose are transformed by glycolysis to two three-carbon
pyruvates, which are either converted to lactate in the cytoplasm (to be
secreted) or decarboxylated via the pyruvate dehydrogenase complex to
acetyl-coenzyme A (Ac-CoA) in the mitochondrial matrix. If the TCA cycle is
operating, then Ac-CoA is oxidized to CO
Neurons require large supplies of cholesterol, especially in synaptic membranes [289, 298, 299], and therefore mitochondrial activity could well have been critical in establishing a neural phenotype in the LEUMCA prior to the later evolution of bilaterian circulatory systems and vertebrate lipoproteins. Having discussed the relationship between glycolysis, neurons, and steroidogenesis, we can consider steroidogenesis in some greater detail.
PGRMC1 membrane trafficking includes LDLR internalization involving a complex
with TMEM97 [43, 140], which has been shown by the Mach group to mediate
A
That specifically PGRMC1-mediated membrane trafficking is involved in AD is highly likely. It was shown to be required for the mechanism of action of synaptorestorative effects of CogRx small molecules [122, 165]. There is every reason to assume that it will also apply to the ‘usual suspect’ ApoE4 [302], as described below.
Neurons express high levels of the proteins that sense cholesterol levels.
Neurons and astrocytes are responsible for most brain cholesterol synthesis
[303]. Reduced cholesterol metabolism leads to A
As hypothesized in the accompanying paper [1], PGRMC1 may be eukaryogenically related to sterol transport, particularly for the mitochondrion. Mitochondrial membranes contain proportionally less cholesterol than other main membranes, however the lower amounts present are required for mitochondrial functions related to proton permeability as well as serving as substrates for steroid and bile acid synthetic pathways [306, 307]. Although it is debated whether higher or lower cellular levels of cholesterol consistently characterize AD, there is much evidence that elevated mitochondrial cholesterol levels are associated with several pathologies, including AD [303, 306, 308, 309].
In primary cultured rat cortical neurons, Hou and colleagues [310] from Hebei in
China showed that oligomeric A
Much mitochondrial cholesterol enters via ER contacts at MAMs, whereas
extracellularly obtained sterol originating from lipoprotein endocytosis enters
from a late endosomal pathway. Both the exogenous LDLR/endocytic and endogenous
synthesis/MAM pathways for cholesterol transport to mitochondria are thought to
involve membrane-membrane contacts with mitochondrial outer membrane and involve
some of the same mitochondrial proteins [303, 307]. PGRMC1 is involved in
lipoprotein [43] and A
It remains unknown how heme transport to the ER occurs [315]. However, a hypothesized positioning of PGRMC1 at cholesterol enriched MAMs would be consistent with its largely sterol-related biology, as well as the known induction of pgrmc1 gene under conditions of low iron [316]. PGRMC1 has long been known to be associated with iron regulation, such as the regulation of hepcidin, the regulatory peptide of the ferroprotein membrane iron exporter [317]. This biology is eerily reminiscent to the CPR loci containing a PGRMC1-related cytb5MY gene, TSPO, two other cytb5 domain genes, a putative ferric reductase-related gene, and an inducible two component regulatory gene [1, 153].
PGRMC1 exerts dramatic effects on mitochondrial form (fusion/fragmentation) and
metabolism [19], and several mitochondrial genes are thought to have co-evolved
with PGRMC1 (or MAPR proteins) [97]. As cited above, PGRMC1 is required for the
localization of mitochondria to MAMs in HepG2 but not HEK293 cells, and interacts
with hexokinase (the first enzyme in the glycolytic pathway that directs glucose
carbon skeletons towards the mitochondrial Krebs Cycle) [29]. Intriguing to this
hypothesis, the MAM is also the site of APP cleaving enzymes BACE and
The further complexity of this field surpasses the scope of this work. Suffice to say here that mitochondrial function and sterol content are altered in AD, and mitochondria have been proposed as therapeutic targets, with promising preclinical results [318, 319]. Considering the pronounced effects that PGRMC1 phosphorylation can exert on mitochondria [19], it would be worthwhile to pursue this question experimentally.
As discussed in the accompanying paper [1], the LEUMCA (the first eumetazoan to
possess the gastrulation organizer and differentiated neurons) inherited Hif-1
from its last common ancestor with placozoans. During mammalian embryogenesis of
the brain, expanding radial glial cells maintain a glycolytic metabolism under
the influence of Hif-1
Hypoxia is recognized as a major factor in AD (associated with usual suspects),
where declining oxygen leads to hypometabolism which promotes, among other thing,
A
AD inflammation is also mediated by the transcription factor nuclear factor
Pericytes have been shown to constrict brain capillaries in response to
A
Altered distribution of 5-methylcytosine at genomic CpG (cytosine-guanosine) sites is associated with normal embryological development and multiple diseases [330, 331], including cancer [331, 332, 333] and AD, where many enhancer CpG sites become hypomethylated [334, 335]. In a mouse model of AD inhibition of histone methyltransferases reverses histone hypermethylation and the repression of genes involved in neuronal signaling. This leads to improved synaptic function and cognitive performance of the mice [336]. This is a complex field, however AD-associated epigenetic changes at least qualitatively resemble the epigenetic changes induced by different PGRMC1 phosphorylation mutations in cancer cells [257]. To note is that PGRMC1 can be a major epigenetic regulator, CpG epigenetic enhancer regulation was adopted by bilaterians in association with the evolution of multiple different cell and tissue types, and as such is based upon the foundations of the gastrulation organizer that coincides with the evolutionary appearance of both PGRMC1 Y139/Y180 and neurons. Mutation of PGRMC1 induced a hypermethylated CpG degree that resembled embryonic stem cells [257], and epigenetics is a major feature of AD [334]. Once more, PGRMC1 biology overlaps with AD phenotype.
We saw in the accompanying paper [1] that memory formation and learning require
sleep, and that sleep seems to have evolved in the LEUMCA, the animal which gave
rise to cnidarians and bilaterians, and was characterized by a gastrulation
organizer, the appearance of the Netrin/DCC system, the first neurons, and a
circadian rhythm involving presumed neural regeneration via sleep. Sleep loss is
associated with AD risk, and sleep is associated with diurnal variations in
synaptic A
Sleep duration is influenced by A
For many years and as discussed above, AD research has focused on a presumed
pathology involving faulty A
ApoE is involved with the lipoprotein clearance pathways of both Tau and
A
Whether or not it is directly associated with AD etiology [190], ApoE4-mediated
A
As an aside here, altered S2R-associated membrane trafficking (which includes
PGRMC1 and/or probably LDLR or LRP1) is involved in the pathology of
These results imply that the PGRMC1/S2R membrane trafficking function targeted by the S2R ligand CT1812 plays a critical role in general synaptic membrane trafficking function (not specialized to AD pathology), which is upstream of a neuronal metabolic switch. Given the pleiotropy of effects caused by PGRMC1, we can expect to encounter perturbations of this functionality to be associated with a variety of non-neuronal cell types and pathologies.
An impaired sense of smell is one of the earliest symptoms of AD, and correlates
with the presence of A
Like PGRMC1, PHBs are proteins that exhibit diverse subcellular localization and
multiple pleiotropic functions [348]. In MIA PaCa-2 cells PGRMC1 is present in
the same protein complexes as both PHBs and alpha-actinin (although direct
protein-protein interactions have not been demonstrated) [37]. PHBs are also
recruited into PGRMC1 protein complexes by progestogen treatment of estrogen
receptor alpha (ESR
PHBs very interestingly share much overlapping biology with PGRMC1, such as
association with aurora kinase, association with alpha-actinin, modulation of the
wingless-integrated (Wnt) signaling and PI3K/Akt pathways [348]. The
mitochondrial PHB1/PHB2 complex that is perturbed in AD is involved in metabolic
(glycolytic/gluconeogenic) regulation, the dynamics of mitochondrial
fusion/fission, and mitochondrial biogenesis [346, 350, 351]. PHB1 has been
discussed as a pleiotropic mediator of genes related to obesity, insulin
resistance and metabolic dysregulation [352], which resembles the metabolic
effects of PGRMC1. It is possible, although unproven, that the suite of symptoms
induced by binding of A
PHB-encoding genes are ancient, being present in both prokaryotes and eukaryotes. The reported eukaryotic distribution (plants, fungi and animals) could be consistent with presence in the last eukaryotic common ancestor (LECA) [356], however this particular question has not been addressed and remains unresolved. Therefore, it is possible that PHBs are not only involved in the PGRMC1-associated pathologies of AD, but also that they are involved in many of the eukaryotic MAPR functions described in the accompanying paper [1]. This requires future study. We will encounter PHBs again in considering cancer biology.
Because the target of the A
Since the putative ancestral MAPR cytb5MY protein from CPR bacteria was probably associated with an inducible redox-related operon that contained genes for TSPO as well as two other heme-binding cytb5 domain proteins and a putative ferric-reductase [1, 153], it is tempting to speculate that PGRMC1 and TSPO share a heme-dependent relationship which is related to redox perturbances in AD. However, that is unproven conjecture and both proteins appear to have acquired multiple functions during eukaryotic evolution [34, 153, 155]. Notwithstanding, our identification that TSPO is required for TMEM97-mediated S2R activity, the knowledge that S2R is the target of CT1812, and the observation that TSPO itself is strongly implicated in AD, highlight the strong overlap between PGRMC1 and AD biology.
PGRMC1 is a single pass transmembrane protein with two positive residues (K44, R47) immediately C-terminal of its transmembrane helix. The positive charge is conserved in chordates [34]. Such positive residues are recognized by the ER membrane protein complex (EMC) and oriented towards to the cytoplasm to determine the orientation that a transmembrane helix is inserted into the membrane as the signal recognition particle ferries the nascent polypeptide/ribosome complex to the ER [361]. Therefore, PGRMC1 and PGRMC2 (which also has conserved positive charges, not shown) are predicted to be type 1 membrane proteins, with a luminal N-terminus and cytoplasmic C-terminus. The Phosphosite database (phosphosite.org) of posttranslational modifications (PTMs) has hundreds of referenced instances of PGRMC1 C-terminal PTMs consistent with a type 1 cytoplasmic C-terminus. However, in several very well characterized instances the PGRMC1 C-terminus is clearly luminal or extracellular, in type 2 topology. These include in pluripotent stem cells (PSCs) [224, 362], in Alzheimer’s neuron synapses [165], in hepatocytes [363], and in lung cancer cells [364]. In the latter, PGRMC1 was associated with the exosome pathway. PGRMC1 was also recently detected as a chondroitin sulfate-linked protein in urine [365]. The glycosaminoglycan was linked via S54 of PGRMC1, and therefore indicates a type 2 topology. It is then critical to understand what regulates membrane topology.
In neurons, Munton et al. [366] observed a fifteenfold higher levels of PGRMC1 in murine brain subcellular post-synaptic density fractions than from synaptic membranes. However, tyrosine phosphorylated PGRMC1 was only observed in the synaptosomal preparations, and not synaptic vesicles or postsynaptic density (see both Supplementary Table 1 and Supplementary Fig. 4a from Munton et al. [366]). Tyrosine phosphorylated PGRMC1 certainly has a cytoplasmic C-terminus, but AD synapses had an extracellular C-terminus [165]. It is possible that either adjacent pre- and post-synaptic neurons have different PGRMC1 topologies, that both topologies are present in single neurons, perhaps at different subcellular localities, or that altered topology is related to AD pathology. Understanding what causes PGRMC alternative membrane topologies is likely to reveal profound insights and should be investigated.
In summary of the above, much of PGRMC1’s biology overlaps with the pathology of AD. The author is unaware of another protein for which this overlap is so comprehensive. This represents evidence in favor of PGRMC1 playing a central mechanistic role (if not the central role) in AD pathology [162]. Stated more theatrically, PGRMC1 may be the metal of the blade of the sword that causes AD.
It is fitting to close consideration of conventional human diseases with cancer biology, which also historically reflects how PGRMC1 phosphorylation was first discovered [301, 367], and its importance was indicated [147]. The relationship between pleiotropic PGRMC1 biology and cancer has been reviewed previously [28, 100, 253, 368, 369], and is considered here to be established. That position is underlined by the recent confirmation [18, 19] of PGRMC1’s predicted [301] role in Warburg glucose metabolism, and the discovery that PGRMC1 tyrosine phosphate acceptor Y180 affects genomic mutation rate and the state of CpG epigenetic modification [257] as well as being required for PI3K/Akt signaling and efficient subcutaneous xenograft tumor growth [19].
Our original observation of disparate PGRMC1 phosphorylation status between
breast cancers with differential ESR
Recently Zhang et al. [19] demonstrated that the mitogenic effects of estrogen and norethisterone on breast cancer cells mechanistically involves the PGRMC1-mediated activation of the PI3K/Akt pathway. Interestingly, PI3K/AKT activation required PGRMC1 S181, and could be blocked by the casein kinase 2 (CK2) inhibitor quinalizarin [372], whereas our study implicated Y180 in PI3K/AKT activity. S181 is adjacent to Y180 and has been proposed to sterically block Y180 phosphorylation when phosphorylated by CK2 [19, 147, 301]. Simultaneous phosphorylation of both Y180 and S181 has never been reported [96] (https://www.phosphosite.org/proteinAction.action?id=5744, accessed 29 September 2022). The mechanistic contributions of Y180 and S181 (and the adjacent T178) towards PI3K/AKT activation require further study.
Until recently no PGRMC1 polymorphic alleles have been associated with cancer.
Allelic variation does exist and has been linked to primary ovarian insufficiency
as discussed in the accompanying paper [1]. The CanProVar 2.0 database [373]
(accessed 20 November, 2021) lists six non-cancer specific variations, and no
cancer-related variants. Osman et al. [374] reported in December 2021
that one of the 6 variants (PGRMC1-rs145582672) is significantly associated with
Hodgkin lymphoma (p-value 8.56
DNA methylation changes alter the level of lineage-specific cell differentiation and may progress cells towards a dedifferentiated stem cell-like status associated with cancer. In accord with the hypothesis that PGRMC1 modulation of actin cytoskeleton modulates migration, and cancer biology, Lee et al. [375] observed reduced cancer cell migration and metastasis formation in PGRMC1 knockout mice, which was associated with reduced abundance of proteins associated with epithelial-mesenchymal transition (EMT). PGRMC1 indirectly leads to MMP-9 stabilization and activity [376, 377], which favors cancer metastasis. Cai et al. [378] observed that the progestogen norethisterone (NET) increases breast cancer cell proliferation and migration in a PGRMC1-dependent manner, whereas the micro-RNA miR-181a suppresses NET-induced proliferation, migration, and enhances apoptotic propensity. In this study, NET increased the abundance of proteins in the PI3K/Akt/mTOR pathway, whereas miR-181a decreased them. Those authors had previously shown that miR-181a suppressed the progestin-dependent growth of breast cancers [379] by reducing the levels of several proteins, including PGRMC1 [380].
In recent work, radiation treatment of uterine endometrioid endometrial
carcinoma patients led to approximately 30-fold reduction of PGRMC1 levels, along
with modulated tumor microenvironment. Together, these were associated with
higher patient disease-free survival, probably via increased infiltration by
FoxP3
While PGRMC1 mutations do not seem to instigate cancer progression (although
PGRMC1 genomic mutations were recently associated with Hodgkin lymphoma [374]),
it is highly likely that its abundance and post-translational modifications
contribute dramatically to tumor biology through the types of biology described
here [28], including the relaxation of epigenetic maintenance of differentiation
status [257] and reversion back along a trajectory towards earlier or scrambled
differentiation states. The case is particularly strong for invasion and
metastasis, which involve the motility of this section topic. As cited above, in
different cancer cell lines PGRMC1 is associated with increased migration and
metastasis associated with markers of EMT, often involving response to
steroids/progestogens [19, 382, 383, 384, 385, 386]. We showed that activated PI3K/Akt activity
associated with Y180 phosphorylation status increased cell migration in scratch
assays [19]. Interestingly, oroxylin A (a Chinese traditional herb) reduced cell
migration of ovarian cancer cells. The effect involved upregulation of peroxisome
proliferator-activated receptor gamma (PPAR
The basis of any functional difference between mammalian PGRMC1 and PGRMC2 proteins remains unresolved. These diverged before the progenitor of cartilaginous fish in vertebrate evolution, have been conserved since [2, 3], and probably therefore participate in at least one mutually exclusive vertebrate cell type-specific function. It remains unknown how such a difference may be relevant to cancer.
As already discussed in Section 2.7, a complex between PGRMC1 and PGRMC2
negatively regulates the transition through the G1/S checkpoint in spontaneously
immortalized granulosa cells. The mechanism involves interaction with GTPase
activating protein binding protein 2 (G3BP2) which itself binds NF-
We have just seen how PGRMC1 and PGRMC2 regulate NF-
Prohibitins also mediate repression by of androgen receptor genes after
treatment with androgen antagonist therapy in prostate cancer by recruiting the
BRG1 core ATPase of the SWI-SNF complex. SWI-SNF destabilizes histone-DNA
interactions to remodel chromatin by the removal of p300-mediated acetylation,
leading to transcriptional repression [355]. Repression also involves direct
deacetylation of the androgen receptor by Sirtuin (SIRT1) [395], which we
will encounter below in relation to aging biology. SIRT1 is involved in
transcriptional repression of ESR
PHBs can activate anti-oxidant response element-containing promoters, including the heme-oxygenase-1 promoter [398], linking their activity with the heme-biology of PGRMC1, which binds and regulates ferrochelatase, the last and rate limiting enzyme in heme synthesis [311]. Thereby, PGRMC1 could regulate both the synthesis and degradation of heme. Recall the putative origins of MAPR proteins from an inducible CPR bacterial cytb5MY locus that encoded two other heme-binding cytb5 proteins, TSPO which can bind heme, and a putative ferric reductase. Heme biology may be an ancient feature of PGRMC proteins.
The PGRMC1/PHB complex could also directly influence the G1/S checkpoint, which has not been investigated. In MCF-7 and T47D breast cancer cells PHB1 interacts with p53 to enhance its transcriptional activation, whereas interaction with E2F suppresses its transcription [399]. PHB1 also interacts with the E2F repressor Rb, however the mechanism of transcriptional repression of E2F via PHB1 and Rb are different [353, 400]. If PGRMC1 activation by progestogens also regulates PHB interaction with these proteins, then it could be involved in PGRMC1-mediated [295, 392] regulation of progression past the G1/S checkpoint, which is dependent on E2F activity, and promote cell cycle block by p53, which induces the pan cdk inhibitor p21 in response to DNA damage. This exciting biology may be mechanistically related to the enhanced DNA mutation rate of the PGRMC1 triple mutation (S57A/Y180F/S181A)-expressing cell line (TM) of Thejer et al. [257], which would imply that PGRMC1 kinases and phosphatases can directly act on the ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related protein (ATR) pathway that induces p53, and which was affected by the TM mutation [257]. We will revisit PGRMC1 and DNA damage in consideration of aging biology, below.
Of relevance to the proposed ancestral association between PGRMC1, heme biology and mitochondrial regulation [1], cyclopamine tartrate (CycT) treatment reduces the synthesis and degradation of heme. In a study using subcutaneous xenograft non-small cell lung cancer (NSCLC) cells, CycT suppressed the oxygen consumption of purified mitochondria, and reduced tumor growth in mice. PGRMC1 was among several genes involved with heme metabolism that were downregulated by CycT. Interestingly bevacizumab (an anti-VEGF mono-anticlonal antibody which inhibits angiogenesis and oxidative phosphorylation) led to even greater reduction in PGRMC1 levels [401]. Note that while hypoxia reduced PGRMC1 levels in this situation, it induced PGRMC1 in ductal carcinoma in situ breast lesions [301]. Clearly, the mechanisms underlying these processes require further investigation.
When reducing potential provided by glutathione is lowered, oxidative damage by
Fenton chemistry is increased which can lead to severe oxidation of membrane
lipids to the extent that membrane integrity is compromised, leading to a
necrotic cell death that has been called ferroptosis. Under these conditions, You
et al. [402] report that attenuation of PGRMC1 levels leads to decreased
cell death, while elevated PGRMC1 levels increased death rates. It was proposed
that PGRMC1 inhibits the “amino acid transport system xc
By the proteomic detection of serum proteins related to oedema in
ESR
PGRMC1 activates the epidermal growth factor receptor (EGFR)-dependent PI3K/Akt
pathway in breast cancer to promote inflammatory responses and tumor progression
[378, 386], and interacts with ESR
Considerable other evidence points to a role of PGRMC1 in inflammation. Its levels correlate with markers of metabolism and inflammation in the hippocampus [409]. PGRMC1 is involved in the inflammatory response of the fetal membrane [410, 411] and the placenta [412]. Glycyrrhizin, a substance with anti-inflammatory properties, binds to PGRMC1 to prevent the formation of dimers and suppress tumor growth [413]. Pgrmc1 knockout mice exhibited altered hepatic metabolism and liver inflammation [229].
In addition to effects on metabolism, inflammation, epigenetics, EMT and migration, and in line with PGRMC multifunctionality, the effects on mitochondria, steroidogenesis, autophagy, and membrane trafficking described elsewhere in this and the accompanying paper [1] are also likely to play prominent cancer roles. Indeed, PGRMC1 biological attributes are associated with most of the “Hallmarks of Cancer” [414] (Fig. 4, Ref. [414]). Substantial progress has been achieved since our initial report of differential PGRMC1 phosphorylation in cancer [301], and that progress points the way towards much needed future work.
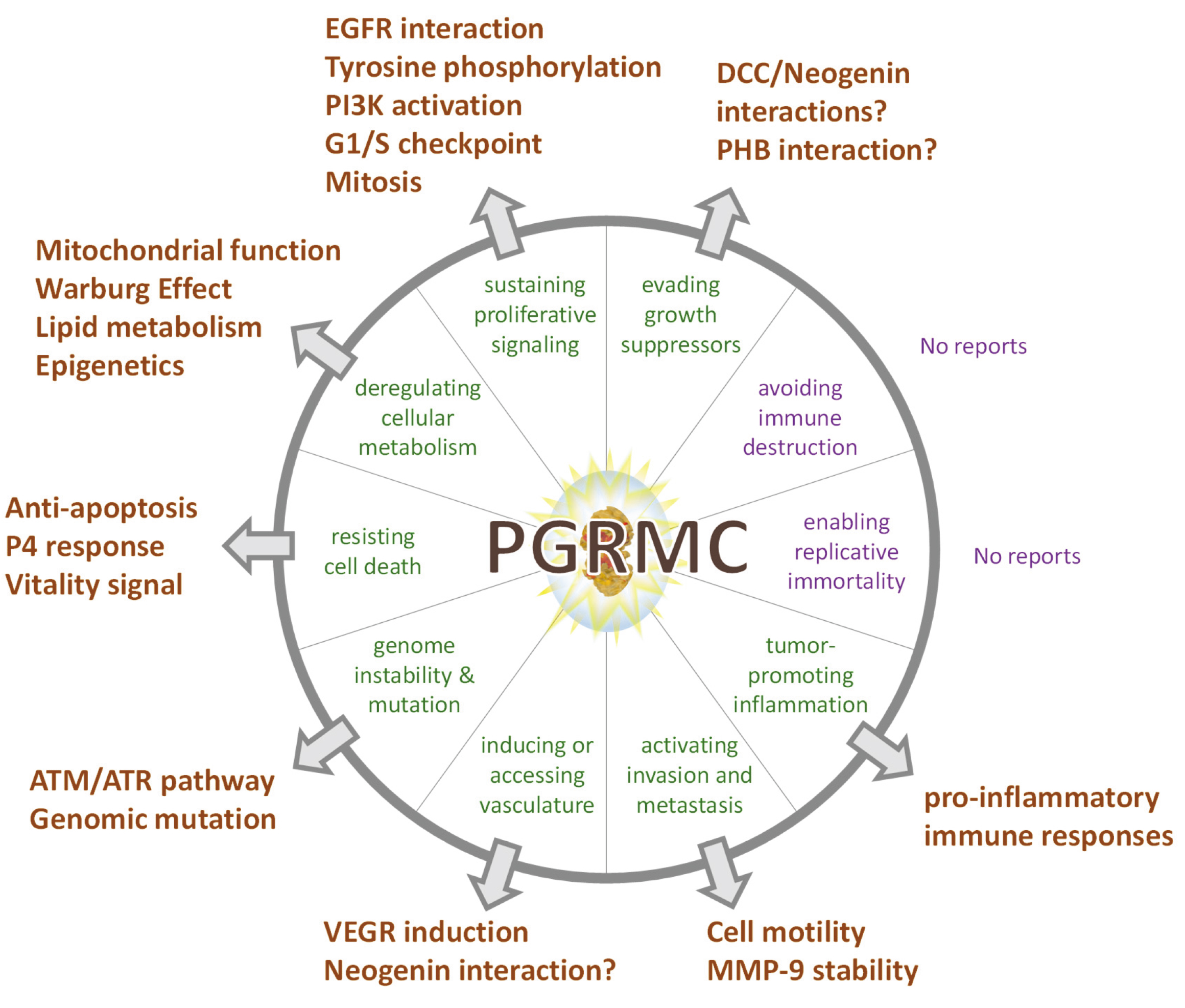
Cancer is one of several ailments associated with aging, including cardiovascular disease, impaired fertility, neuropathologies such as AD, diabetes mellitus, and many more. Quite interestingly, PGRMC1 biology features prominently in many of these diseases. Sinclair argues that age-related pathologies are all symptoms or hallmarks of the aging process, and that by slowing or reversing aging, all these diseases may be suppressed by an increase of whole organism vitality associated with biological youth.
“In this way of thinking, cancer, heart disease, Alzheimer’s, and other conditions we commonly associate with getting old are not necessarily diseases themselves but symptoms of something greater” [415].
Cancer can be viewed as one of several ailments that could be avoided by reversing the damage caused by the aging process, which is attracting considerable interest [416]. This includes the identification of the Horvath clock system of epigenetic regulation of aging, which can be used to measure effective biological age [417, 418], or can even be reversed [419, 420]. The Horvath aging clocks measure the methylation status of suites of CpG loci, which change in a predictable manner as the organism ages.
PGRMC1 regulates chromosomal epigenetics via the Y180 motif [257] which was
acquired by and has been conserved in bilaterians since the LEUMCA and the origin
of the gastrulation organizer [34]. This predated the evolution of differentiated
tissues, and therefore the differentiation process that is affected by aging.
Some proteins, like Sox2, involved in adult neural aging and implicated in AD are
also critical in maintaining PSC pluripotency [421], which is part of the pathway
leading to gastrulation organizer activity. Could the apparent overlap between
PGRMC1 functions, PGRMC1 tyrosines, and the evolution of the gastrulation
organizer and bilaterian aging be more than coincidence? Might PGRMC1 be a
fulcral component of the aging process, or of the overarching systems that can
reverse it? It must be categorically stated that this is speculation. However,
such an association could explain much about PGRMC1’s long-discussed
multifunctional pleiotropy [28, 98, 147]. For instance, consider that loss of
brown adipose function occurs during aging [422], active brown adipose can
enhance longevity [423], and the PGRMC2 knockout mouse with impaired heme
chaperoning and activated heme-responsive transcriptional repressor nuclear
receptor subfamily 1 group D member 1 (Rev-Erb
As described above, sleep is thought to have originated in the LEUMCA, the first
animal to have PGRMC1 Y139 and a Y180-containing C-terminus. We have
seen that PGRMC1 is important in neurons, and the brain’s neurons control sleep.
In fact, much of the synaptic remodeling and memory formation that is defective
in AD patients occurs during sleep [424, 425, 426, 427, 428, 429, 430, 431]. Optimal sleep and circadian clock
activity are intimately associated with aging processes [428, 432, 433, 434, 435, 436, 437, 438, 439], PGRMC1
Y180 stimulates GSK-3
Altered mitochondrial metabolism and ketogenic metabolic profile (analogous to glucagon-induced rather than insulin-induced metabolism) in the brain accompanies reproductive senescence [443]. Such metabolic shifts in female rat hypothalamus are associated with alterations in P4 exposure, which can be addressed by post-menopausal hormone therapy [409]. In an in vivo model of ischemia, which can lead to senescence, the miRNA let-7i negatively regulates PGRMC1 levels, which in return reduces the protective effects of P4 on ischemia and vital neural functions, such as synaptogenic ability [239, 241]. Thereby, PGRMC1 is implicated in the P4-modulated control of cell metabolism and vitality that controls the propensity of neurons to enter the senescent state. A similar vitality-bestowing functionality was described by Sakamoto et al. [114] for PGRMC1 (under the synonym 25-Dx; 25 kDa protein induced by dioxin) in rat Purkinje cells.
In female reproductive tissues, Feng et al. [444] showed that PGRMC1 was responsible for the P4-dependent protection against senescence-inducing oxidative stress in chorion cells during pregnancy. Clarke et al. [445] showed that conditional ablation of pgrmc1 from female reproductive tissues promoted premature reproductive senescence, which arguably could be associated with accelerated aging of those tissues. In a recent study [446], PGRMC1 attenuation in endometrial stromal cells led to the increased expression of the transcription factor forkhead box protein O1 (FOXO1), and the appearance of senescence associated markers which could be reversed by attenuation of FOXO1 levels. Since senescence is an attribute of aging, and PGRMC1 is strongly implicated in senescent biology, these observations promote a strong argument that PGRMC1 is directly involved in aging biology.
Metformin inhibits the 5’-AMP-activated protein kinase (AMPK)/mTOR pathway and activates phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase (PTEN) to inhibit the PI3K/Akt pathway [447, 448, 449, 450], which are important in the metabolic regulation and pluripotency maintenance of PSCs [451]. It is associated with reduced cancer risk and/or attenuation of cancer symptoms in a variety of cancers [452, 453, 454, 455, 456, 457, 458, 459, 460], as well as improvements in many inflammatory and age-related diseases, as well as lifespan and other hallmarks of aging [447, 448, 449, 461, 462, 463].
These metformin-improved age-related hallmarks include the accumulation of
hyperphosphorylated Tau neurofibrillary tangles in AD, which are cleared by
activating autophagy [464, 465]. Importantly, this is a potential common point
with PGRMC1 biology. As well as being a metabolic modulator like metformin,
PGRMC1 modulates autophagy via the mTOR pathway [254] and is also thought to be
part of the S2R complex targeted by CogRx anti-AD drug CT1812 which causes
reduction of hyperphosphorylated Tau in AD patients [166, 169]. In this system,
hyperphosphorylated Tau is downstream of the S2R/PGRMC1 complex. It is, in a
manner, irrelevant to treatment if we do not understand how Tau processing is
impaired, if we know we can alleviate it by targeting the S2R complex. It is
formally unclear whether PGRMC1 is present in the CT1812-targeted S2R complex
present on the membranes of AD synapses loaded with A
A PubMed search for PGRMC1 and metformin returned no results (23 November 2022). Testable predictions of the hypothesis generated here are that metformin induces post-translational changes in PGRMC1 as part of its mode of action, or that PGRMC1 mutants will interfere with metformin function.
Sinclair and colleagues have demonstrated that the inability to repair DNA
damage in a sirtuin and NAD-dependent process leads to epigenetic changes
associated with aging [466, 467, 468, 469, 470]. In yeast, nucleolar disruption is associated
with the production of DNA damage and extrachromosomal ribosomal DNA circles
which lead to cell aging, in a process which can be inhibited by translocation of
sirtuin enzymes from the nucleolus [471, 472]. The basic mechanism is conserved
in mammals, where sirtuins also relocate from the nucleolus to sites of DNA
damage [473] in a process associated with aging [415, 474, 475]. PGRMC1 interacts
with prohibitins to relieve PHB-mediated transcriptional repression of
ESR
The yeast Dap1 homolog of PGRMC1 is a DNA damage response protein [146]. It is unclear whether Dap1 can occupy the nucleolus. PGRMC1 can localize to the nucleolus of granulosa cells and oocytes, where it is required for nucleolar localization of nucleolin, an inducer of chromatin decondensation. Nucleolin was released from the nucleolus by RNAi targeted depletion of PGRMC1, or by peroxide-induced stress [477]. Under the synonym of 25-Dx, one of the original identifications of PGRMC1 was as a gene induced in rat liver after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), also known as dioxin [478], which induces DNA damage [479]. Doxorubicin, a standard of care chemotherapeutic for cancer patients also induces DNA damage [480], and elevated PGRMC1 expression is associated with doxorubicin resistance [385, 481]. PGRMC1 associates with both CYP2D6 and CYP3A4, the two CYP450s which hydroxylate and inactivate doxorubicin [40].
This represents one of the best characterized aspects of PGRMC1 function. In a variety of cell systems, overexpression of PGRMC1 leads to enhanced susceptibility to doxorubicin treatment, whereas subsequent P4 treatment has a PGRMC1-dependent protective effect [385, 482, 483]. Therefore, the best characterized DNA damage-associated function in mammalian cells is associated with PGRMC1’s CYP450 biology. When we assayed for the effects of PGRMC1 phosphorylation mutants on doxorubicin-induced death, all mutants exhibited wild-type activity with respect to doxorubicin toxicity, indicating that PGRMC1’s Y180 motif is not required for this function [257] (Fig. 5, Ref. [19, 257]).
 Fig. 5.
Fig. 5.PGRMC1 phosphorylation status does not affect P4-dependent resistance to doxorubicin toxicity. (A) Schematic diagram of the PGRMC1 mutants employed in panel B. Reproduced unchanged from [19] under a Creative Commons Attribution 4.0 International License (CC BY 4.0). A copy of the license can be viewed at: https://creativecommons.org/licenses/by/4.0/. The license permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. (B) Boxplots of area under the curve (AUC) results for respective transfected cell lines expressing the PGRMC1 mutant proteins from panel A. MP, parental MIA PaCa-2 cell line. WT, DM and TM are as per panel A. For further details see [257], from which the panel is reproduced under a CC BY 4.0 Creative Commons license under conditions as per panel A. The panel is unchanged except the Y axis is relabeled to “AUC (relative cell numbers)”, representing cumulative percentage of cells relative to reference time zero as described in [257].
When we mutated different combinations of PGRMC1 S57, S180 and Y180 in stably transfected MIA PaCa-2 pancreatic cancer cells, our proteomics pathways analysis included predicted modulation of the ATM/ATR DNA repair pathways related to Ataxia Telangiectasia [257], a pathology that can be rescued by NAD replenishment in mouse models [469]. This prompted us to examine DNA mutation rates, where we detected significant differences between mutant cell lines, indicating that PGRMC1 affected genomic sequence stability [257]. This was despite finding no differences in PGRMC1-dependent resistance to doxorubicin-induced death, as discussed above. Therefore, the PGRMC1 phosphorylation mutants have identified the existence of separable PGRMC1 functions, providing the beginnings of a functionally stratified framework, where specific different PGRMC1 functions can be identified, characterized, and in the future be specifically pharmacologically addressed.
A PubMed search for PGRMC1 and sirtuin (23 November 2022) returned one result. where SIRT3 levels were lowered by oxidative stress, and further by cells with a PGRMC1 knockdown. Therefore, PGRMC1 served to maintain SIRT3 levels [444].
NAD synthesis leads to decreased ribosomal RNA (rRNA) transcription at the nucleolus. Depletion of the SIRT1-associated (and NAD-synthesizing) enzyme nicotinamide mononucleotide adenylyl transferase (NMNAT1, Fig. 6A, Ref. [19, 257]) increases rRNA synthesis as well as causing sensitivity to nutrient paucity and DNA damaging agents [484]. This sensitivity is at least superficially reminiscent of the sensitivity that is caused by over-expression of PGRMC1, which can be rescued and overcompensated into elevated viability by the presence of P4 as discussed above. Overarching PGRMC1 effects on ribosomal biosynthesis would be compatible with results observed for different phosphorylation mutants of PGRMC1, where pathways enrichment analysis predicted changes in proteins involved with tRNA aminoacylation, translation/elongation and T complex chaperonins [19]. Future research should investigate the contribution of PGRMC1 to NAD levels.
 Fig. 6.
Fig. 6.PGRMC1 Y180 phosphorylation is implicated in NAD
homeostasis. Panels A-C of this figure are reproduced from Thejer
et al. [257], with modifications, under a Creative Commons Attribution
4.0 International License (CC BY 4.0) as described in the legend to Fig. 5.
Changes are described in the legend descriptions. (A) Model of the NNMT and
1-methylnicotinamide (1-MNA) pathway. The panel is modified from [257] by the
addition of NAMPT (nicotinamide phosphoribosyl transferase), NMNAT1
(nicotinamide/nicotinic acid mononucleotide adenylyl transferase 1), and the
black and white arrows also depicted in B and C. (B) NNMT abundance is increased
by mutation of PGRMC1 Y180. Quantification of NNMT mRNA levels in WT, DM and TM
cells (nomenclature follows Fig. 5A) by RT-PCR. ANOVA post-hoc Tukey’s HSD
p
Interestingly in this respect the PGRMC1 S57A/Y180F/S180A triple mutant (TM, Fig. 5A) — which differs from the S57A/S180A double mutant (DM, Fig. 5A) only by the phosphate accepting oxygen atom of Y180 — exhibited elevated levels of nicotinamide-N-methyltransferase transferase (NNMT) mRNA and decreased levels of nicotinamide phosphoribosyl transferase (NAMPT) protein, which acts upstream of NMNAT1 in NAD salvage (Fig. 6). We would therefore predict from these results that PGRMC1 Y180 phosphorylation enhances NAD salvage from nicotinamide to elevate NAD levels. This is relevant to the prediction made in Section 2.11 above, that PGRMC1 tyrosines could be involved in sleep and circadian regulation. NAMPT is the rate-limiting enzyme of NAD synthesis in a feedback-regulated circadian metabolic pathway [485, 486].
Ramsay et al. [485] proposed that circadian regulation of NAD may be part of the mechanism that regulates a core clock. Those daily cycles could also form part of the clockwork mechanism of the epigenetic Horvath clock [487], which seems to accurately reflect the biological age of animals. It can apparently be rewound to reverse aging symptoms, and therefore its control of gene expression may be the manifestation of biological age [420]. The ability of the PGRMC1 TM mutation to convert the epigenetic state to a hypermethylated genome [257] suggests that Y180 could have direct access to the clockwork mechanism, although how it may influence the clock would require characterization.
Under the synonym Visfatin, NAMPT can be secreted as a cytokine under inflammatory and some other conditions [488, 489]. It would be interesting to see whether this correlates with PGRMC1 subcellular translocations and whether secretion correlates with type 2 PGRMC1 membrane topology.
As argued above, Y180 which regulates genomic CpG status was acquired by evolution coincidentally with the gastrulation organizer, neurons, sleep, and a variety of other eumetazoan synapomorphies. The activity of the gastrulation organizer and the eumetazoan adoption of a pre-existing system of CpG methylation to define tissue-specific gene expression are required for embryological differentiation. This produces multiple specialized cell types via a series of orchestrated progressions through lineage-specific epigenetic states which evolved after the gastrulation organizer and because of its activity, as discussed above. Furthermore, it is this eumetazoan level of epigenetic information which is thought to become corrupted during the aging process and can be modulated in an NAD-dependent manner [415, 416, 419, 469, 490].
We found differences in 1-methylnicotinamide (1-MNA) levels caused by PGRMC1 phosphorylation site mutants [257]. 1-MNA was more abundant in TM cells expressing a Y180F mutation, and as discussed above these cells also expressed higher levels of NNMT which produces 1-MNA, as well as lower levels of NAMPT which catalyzes the competing NAD salvage reaction [257]. These differences were associated with pronounced and specific effects on genomic CpG methylation, which was accompanied by pleiotropic changes in morphology, metabolism, and motility [19, 257].
Importantly for aging biology, these PGRMC1-dependent events affected the methylation status of CpG sites that are related to aging, as assayed by different manifestations of epigenetic aging clocks. There is a caveat that the MIA PaCa-2 cells used in the study are a genomically unstable cell culture line (one collaborating cytologist could not accurately identify homologous chromosomes by fluorescent spectral karyotyping from adjacent cells in the same culture: not shown), and genome packaging is certainly different than in vivo human cells. However, PGRMC1 phosphorylation mutants affected each of the DNAmPhenoAge Clock (Fig. 7A, Ref. [257]), DNAmAge Clock [418, 491] (Fig. 7B), and DNAmAgeSkinBlood Clock [492] (Fig. 7C). While the ‘estimated age’ Y axis of the graphs in Fig. 7 is meaningless for this cell line, the results do suggest that PGRMC1 phosphorylation status can affect epigenetics related to the aging clock. Future experiments should urgently address whether PGRMC phosphorylation sites in the T178/Y180/S181 motif, which was acquired before the evolutionary origins of bilaterally symmetrical animals, can exert corresponding influence on clock-related CpG sites during mammalian embryogenesis and adult life. Indeed, taken together, the hypothesis can be formulated whereby circadian regulation of NAD levels [493] by PGRMC1 could contribute to NAD-dependent sirtuin demethylase activity and diurnal metabolic regulation.
 Fig. 7.
Fig. 7.PGRMC1 phosphorylation affects CpG loci relevant to aging. Differences at different indicative sets of selected genomic CpG sites according to three different Clock Foundation standard clocks, using the publicly available methylomics data from [257] with analysis provided courtesy of J. Gordevicius, R. Brooke, and S. Horvath, The Epigenetic Clock Development Foundation. (A) DNAmPhenoAge. (B) DNAmAge. (C) DNAmAgeSkinBloodClock. Y axes represent clock-estimated ages. Note that none of the clocks was designed to estimate cell line ages. For exogenous p-PGRMC status of stable transfected cell types WT, DM, TM see Fig. 5A. MP is the parental MIA PaCa-2 cell line [257].
In this respect cnidarians, and therefore most probably the LEUMCA, exhibit sleep like behaviour [9, 494, 495]. The gastrulation organizer of the LEUMCA also generated the first synapsed nerves, and sleep is required for neural regeneration. Since the combination of PGRMC1 Y139 and a Y180-containing C-terminus also appeared first in the LEUMCA, there is a correlation between novel eumetazoan PGRMC1 functions and the onset of circadian rhythm-directed sleep. Whether the correlation is due to a functional dependence requires future exploration.
Mammalian embryogenesis produces hypomethylated naïve PSCs, which mature into hypermethylated primed PSCs, which in turn undergo sequential division and differentiation cycles to produce the differentiated cell types of the adult body. Tissue-specific multi-potent stem cells retain the ability to replace cells with certain lineage-restricted tissues in response to damage [496].
It has become clear that what used to be considered as terminally differentiated states are quite plastic, and terminology is adapting as we understand the phenomenon. Reprogramming is the process where differentiated stem cells can be induced to return to a pluripotent state (induced pluripotent stem cells: iPSCs). These have the propensity to cause cancer in adult cells [496, 497]. Transdifferentiation is the process of converting one specialized cell type to another. Transdifferentiation should be associated with minimal risk of cancer. For instance, an epithelial cell could be transdifferentiated to an advanced neural precursor in vitro and then into neurons in situ to treat neurodegenerative disorders [498, 499].
Sinclair [415] has described the targeted partial reversion of a cell to an earlier differentiation state as exdifferentiation. In this sense, fully differentiated neurons can be exdifferentiated to an earlier neural progenitor stage to repair a damaged optic nerve [420]. For this author, at least, it remains unclear whether in that instance the exdifferentiation protocol may not have reverted the cells to a primed pluripotent state which was immediately directed back along the somatic neurogenic differentiation pathway by extracellular matrix components of the environment. Aged cells could also in future potentially be exdifferentiated to younger versions of themselves, to extend lifespan [415]. The emergence of small molecules with reprogramming properties has enormous potential implications for this entire field of related processes [500]. Be that as it may, the exdifferentiation process resembles a reversion back at least towards pluripotency, and therefore to understand it we need to consider how differentiation works and whether PGRMC1 could be involved.
The induction of neuronal regenerative tissue repair and remodeling response after central nervous system damage requires both PGRMC1 protein [103, 501, 502] and, at least in response to chemical ischemia of the brain, also involves alteration of its phosphorylation status [503]. Such induction and modification probably occur at the same time, which has not been examined. Ischemic neural damage and AD share common properties, at least in part through activity of the Wnt signaling pathway and its influence on cell differentiation status [504]. PGRMC1 modulates Wnt activity [505] and cell fate determination [224] in PSCs.
The regenerative process involves the activation of quiescent stem or somatic
cells to repair tissue [506], by the activation of regeneration enhancers, which
dedifferentiate cells and can transdifferentiate them into other cell types.
Unsurprisingly, there is overlap but not identity between regenerative and
developmental enhancers [507]. PGRMC1 function is implicated above with stem cell
activation and differentiation processes, and these are related to aging biology.
Furthermore, PGRMC1 is required for the maintenance of primed PSC cell
pluripotency in a process that involves the p53 and Wnt/
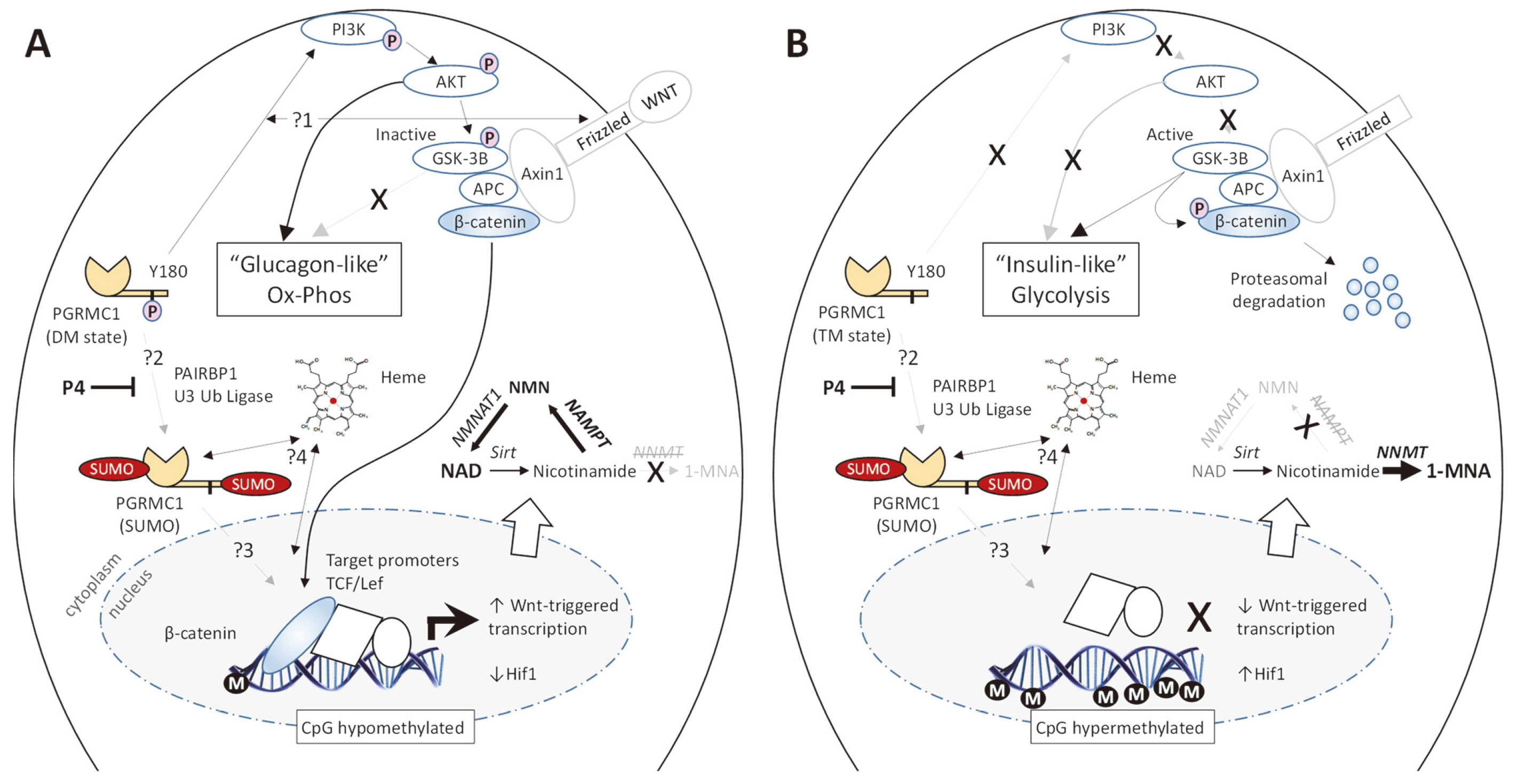 Fig. 8.
Fig. 8.A hypothetical model for the role of PGRMC1 in Wnt signaling,
NAD homeostasis, and epigenomic maintenance. The figure depicts only the biology
proposed to be associated with PGRMC1. The testable model has not been
experimentally validated. It is anticipated, if the model is valid, that the
indicated pathways will act in concert with e.g., the Wnt, LIF/Stat3, Notch, FGF,
and BMP pathways, and their effects on pluripotency transcription factors [509].
Compare the activity of the NNMT pathway to that reported for primed human PSCs,
which reported histone hypermethylation [224]. Binding of sumoylated PGRMC1 to
TCF/Lef binding sites follows [508]. ?1, uncertain relationship between effects
of PGRMC1 phosphorylation and Wnt ligand. ?2, uncertainty which conditions lead
to PGRMC1 sumoylation, therefore depicted in A and B. ?3, effects of sumo-PGRMC1
on
In the study of Kim et al. [224] the PGRMC1 C-terminus was extracellular [362], and so Y180 would not have been accessible to kinases, however Kim et al. [224] did propose that a subset of the PGRMC1 population was cytoplasmic. The overall interpretation of PGRMC1 knockdown could be confounded by interference in the system from PGRMC2, which interacts with PGRMC1 [295, 513] and could function aberrantly upon PGRMC1 knockdown: I.e., functions due to “loss of PGRMC1” could be caused by loss of restraints on PGRMC2.
We discovered [257] that PGRMC1 mutation of PGRMC1 Y180 induced nicotinamide-N-methyl transferase (NNMT), the enzyme which produces 1-MNA (Fig. 6). The association of this pathway with aging biology has been discussed previously, to which the reader is referred [257]. Taken together, it appears that PGRMC1 phosphorylation of Y180 could regulate embryonic stem cell status. However, this has not been demonstrated, and is a testable prediction.
Lu et al. [420] describe the recovery of a youthful epigenetic state in neurons by expression of three Yamanaka transcription factors (Oct4, Sox2 and Klf4) (OSK), and discuss the possible existence of an “Observer” function that enables cells to reactivate youthful epigenetic status. The ability of PGRMC1 phosphorylation mutants to apparently move the epigenetic state towards at least a pseudo-youthful hypermethylated condition (the TM containing Y180F mutation of Fig. 5 is hypermethylated to a similar extent as primed PSCs [257]) suggests that PGRMC1 could access “Observer” function. Hypothetically, the gastrulation organizer function may be related to the observer mechanism and mediate its effects at the level of chromosomal packaging which requires a PGRMC1-dependent process to maintain fidelity. Of course, a mechanistic explanation as to how PGRMC1, and especially the Y180 motif, modulates the epigenetic landscape is lacking and urgently required.
To add a hopefully novel contribution to the nature of the observer, the ability of Yamanaka factors to induce pluripotency from somatic cells implies that the promoters active in the pluripotent state exert a dominant epigenetic effect over ‘downstream’ chromosomal regions (regions which should be activated or repressed at a later stage in development because of these transcription factors). The primed embryonic PSC condition of expression corresponds to the LEUMCA epithelial cells prior to the induction of the gastrulation organizer.
Transcripts of the target genes of Yamanaka OSK factors are significantly enriched in developmental processes. Lu et al. [514] omitted the Myc Yamanaka factor. The targets of c-Myc in primed PSCs are mainly enriched in metabolism processes that are reduced in murine naïve PSCs. This is interesting because genes associated with metabolism are typical for endoderm, which is the most ancient germ layer and reflects general feeding functions. Whereas endoderm gene expression most closely resembles the ancestral monocellular state, the gametes, zygote, naïve PSCs and their derived germline cells represent highly innovative metazoan inventions [515]. Therefore, the OSK factors probably induce pluripotency by avoiding endodermal induction.
Yamanaka factors activate Nanog, Notch, Wnt, MAPK, p53, TGF-
The expression of genes is regulated by super-enhancers and insulators which govern the availability of proximal gene promoters to the transcriptional machinery. Classical enhancers bind transcription factors in the relative vicinity of the transcription start site and are enriched for the histone modification mono-methylation at lysine 4 of histone (H3H3K4me1). They can be classified into poised or active states by the respective increased abundance of either mutually exclusive lysine 27 methylation (H3K27me3) or acetylation (H3K27Ac) of histone 3, where acetylation is thought to protect against methylation [516] (for review: [517]). Therefore, histone methylases and deacetylases play critical roles in transcriptional regulation [518]. Active enhancers bind transcription factors with transactivation domains which recruit co-activators such as transcriptional activator CREB binding protein (CBP) or P300 to in turn recruit RNA polymerase II [519, 520]. In this respect, the PGRMC1-modulated NAD/1-MNA pathway (Fig. 6) affects both the level of NAD required for histone deacetylases such as Sirtuins and impinges on bulk levels of S-adenosyl methionine (SAM), the donor for cellular methylation reactions, such as of CpG or histones.
Active super-enhancers are bound by a high density of lineage-specific transcription factors, and exist in insulated chromosomal regions looped between insulators, which contain DNA binding sites for CCCCTC binding factor (CTCF). Together with cohesin, CTCF tethers chromosomal DNA into large loops. Such loops, or Topologically Associated Domains (TADs) exist between two CTCF sites which can be separated by a great many base pairs. It may be that the colocalization of active promoters and their associated RNA polymerase complexes may order nuclear architecture, and physical three-dimensional proximity to such a center may drive the rate of transcription from any promoter. By recruiting or dismissing CTCF binding sites, loops can be lengthened or shortened, thereby changing the activity of genes that resultingly find themselves in different TADs [521, 522, 523]. The order and proximity of genes separated by CTCF binding sites on a given chromosome is therefore important in how sets of genes can be either mutually or reciprocally regulated, or both, in different epigenetic circumstances.
The activity of such lineage specific super-enhancers is very probably, in the author’s opinion, closely related to the observer function. This probably involves achieving a transcriptional state where key master combinations of transcription factors are expressed that determine the root identity of a group of related more highly differentiated lineages, as achieved by establishing the activity of a certain set of super-enhancers. In this model, those super-enhancers will impose correct control of the CTCF-directed chromosomal regions (i.e., TADs). Importantly, this should involve activating/silencing of genes which should be off/on in vital/younger cells, but which may have become inappropriately regulated by the aging process. In this context, it is notable that in human granulosa cells nuclear PGRMC1 is localized in a punctate pattern, in physical proximity with the classical PGR [513]. It would be interesting to know whether CTCF is colocalized in these puncta, and to what extent sumoylation of nuclear PGRMC1 [18, 29, 393, 524] contributes to epigenetics.
The best studied differentiation system is the naïve to primed PSC transition, where certain naïve-specific enhancers are deactivated during the transition to the primed state. Enhancers that become active in the primed state were detectable in naïve cells (in a ‘poised’ state: H3K27me3), became active in primed cells (H3K27Ac), and often retained activity into the somatic state, frequently contributing to tissue-specific super-enhancers [525].
How alterations in CpG methylation affect changes in such higher order chromatin structures during development has been debated [526], however there is strong evidence that CpG methylation may be involved in the irreversible silencing of genes, such as in genomic imprinting. Studies from parental genomic imprinting sites suggest that CpG status affects the size of CTCF/condensin-structured TADs. Thereby, CTCF sites can change from being actively tethered at the base of a loop or being untethered. Intervening genes can be activated or silenced as a result [527].
Bestor et al. [526] argue that genes are sequentially activated and repressed solely by protein- and RNA-based mechanisms (including histone modifications) during developmental processes, whereas DNA methylation mediates permanent gene inactivation that originates in the germline and which usually does not change over a single lifetime. However, DNA methylation has been implicated in modulating alternative enhancers, splicing sites, and intragenic promoters. Furthermore, changes in DNA methylation in multiple disease states, as well as during development and with aging make a compelling case that changes in CpG methylation are important in gene regulation [528]. Active super-enhancers are associated with increased H3K27Ac (active state), and decreased DNA methylation. These are maintained by the presence of RNA Polymerase II-transcribed long non-coding extended RNAs (eRNAs) which participate in maintaining open chromatin structure. eRNAs require the activity of Tet (ten-eleven translocation) family methylcytosine dioxygenase DNA demethylases (which remove DNA methylation) to produce hypomethylation, which strongly associate DNA CpG hypomethylation with the establishment of active enhancers [529]. See also the references above to the Horvath clock above for functions of CpG methylation. PGRMC1 Y180 which affects CpG status was obtained in evolution at precisely the time when the reversible transition between naïve and primed PSC cell types probably evolved, which is formative to activity of the gastrulation organizer inherited by the LEUMCA.
In a possibly interesting connection between PGRMC1 and genomic methylation changes between naïve and primed PSCs, the orphan classical steroid receptor estrogen-related receptor beta (Esrrb: also known as Steroid hormone receptor ERR2, or just ERR2) binds to silenced enhancers in primed cells, displacing nucleosomes and opening up chromatin to cause epigenetic reversion to the hypomethylated naïve state [530]. We previously noticed that the abundance profile of mitochondrial proteins induced by the presence of PGRMC1 S57A/S181A DM (Fig. 5A), which is thought to permit easier Y180 phosphorylation by relieving steric hindrance of adjacent phosphorylated residues, overlapped strongly with proteins predicted to be induced by Esrrb/ERR2. The overlap profile was not perfect, and we concluded that PGRMC1 and Esrrb/ERR2 regulated overlapping sets of proteins, possibly related to shared steroid biology [19].
Given the undeniable relationship between PGRMC1 and steroid biology, it will be interesting to see whether PGRMC1 is responsible for production of the orphan ligand of Esrrb/ERR2 which is required for the naïve state. Note: (1) that we investigated Esrrb/ERR2 independently of PSC biology or aging, and (2) that classical (nuclear) steroid sensing first appeared in the LEUMCA, while receptors like Esrrb/ERR2 probably arose with the urbilaterian [2]. Conceivably, the naïve to primed PSC transition is partly due to PGRMC1-dependent changes in the levels of an unknown steroid ligand of Esrrb/ERR2. Recall again in this context the discussion above where PGRMC1 was required for the pluripotency of primed PSCs [224], while the NAD/NNMT/1-MNA pathway maintains the naïve state [505]. Esrrb/ERR2 may well be an intermediary in the mechanism. Of course, this is speculation that would require experimental validation.
Note also in this context, as detailed above, that PGRMC1 seems intimately related to mitochondrial function, and the overt regulation between glucose or lipid/amino acid (TCA cycle) respiration, and that primed cells exhibit increased mitochondrial respiration. PGRMC1 tyrosine phosphorylation (and the extended C-terminus that holds the Y180 motif) were acquired at the same time as the gastrulation organizer, sleep, and the foundational platform for cell differentiation mechanisms, making it at least plausible that PGRMC1 function is related to the differentiation processes that ultimately drive the aging process, and the mechanisms that can slow it. I.e., PGRMC1 acquired new functions at just the right time in animal evolution to affect aging, which evolved later (after the evolution of all those differentiated tissue types which age). This is another untested hypothesis, with testable predictions that, like metformin, resveratrol, and NAD supplements [415], PGRMC1 phosphorylation mutants, post-translational modifications, and/or pharmacological interference with PGRMC1 functions, could affect healthspan and lifespan.
The previous discussion directly connects PGRMC1’s biology with all the main AD biomarkers, with multiple aspects of tumor biology (migration, EMT, epigenetic plasticity, survival, etc.), and with key feature of other diseases of aging, such as diabetes, as well as with several key pathways of aging itself. The common denominator could well be the perturbation of foundational level metabolic switches reflecting the proposed original eukaryotic function of PGRMC as a metabolic regulator [1, 153], and the adaptation of this PGRMC-based eukaryotic cellular foundation towards specialized multicellular body plans and differentiated cell types.
Fluctuations in levels or properties of the leading accepted markers for AD could be directly explained by a new model centrally involving PGRMC1 and its associated sterol and heme biology at the interface of ER and mitochondrial integration in the pathogenesis of AD. Therefore, PGRMC1 and the regulatory apparatus surrounding it may represent one of the best current directions in which to search for improved AD cures, and possibly for many other age-related illnesses.
No doubt, as stated here the model is incomplete and should be substantially refined. However, this new compass bearing has every prospect of plotting a better course to, e.g., successful AD therapies than has commanded the helm for three unsuccessful Amyloid Cascade Hypothesis-directed decades. Therein lies the justification for a PGRMC-based model of AD, and perhaps eumetazoan aging, which is nothing short of the heuristic core of a new paradigm for some of the most urgent medical issues facing modern human society. Because of the foundational level regulation proposed for PGRMC in animal biology [1], these PGRMC functions are expected to be manifest across multiple clinical indication areas.
The biology being discussed here, involving PGRMC phosphorylation and other modifications, will not be detected by many modern routine analytical techniques: especially transcriptomic-based ones. Since we discovered differential PGRMC1 phosphorylation in breast cancers [301], only Sabbir [18] from the rest of the scientific community has systematically attempted to monitor PGRMC1 phosphorylation status, which he found to change in response to P4 treatment, and where he cited the author’s publications as rationale. (See also Peluso and Pru [393]). Sabbir and the group of Peluso and collaborators have investigated sumoylation and ubiquitination [18, 29, 108, 524]. There is an urgent need to develop reagents and methods to assay the state of PGRMC post-translational status, membrane topology, and subcellular location in a variety of different organisms and their various cell types during embryological differentiation stages and adult biology.
In terms of eumetazoan biology, Archimedes may have recognized in PGRMC the fulcrum that provided sufficient evolutionary leverage to enable development of the body architecture and organization of the eumetazoan animal world, and to topple its metabolic organization in modern disease states. It appears that the PGRMC research journey will continue to illuminate many more important mechanisms for human health in the future.
1-MNA, 1-methylnicotinamide; A
MAC conceptualized and wrote this work.
Not applicable.
I am grateful to Tim Cahill (R.I.P.) for Latin language assistance.
This research received no external funding.
Michael A. Cahill is scientific advisor to and minor shareholder (
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.