1. Introduction
The plant Lepidium meyenii Walp. (maca) originated in the Andes where
the temperature is low at high altitudes between 3700 and 4450 m, and it has
traditionally been used to enhance fertility [1, 2]. The major active ingredients
in maca are polysaccharides, macamides, macaenes, and alkaloids [3]. In 2011, the
Chinese government classified maca as a functional food, and this plant is
well-known for being nutrient-rich and for promoting fertility in humans [4]. In
many studies, maca has been shown to scavenge free radicals and protect cells
against oxidative stress [3]. Oxidation can produce energy to promote essential
biological processes in many organisms. However, reactive oxygen species (ROS)
are often overproduced during oxidation, resulting in an imbalance in the
antioxidant defense system [5]. Furthermore, excessive oxidative stress plays a
vital role in the aging process [6, 7]. Maca is a source of macamides and
polysaccharides, which can combat oxidative stress and damage in human
erythrocytes [8]. In addition, previous studies have indicated that maca
polysaccharide (MP) effectively scavenges 1,1-Diphenyl-2-picrylhydrazyl (DPPH)
and peroxyl radicals to protect erythrocytes against HO-induced
hemolysis by inhibiting the generation of malondialdehyde (MDA) [9].
D-Galactose (D-gal) is commonly used to establish aging models because it can
induce oxidative damage [10, 11, 12]. Small amounts of D-gal can be metabolized and
utilized [13]. However, exogenous supplementation with excessive D-gal causes
large amounts of D-gal to accumulate; the resulting high D-gal levels alter
oxidase activity and lead to the production of large amounts of oxidation
products, which further affect physiological structure and function [14].
ROS, which include oxygen ions, peroxides, and oxygen-containing free radicals,
are potentially dangerous byproducts of normal aerobic cellular metabolism in
organisms [15, 16]. Increasing evidence has shown that excessive ROS levels
during neuronal cell apoptosis are related to various chronic neurodegenerative
disorders [17], as neuronal cells are thought to be more sensitive to oxidative
stress than cells in other tissues [18]. Exogenous supplementary antioxidants may
protect cells from the damaging effects of ROS by scavenging free radicals [19, 20]. Increasing attention has been given to identifying effective and safe
natural antioxidants due to the carcinogenicity of synthetic antioxidants.
Polysaccharides are widely distributed in various organisms, and polysaccharides
isolated from many kinds of plants have been shown to exhibit strong antioxidant
activity against free radicals [21, 22]. Thus, these compounds should be further
investigated as potential novel antioxidants.
In the present study, MP was extracted from maca. As SH-SY5Y cells are sensitive
to the apoptosis- and cytotoxicity-inducing effects of HO [23], the
effects of MP on HO-treated human neuronal SH-SY5Y cells were
evaluated in vitro. For in vivo experiments, model mice with
D-gal-induced oxidative stress-related aging were administered MP, and the
effects of MP on brain tissue were observed. This study aimed to provide a
theoretical basis for further research on MP as a medicine and to provide a
material basis for further research on maca.
2. Materials and methods
2.1 Reagents and materials
Maca was purchased from Lijiang Green Hanson Biotechnology Development Co., Ltd.
(Lijiang, China). Dulbecco’s modified Eagle’s medium/Ham’s nutrient mixture F-12
(DMEM/F-12, 1:1) was purchased from HyClone Laboratories (Logan, UT, USA). The
human neuroblastoma cell line SH-SY5Y was obtained from the Cell Bank of the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai, China), while
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), trypsin-EDTA,
fetal bovine serum (FBS), penicillin and streptomycin were purchased from
Gibco-BRL (Grand Island, NY, USA). Radioimmunoprecipitation assay (RIPA) buffer
was purchased from Shanghai Wellfeng Biotech Co., Ltd. (Shanghai, China). A
propidium iodide (PI)/RNase staining buffer kit was purchased from Becton
Dickinson and Company (Franklin Lakes, NJ, USA), and an Annexin V Cell Apoptosis
Analysis Kit and 2,7-dichlorofluorescein diacetate (DCFH-DA) were purchased from
Sungene Biotech Co., Ltd. (Tianjin, China). D-Gal was obtained from Life
Sciences. A bicinchoninic acid (BCA) protein concentration determination kit,
GSH-Px assay kit, MDA assay kit, and LDH assay kit were obtained from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China). Rabbit anti-caspase 3
(#8231) and rabbit anti-cleaved caspase 3 (# 8231) antibodies were obtained
from Cell Signaling, and anti-P53 (mouse monoclonal, AP062), anti-tubulin (mouse
monoclonal, AT819), HRP-labeled goat anti-mouse and rabbit IgG (H + L) (A0412)
antibodies were obtained from Beyotime Biotechnology (Shanghai, China). All other
chemicals were of analytical grade and were obtained from Nanjing Jiancheng
Bioengineering Institute (Nanjing, China).
2.2 Identification of maca
The content of N-benzyl-hexadecanamide in maca was determined by SPD-M20A
high-performance liquid chromatography (Shimadzu, Japan). N-Benzyl-hexadecanamide
(20 mg; China Food and Drug Administration) was used as a reference substance,
and 5 mg was weighed into a 20 mL volumetric flask, shaken and completely
dissolved as a reference substance stock solution. An Accucore™
C18 column (particle size 5.0 m; size 4.6 250 mm; Thermo
Fisher Scientific, Inc. (Shanghai, China) was used, and the mobile phase solvents
included mobile phase A (0.005% trifluoroacetic acid in water) and mobile phase
B (0.005% trifluoroacetic acid in acetonitrile). The gradient conditions were as
follows: 0–35 min, 55–95% phase B; 35–40 min, 95–100% phase B; flow rate,
1.0 mL/min. The injection volume was 20 L, the column temperature
was 30 C, and the detection wavelength was 210 nm [24].
2.3 MP extraction
A dried maca block was crushed, and the powder was extracted twice with
distilled water (1:20, w:v) at 95 C for 2 h each time. The solutions
were combined and filtered, centrifuged, concentrated, and adjusted to pH 6.5
with phosphate buffer. Two milliliters were incubated in a 60 C water
bath for 3 h after thorough mixing. Then, 1 mL of amylase was added, and the
mixture was incubated for another 2 h. After enzymatic hydrolysis, the solution
was placed in a boiling water bath for 10 min to inactivate the enzyme, cooled to
room temperature and centrifuged; the supernatant was then deproteinized using
the Sevage method [25]. Subsequently, the deproteinized supernatant was added to
95% ethanol at a ratio of 1:5 and vigorously stirred, and the mixture was placed
at 4 C overnight. After centrifugation, the precipitate was washed
successively with absolute ethanol, acetone and ether and then vacuum-dried at 40
C to obtain the crude MP. The carbohydrate content in the MP was
determined by the phenol-sulfuric acid method [26].
2.4 Effects of MP on D-gal-induced brain impairment in mice
2.4.1 Animals
Healthy ICR male mice (8 weeks, 29–32 g, SPF grade) were purchased from Hunan
Slake Jingda Experimental Animal Co., Ltd. (certificate number: SCXK (XIANG)
2019-0004). The mice were allowed to adapt for 7 days in an environment with an
ambient temperature of 19–21 C and a relative humidity of 50–60%
under a 12 h/12 h light/dark period, and all mice were allowed to eat and drink
freely.
2.4.2 Animal models
Fifty ICR mice were randomly divided into 5 groups (10 mice in each group). The
mice in group 1, which served as the control group, received saline (0.9% NaCl)
daily by intraperitoneal (i.p.) injection and received distilled water without MP
orally. The mice in groups 2–5 received 500 mg/kg D-gal daily by i.p. injection
for eight weeks [27]. The D-gal-treated mice in group 2, which served as the
D-gal group, also received distilled water without MP. The mice in groups 3–5
received 75, 150 or 300 mg/kg MP in distilled water orally for eight weeks. After
8 weeks, the animals were sacrificed, and brain tissue samples were collected.
Mouse hippocampal dentate gyrus samples from the first three mice in each group
were used for transmission electron microscopy (TEM) analysis, and the brain
cortices of each group of mice were used to detect GSH-Px activity and MDA
content.
2.5 Determination of activity and MDA content in mouse brain tissue
Brain cortex samples were obtained from each group. Subsequently, 100 mg of
cortex tissue from each mouse was placed in a glass homogenization tube, after
which 1 mL phosphate buffered solution (PBS) was added, and the sample was
homogenized on ice for 10 min to produce a 10% brain tissue homogenate. Finally,
the appropriate kit was used to perform the test according to the instructions.
MDA content was measured by the thiobarbituric acid reactive substance (TBARS)
method [28]. Briefly, the homogenate was mixed with 3 mL of HPO
solution (1%, v/v), after which 1 mL of thiobarbituric acid solution (0.67%,
w/v) was added. The mixture was incubated at 95 C in a water bath for
45 min. The colored complex was extracted into N-butanol, and the absorption at
532 nm was measured using tetramethoxypropane as a standard. MDA levels are
expressed as nmol/mg protein. GSH-Px activity was determined by quantifying the
catalyzed reaction rate of HO and GSH [29]. The enzymatic reaction in
tube containing nicotinamide-adenine dinucleotide phosphate (NADPH), reduced GSH,
sodium azide and glutathione reductase was initiated by the addition of
HO. The change in absorbance at 340 nm was monitored. GSH-Px activity
is expressed as U/mg protein.
2.6 TEM analysis
The mouse hippocampal dentate gyrus samples were fixed in a 4% paraformaldehyde
solution (4 C) and then moved to 1% osmium tetroxide, after which
ethanol and acetone were used for gradual dehydration. Then, the samples were
embedded in epoxy resin 618, sliced into ultrathin sections, double-stained with
uranyl acetate and citric acid, and finally observed and imaged with a
transmission electron microscope (Hitachi 7100, Hitachi, Ltd., Tokyo, Japan).
2.7 Effects of MP on HO-induced oxidative impairment in
SH-SY5Y cells
2.7.1 Cell culture and treatment
Cells were cultured in 25 cm flasks in DMEM/F-12 (DMEM:F12 = 1:1)
supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 g/mL
streptomycin at 37 C under 5% CO in an incubator [30]. The cell
culture medium was replaced every three days. Once the cells reached 80–90%
confluence, they were subcultured. SH-SY5Y cells were seeded in well plates at a
density of 8 10 cells/mL and cultured for 24 h.
2.7.2 Measurement of cell viability
Cells were cultured in 96-well plates at a density of 8 10
cells/mL in a volume of 150 L per well for 24 h before treatment. The
cells were cultured in different concentrations of HO (100–800
M) for 6 h, and then an MTT reduction assay was used to evaluate the
effects of the different concentrations of HO on the viability of the
SH-SY5Y cells. The appropriate concentration of HO was determined for
modeling (cell survival rate 50–60%).
Other cells were cultured in 96-well plates at a density of 8
10 cells/mL in a volume of 150 L per well for 24 h before treatment.
The SH-SY5Y cells were pretreated with various concentrations of MP (25, 50 and
100 g/mL) for 24 h and then exposed to 300 M HO for 6
h. Subsequently, 15 L of MTT (5 mg/mL in PBS) was added to each well, and
the cells were incubated at 37 C for 4 h. Then, the supernatant was
carefully aspirated, 150 L of dimethyl sulfoxide (DMSO) was added to each
well to dissolve the precipitate, and the absorbance was measured at 490 nm with
a microplate reader [31, 32, 33]. Cell viability is expressed as the percentage of
control cells.
2.8 Measurement of LDH activity
Cells were cultured in 6-well plates at a density of 8 10
cells/mL in a volume of 2 mL per well for 24 h before treatment. Then, the cells
were incubated with MP for 24 h, 300 M HO was added to the
culture medium, and the cells were cultured for another 6 h. After the cells were
treated as described previously, the cell morphology was observed, and images
were obtained under an inverted microscope (Olympus CKX53). The cell supernatant
was collected, and a kit was used to measure LDH activity according to the
manufacturer’s instructions.
2.9 Flow cytometry detection of apoptotic cells
A commercial Annexin V-FITC detection kit was used to observe the effect of MP
on HO-induced apoptosis. In accordance with the manufacturer’s
instructions, cells in a 6-well plate that had been treated as described
previously were digested with 0.25% trypsin, collected and processed into a
single-cell suspension. The cells were then stained following the manufacturer’s
protocols. Flow cytometry was performed on a Beckton Dickinson FACScan (Franklin
Lakes, NJ, USA) and analyzed using CellQuest Pro software (version 3.3, Beckton
Dickinson).
2.10 Determination of intracellular ROS levels
The oxidation-sensitive fluorescent probe DCFH-DA was used to evaluate the
generation of ROS. Cells in six-well plates were used after being treated as
described previously. Briefly, the cells were incubated with DCFH-DA (final
concentration 10 mol/L) in the dark at 37 C for 30 min, and then
the relative fluorescence intensity was measured using an excitation wavelength
of 485 nm. The cells loaded with DCFH-DA were examined by flow cytometry. The
measured fluorescence value is expressed as a percentage of the fluorescence of
the control cells.
2.11 Determination of cell cycle status
A PI/RNase staining buffer kit was used to assess the effects of MP on
HO-induced changes in the cell cycle. Cells in 6-well plates were
collected after being treated as described previously. The pellets were then
resuspended in ice-cold 70% ethanol and fixed at 4 C for 24 h. Then,
the cells were washed and resuspended in 0.5 mL of PI/RNase staining solution.
The cells were stained at room temperature in darkness for 30 min. The
percentages of cells in the G0/G1, S, and G2/M phases of the cell cycle were
determined by flow cytometry, and the data were analyzed by utilizing Mod FitLT
software (BD), version 2.0 (Software House, USA).
2.12 Western blot assay of signaling proteins
The expression levels of P53, caspase 3 and cleaved caspase 3 in SH-SY5Y cells
were examined by western blot analysis. After being treated as described
previously, cells were trypsinized and collected. Then, the pelleted cells were
lysed in 100 L of RIPA buffer on ice for 15 min, after which the lysates
were centrifuged at 12,000 g for 10 min at 4 C. Subsequently,
the supernatants were collected, and the protein concentrations were determined
with a BCA protein assay kit. After boiling the samples in loading buffer for 5
min, the proteins were separated by 12% sodium dodecyl sulfate–polyacrylamide
gel electrophoresis (SDS–PAGE) at 100 V for 60 min and then transferred to
nitrocellulose membranes using a transfer apparatus for 50 min at 300 mA. Then,
the membranes were rinsed in PBST (PBS with 0.1% Tween 20), blocked with 5%
nonfat dry milk in PBST at room temperature for 60 min and probed with the
following primary antibodies on a platform shaker overnight at 4 C:
mouse anti-P53 mAb (1:1000), rabbit anti-caspase 3 (8G10) mAb (1:1000), rabbit
anti-cleaved caspase 3 mAb (1:1000) and mouse anti-tubulin mAb (1:1000). The
membranes were washed six times for 5 min each using PBST. After that, the cells
were incubated with the appropriate HRP-conjugated secondary antibody at room
temperature for another 1 h and washed again six times in PBST buffer. The
membranes were then incubated with enhanced chemiluminescence (ECL) substrate
solution (Thermo Scientific, Waltham, MA, USA) for 5 min according to the
manufacturer’s instructions and visualized with radiography film.
2.13 Statistical analysis
The results are reported as the mean SD. Statistical evaluation was
performed with Student’s t-test when two groups were compared, and
p 0.01 or p 0.05 was considered to indicate a
significant difference.
3. Results
3.1 Phytochemical analysis of maca
High Performance Liquid Chromatography (HPLC) analysis of the maca composition
revealed the presence of N-benzyl-hexadecanamide (retention time [t] =
36.37 min, peak 1; Fig. 1) at a concentration of 0.077%.
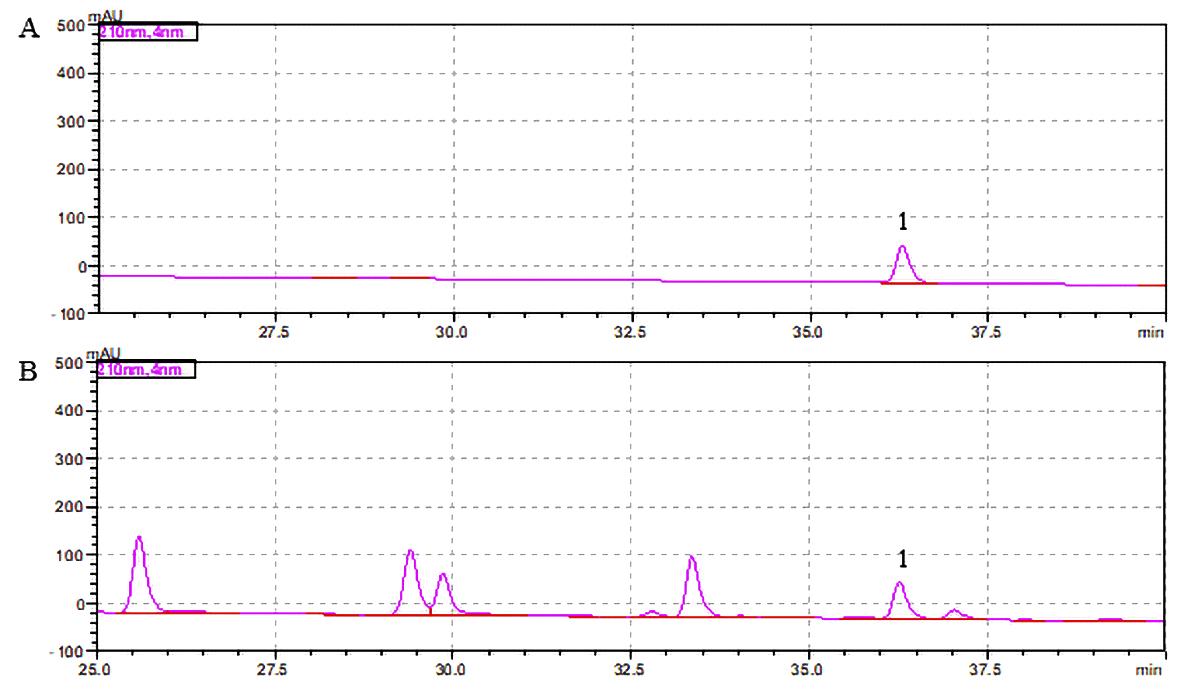 Fig. 1.
Fig. 1.
Maca extract chromatogram. (A) Standard. (B) Maca
extract. The number 1 indicates N-benzyl-hexadecanamide.
3.2 Extraction of polysaccharides from maca
Using the formula polysaccharide yield (%) = polysaccharide weight in the
extract (g)/weight of maca (g), the yield of MP was calculated to be 13.36%. The
purity of carbohydrates in MP was determined by the phenol-sulfuric acid method
to be 71.54%.
3.3 Effect of MP on GSH-Px activity and MDA content in mouse brain
tissue
The GSH-Px enzyme activity in brain tissue was significantly different among the
groups of mice (F = 16.27, p 0.01). In the presence of
D-gal, the GSH-Px enzyme activity of the D-gal group was 492 58 U/mg
protein, while that of the control group was significantly higher at 727 83 U/mg protein (p 0.01). Compared to that in the D-gal group, the
enzyme activity in the low-, medium- and high-MP groups was significantly higher
(p 0.05 or p 0.01). The MDA content in brain tissue
also significantly differed among the groups (F = 35.24, p
0.01). There was a significant difference in MDA content between the D-gal and
control groups, and in all MP administration groups, the MDA content was
significantly lower than that of the D-gal group (p 0.01, Table 1).
Table 1.Effects of MP on the activity of related enzymes in mouse brain
tissue ( s, n = 10).
| Group |
GSH-Px (U/mg protein) |
MDA (nmol/mg protein) |
| Control |
727 83 |
0.9 0.11 |
| D-gal |
492 58 |
1.6 0.15 |
| D-gal + MP (75 mg/kg) |
573 71* |
1.1 0.09** |
| D-gal + MP (150 mg/kg) |
610 49** |
1.2 0.12** |
| D-gal + MP (300 mg/kg) |
652 28** |
1.1 0.13** |
| F |
16.27 |
35.14 |
| p |
0.01 |
0.01 |
| Note: Compared with the control group, p 0.01; compared
with the D-gal group, **p 0.01, *p 0.05. |
3.4 Effects of MP on the brain cortex ultrastructure in
D-gal-treated model mice
Pathological changes in the brain cortex ultrastructure in mice were observed by
electron microscopy. The control mice showed clear structures; large, round
nuclei; complete, smooth nuclear membranes; uniform chromatin distributions; and
abundant cytoplasm (Fig. 2A). In contrast, the D-gal group exhibited pyknosis, an
incomplete nuclear membrane structure, cracks in the perinucleus, aggregated
nuclear chromatin, and an expanded cytoplasmic endoplasmic reticulum (Fig. 2B).
The cell structure in the low-, medium- and high-MP groups tended to be normal,
with large, round nuclei; a complete nuclear membrane structure; and a uniform
chromatin distribution (Fig. 2C–E).
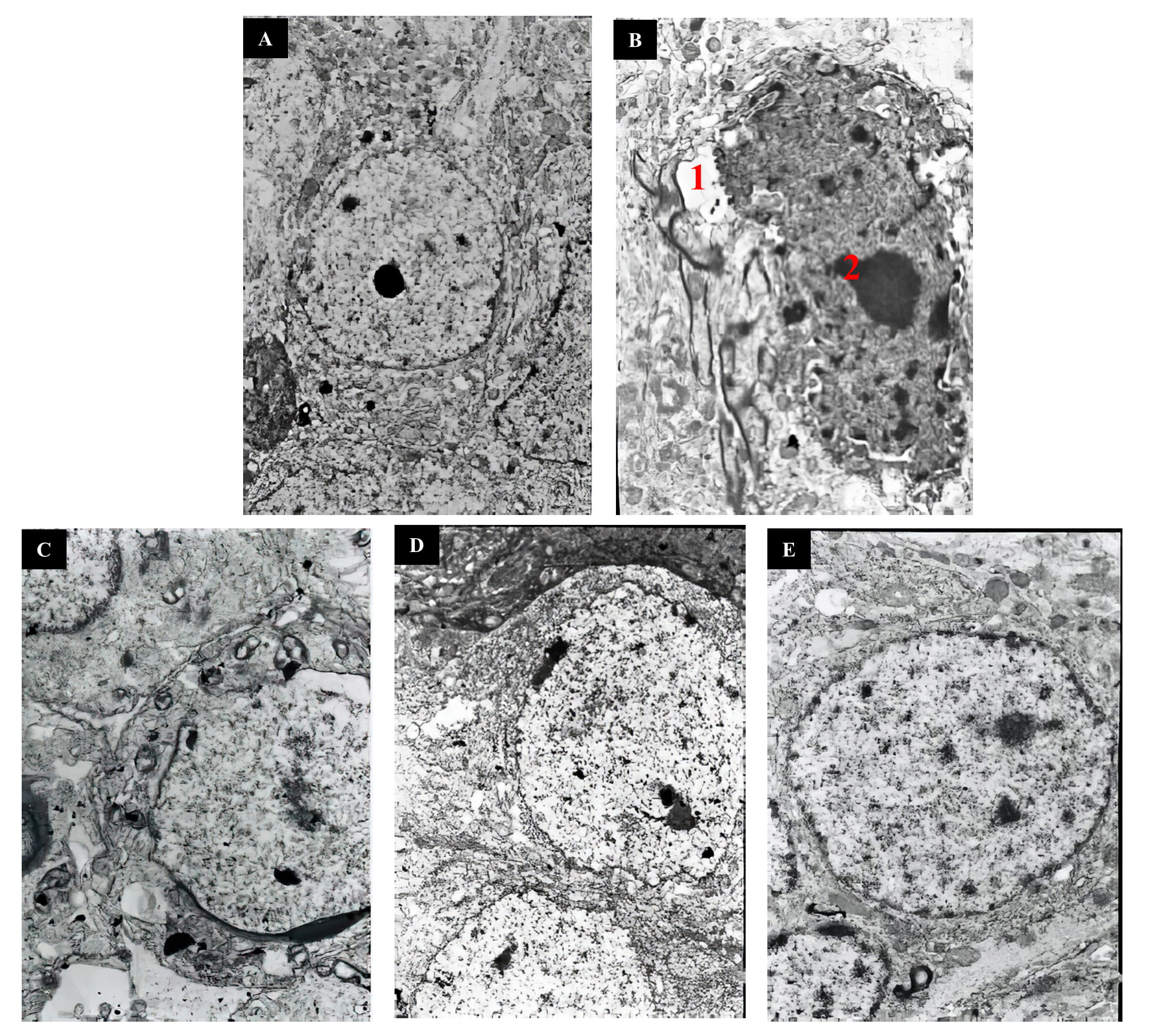 Fig. 2.
Fig. 2.
Ultrastructural pathology of mouse brain slides. (A)
Control group. (B) D-gal group. (C) D-gal + MP (75 mg/kg) group. (D) D-gal + MP
(150 mg/kg) group. (E) D-gal + MP (300 mg/kg) group. Original magnification:
4000 (A), 4500 (B), 4500 (C), 3500 (D),
and 4500 (E). Notes: 1: incomplete nuclear membrane structure, 2:
aggregated nuclear chromatin.
3.5 Protective effect of MP against HO-induced SH-SY5Y
cell injury
SH-SY5Y cells were cultured with HO (100-800 M) for 6 h, and
cell viability was detected by MTT assay. The results showed that all assayed
concentrations induced significant decreases in cell survival in a dose-dependent
manner (p 0.01). In the presence of 300 M HO,
cells exhibited only 53.0 4.58% (mean SD, n = 8) of the
viability of control cells (Fig. 3A). Therefore, 300 M HO
treatment for 6 h was used to induce SH-SY5Y cell injury in the following
experiments. As shown in Fig. 3B, the cell viabilities were compared among the
groups, and significant differences from the viability of the HO
group were observed (F = 93.39, p 0.01). Specifically, cell
viability was significantly increased in the low-, medium- and high-MP groups
(p 0.05 or p 0.01). The effects of MP were also
confirmed through morphological observations (Fig. 3C). LDH activity was found to
differ significantly among the groups (F = 1350, p 0.01).
As shown in Fig. 3D, a significant increase in LDH leakage was observed after 6 h
of exposure to 300 M HO, indicating an increase in cell
toxicity, while MP treatment significantly attenuated this increase in LDH
outflow (p 0.01).
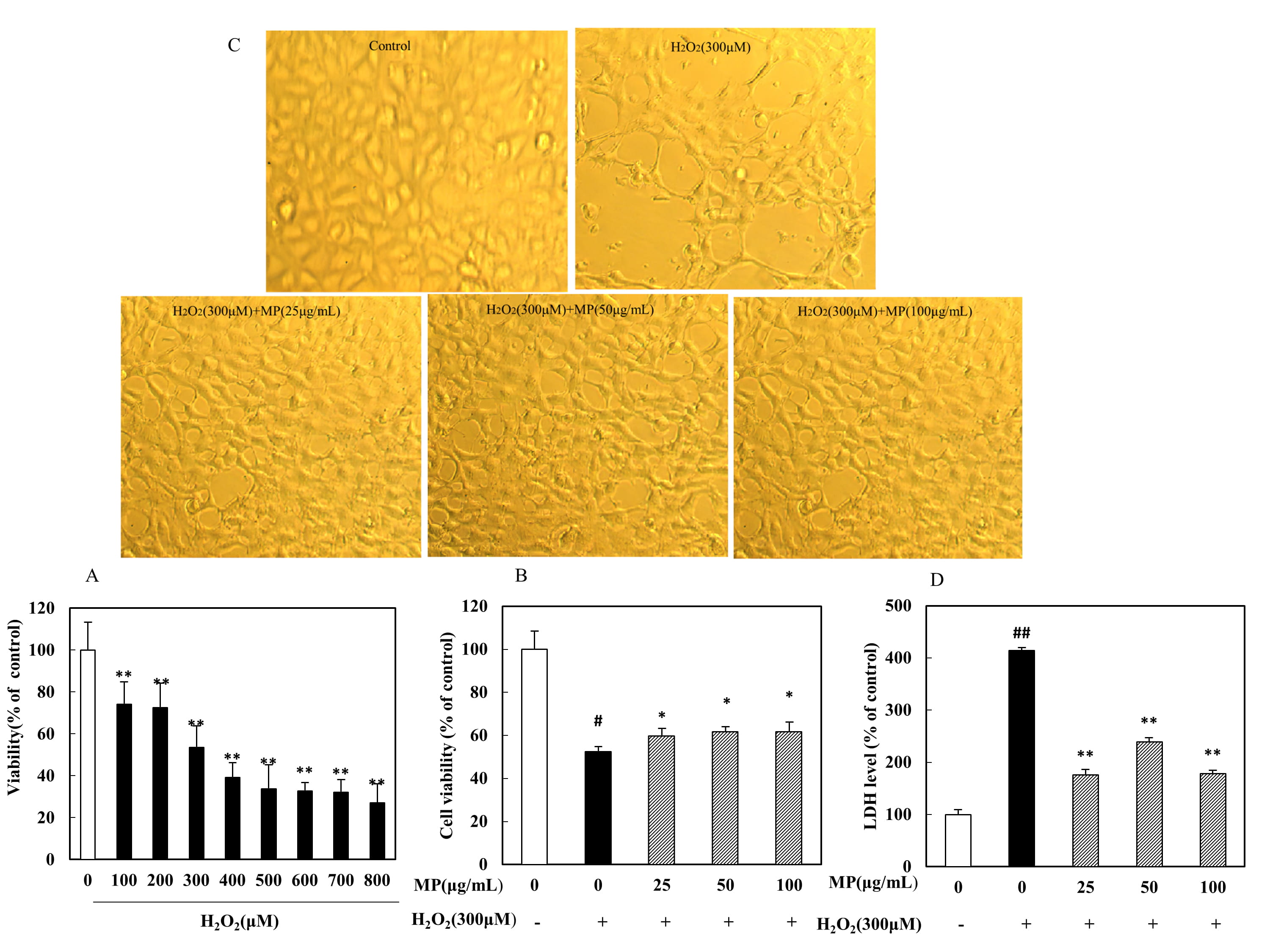 Fig. 3.
Fig. 3.
Protective effect of MP against HO-induced SH-SY5Y
cell injury (n = 8). (A) Dose-dependent toxic effects of different
HO concentrations on cells. (B) Viability of SH-SY5Y cells. (C)
Morphological alterations in SH-SY5Y cells. (D) Release of LDH. All data are
presented as the means SDs from three independent experiments performed
in triplicate. Note: p 0.01 versus the control group;
*p 0.05, **p 0.01 versus the HO group.
3.6 Protective effect of MP on SH-SY5Y cells against
HO-induced apoptosis
The percentage of apoptotic cells was measured by flow cytometry using double
Annexin V and PI staining. The apoptosis rate significantly different among
groups (F = 219.6, p 0.01). As shown in Fig. 4, 6.9
0.78% of cells were apoptotic in the control group. After 300 M
HO treatment, the percentage of positive cells increased to 34.63
1.34%, significantly higher than the value in the control group
(p 0.01). However, this increase was significantly attenuated by
pretreatment with 50 and 100 g/mL MP (p 0.01; Fig. 4A,B).
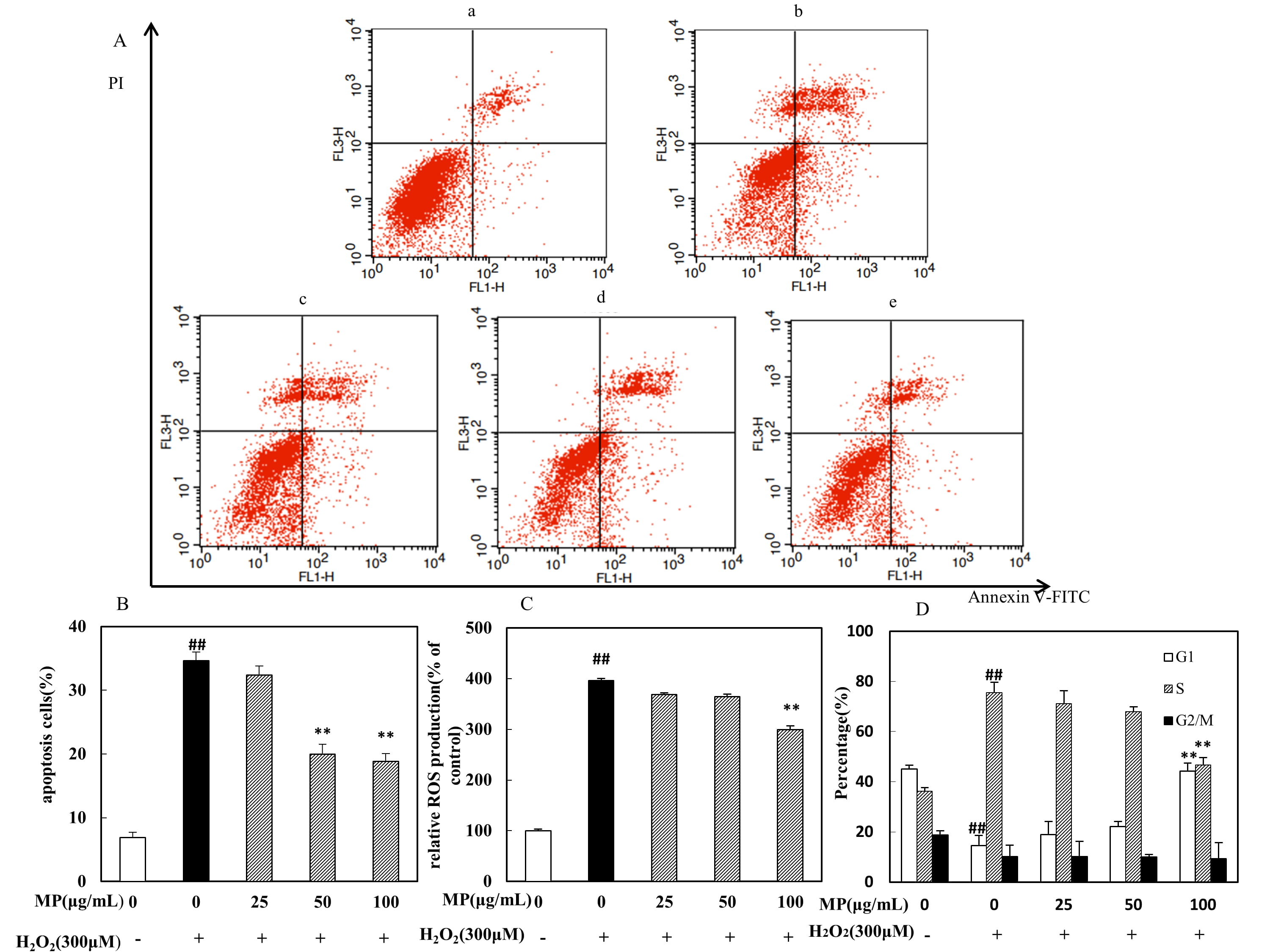 Fig. 4.
Fig. 4.
Effects of MP on SH-SY5Y cells under HO-induced
oxidative stress, as determined by flow cytometry (n = 3). (A,B)
Neuroprotective effects of MP in SH-SY5Y cells. (C) Intracellular ROS levels in
SH-SY5Y cells. (D) Cell cycle arrest in SH-SY5Y cells. All data are presented as
the means SDs from three independent experiments performed in triplicate.
p 0.01 versus the control and **p 0.01 versus
the HO group. Note: In Fig. 4A, (a) control group, (b) HO
group, (c) HO + MP (25 g/mL) group, (d) HO + MP
(50 g/mL) group, and (e) HO + MP (100 g/mL) group.
3.7 MP inhibits the HO-induced generation of ROS
The levels of intracellular ROS generated after HO treatment were
quantified by DCFH-DA fluorescence analysis and found to differ significantly
among groups (F = 85.69, p 0.01). The cells incubated with
300 M HO showed significantly higher intracellular ROS levels
than the untreated cells. As shown in Fig. 4C, cells pretreated with MP (100
g/mL) showed significantly lower intracellular ROS levels than
HO-treated cells without pretreatment (p 0.01),
indicating that MP was able to block (attenuate) the production of (and/or
scavenge) ROS.
3.8 MP relieves HO-induced cell cycle arrest
The periodic distribution of cells in the G1, S, and G2/M phases was found to
differ significantly among groups (F = 75.01, p 0.01;
F = 48.1, p 0.01; F = 1.675, p 0.01).
Compared to that in the control group (36.1%), the percentage of S-phase cells
in the HO group was significantly higher (75.43%), while those in
the low-, medium- and high-MP groups were not as high at 70.93%, 67.95%, and
46.46%, respectively; however, the percentage in the high-dose group was
significantly elevated (p 0.01). Therefore, a specific concentration
of MP was able to alleviate the S-phase cell cycle arrest caused by
HO (Fig. 4D). The influence of MP on the distribution of cells in
other cycle phases was likely related to the effects on cell cycle arrest in the
S phase.
3.9 Effect of MP on related protein expression
We investigated the ability of MP to modulate the activation of two signaling
proteins. The results showed that SH-SY5Y cells treated with 300 M
HO exhibited significant increases in the levels of P53 and cleaved
caspase 3, while cells pretreated with MP (50, 100 g/mL) showed
significantly reduced expression of these two proteins (p 0.01)
(Fig. 5).
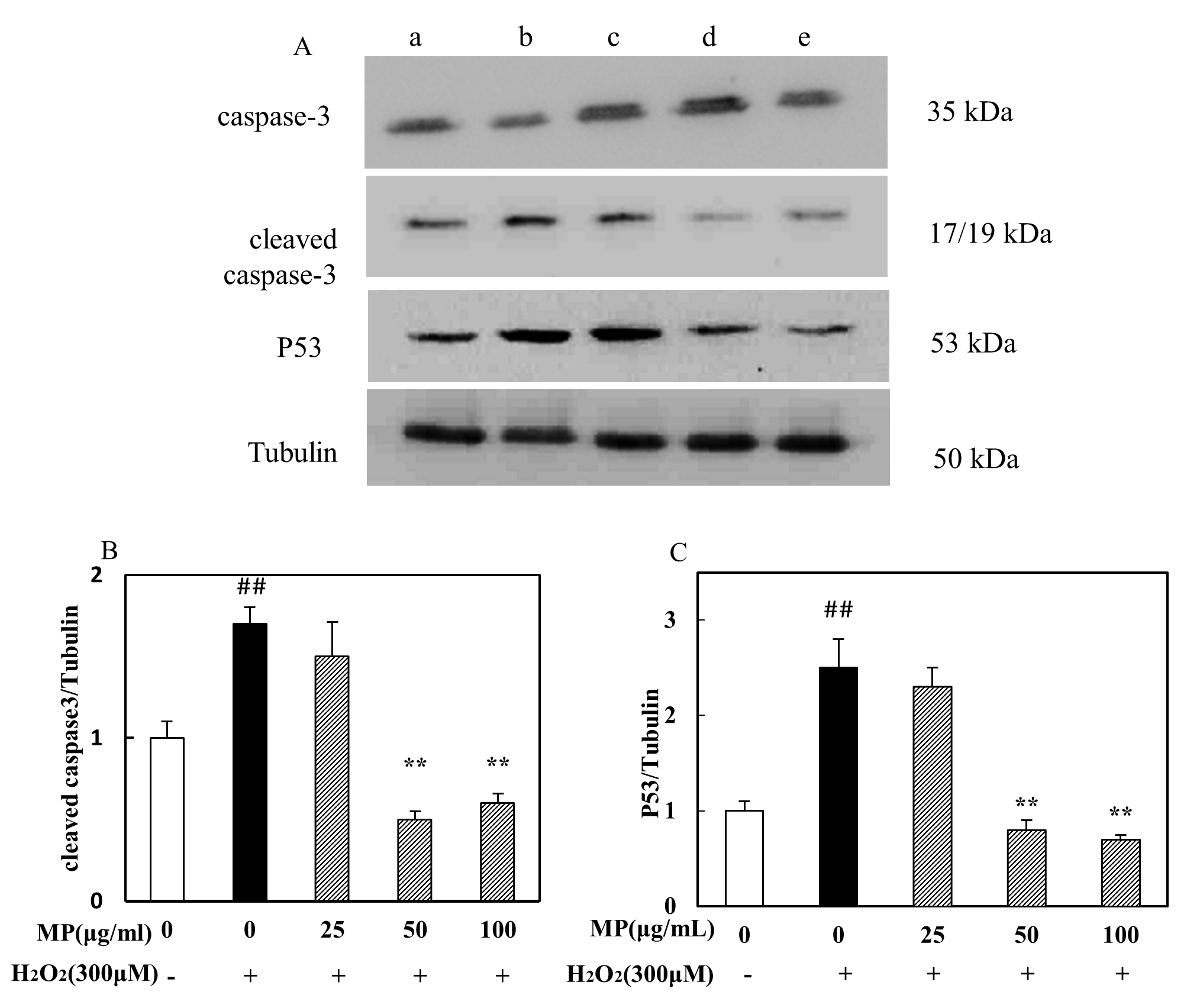 Fig. 5.
Fig. 5.
Effects of MP on cleaved caspase 3 and P53 protein expression in
HO-treated SH-SY5Y cells, as assessed by western blot analysis (n =
3). (A) The expression level of caspase-3, Cleaved caspase-3, P53. (B)
The relative expression of cleaved caspase3. (C) The relative expression of P53.
All data are presented as the means SDs from three independent
experiments performed in triplicate. p 0.01 versus the
control; *p 0.05, **p 0.01 versus the HO
group. Note: In Fig. 5A, (a) control group, (b) HO group, (c)
HO + MP (25 g/mL) group, (d) HO + MP (50
g/mL) group, and (e) HO + MP (100 g/mL) group.
4. Discussion
N-Benzyl-hexadecanamide is a relatively abundant macamide component in maca, and
chromatography showed that the N-benzyl-hexadecanamide content in the present
study was similar to that described in previous studies [34]. Research on
neurological diseases has shown that oxidative stress is associated with
neurological deterioration in many neurodegenerative disorders, including
Alzheimer’s disease and Parkinson’s disease [35]. Therefore, removing excess ROS,
inhibiting ROS production and preventing cell death induced by excessive
oxidative stress are effective means of protecting the nervous system. In recent
years, great efforts have been made to find safer and more efficient natural
antioxidants with neuroprotective potential. Polysaccharides, which are major
bioactive components in various plants, have received increasing attention due to
their antiviral, anticancer, anti-inflammatory, and antioxidant activities and
their neuroprotective effects [36].
In organisms, ROS with unpaired electrons and other ROS can be removed by GSH-Px
and other enzyme systems. The level of MDA, a lipid peroxide formed by oxidative
stress, directly reflects the degree of oxidation in the body [37]. In the
present study, MP significantly increased GSH-Px activity and reduced MDA levels.
According to the TEM results, the aggregation of neuronal chromatin in the
hippocampal dentate gyrus was significantly alleviated in the MP administration
group compared with the D-gal group, and the cell structure tended to be normal
in the MP group. Therefore, MP effectively alleviated D-gal-induced oxidative
stress in mice.
HO is a commonly used oxidative stress inducer [38]. We sought to
determine the protective effect of MP against HO-induced injury in
SH-SY5Y cells, as HO causes apoptosis in SH-SY5Y cells through
oxidative stress. We confirmed that treating cells with HO resulted
in a dose-dependent loss of cell viability. Pretreatment with different
concentrations of MP greatly increased cell viability, which was further
confirmed by morphological observations and an LDH release assay. LDH is present
in all cells in the human body, and when cells are injured by HO, it
is quickly released into the cell culture medium [39]. Thus, stronger LDH
activity in the culture supernatant indicates a greater number of apoptotic or
damaged cells. The results indicated that MP had a protective effect against
HO-induced cell damage.
Increases in the levels of intracellular free radicals or suppression of
intracellular antioxidant defense causes oxidative stress [7]. This study found
that pretreatment of SH-SY5Y cells with MP significantly reduced the apoptosis
rate. This result was consistent with inhibition of the activated fragments of
cleaved caspase 3 when cells were treated with MP. The expression of cleaved
caspase 3 is low in normal cells, but it is significantly increased in tissue
injury models. Intracellular ROS, mainly O, hydroxyl free radicals
(OH) and HO, are highly active molecules whose content can directly
reflect the degree of oxidative stress in cells. MP blocked (attenuated) the
production of (and/or scavenged) ROS, which indicates that MP has a certain
ability to scavenge free radicals and exert an antioxidant effect; these effects
may be related to its neuroprotective effect. MP can also alleviate cell cycle
arrest caused by HO. The cell cycle refers to the entire process of
cell proliferation. Protein synthesis occurs in each phase of the cell cycle,
while DNA synthesis occurs in the S phase, and RNA synthesis occurs in the G1, S,
and G2 phases. When cells are damaged by ROS, they repair DNA damage by inducing
cell cycle arrest in the S phase. The HO-treated cells showed
significantly more cell cycle progression than the untreated cells, which were
arrested in the S phase. An increase in the proportion of cells in the S phase
has been reported to be an indicator of HO-induced oxidation of
cellular targets [40], which is in accordance with the altered cellular redox
status detected in exposed cultured cells. Progression to the S phase may also be
an adaptive response to oxidative stress. The cell cycle arrest in the current
study was significantly attenuated by pretreatment with MP. Cell cycle arrest may
be associated with apoptosis, and HO has been reported to activate
caspase 3 [41]. As shown in Fig. 5, MP significantly suppressed the activity of
caspase 3. In addition, P53 protein expression was increased in
HO-treated cells, but MP significantly suppressed P53 expression.
Clarifying the underlying mechanism of nerve cell damage caused by oxidative
stress and elucidating the protective effect of MP on nerve cells will provide an
improved theoretical basis for research and further development of the MP
mechanism. In this study, we evaluated the antioxidant activity of MP in
vivo and in vitro. In summary, MP alleviated brain tissue injury, as in
MP-treated mice, the structure of the hippocampus was relatively normal, the
nuclei were round and large, the structural integrity of membranes was retained,
GSH-Px activity was increased, and MDA levels were reduced. The results also
showed that MP was able to block the production of ROS and ameliorate
HO-induced apoptosis in SH-SY5Y cells. The protective effects of MP
were attributable to the modulation of endogenous antioxidant enzymes and ROS
scavenging as well as to the modulation of endogenous apoptosis-related protein
expression. We have elucidated the underlying cellular mechanisms associated with
the protective effects of MP against cellular damage, which are as follows: (1)
increases in the activity of antioxidant enzymes, (2) blockade of ROS production,
(3) attenuation of cell cycle arrest, and (4) blockade of P53 and cleaved caspase
3 protein expression.
5. Conclusions
MP have protective effects on neuronal oxidative damage models in vivo and in
vitro, increased GSH-Px activity, reduced MDA levels, and attenuated the cell
damage induced by HO. Furthermore, MP protected neuronal cells from
oxidative stress through a mechanism including a decrease in LDH leakage and
reversal of HO-induced cell morphological damage. MP treatment
alleviated the HO-induced increases in ROS levels, inhibited
apoptosis, relieved cell cycle arrest, and downregulated cleaved caspase 3 and
P53 protein expression. MP is a novel antioxidant with neuroprotective effects.
Abbreviations
Maca, Lepidium meyenii Walp.; MDA, malondialdehyde; LDH, lactate
dehydrogenase; ROS, reactive oxygen species; DMSO, dimethyl sulfoxide.
Author contributions
YZhou and LZ—Experimental plan design, manuscript writing,
participation in experimental practical work, statistical analysis of data.
HL and WX—Preparation and quantification of MP extract.
JL—Animal experiment practice work. YZhang—Practical
operation of cell experiments. YL and CW—Review of
the experimental plan and key revisions.
Ethics approval and consent to participate
The Animal Ethics Committee of Changsha Medical University approved the animal
experiments in this study (approval number: 2019042).
Acknowledgment
Not applicable.
Funding
The work was supported by the Natural Science Foundation of Hunan Province
(2019JJ50694, 2020JJ5924, 2020JJ3060), the Administration of Traditional Chinese
Medicine of Hunan Province (2021075, 202098), the Hunan Provincial Education
Commission Foundation (17A026, 18A497, 19C0194, 20C0198), the National Natural
Science Foundation of China (81902308), the Provincial Clinical Medical
Technology Innovation Project of Hunan (2020SK53710, 2020SK53709), and the
Funding by young backbone teachers of Hunan province training program foundation
of Changsha Medical University (Hunan Education Bureau Notice 2021 No.29 -26).
Conflict of interest
The authors declare no conflict of interest.






 , Chenggong Wang 3,*
, Chenggong Wang 3,*