









 , Dhivya Shanmugarajan 2, Dinesh Babu 3, Arvind Kumar Goyal 4, Hasan Soliman Yusufoglu 5, Kora Rudraiah Sidhalinghamurthy 1
, Dhivya Shanmugarajan 2, Dinesh Babu 3, Arvind Kumar Goyal 4, Hasan Soliman Yusufoglu 5, Kora Rudraiah Sidhalinghamurthy 11 Department of Biochemistry, Bangalore University, Bengaluru, 560029 Karnataka, India
2 DBT-BIF Facility, Department of Biotechnology, Maharani Lakshmi Ammanni College for Women, 560012 Bangalore, India
3 Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, AB T6G 2E1, Canada
4 Centre for Bamboo Studies, Department of Biotechnology, Bodoland University, Kokrajhar, 783370 Assam, India
5 College of Pharmacy, Prince Sattam Bin Abdulaziz University, 16278 Al-Kharj, Saudi Arabia
Abstract
Introduction: Breast cancer is the most common type of cancer globally
and its treatment with many FDA-approved synthetic drugs manifests various side
effects. Alternatively, phytochemicals are natural reserves of novel drugs
for cancer therapy. Punica granatum commonly known as pomegranate is a
rich source of phytopharmaceuticals. Methods: The phytoconstituents
of Punica granatum leaves were profiled using GC-MS/MS in the present
work. Cytoscape-assisted network pharmacology of principal and prognostic
biomarkers, which are immunohistochemically tested in breast cancer tissue, was
carried out for the identification of protein target. Followed by, rigorous
virtual screening of 145 phytoconstituents against the three ER isoforms
(
Keywords
- Breast cancer
- Docking
- MD simulations
- GCMS/MS profiling
- Natural compounds
- Pomegranate
- Estrogen receptor
- ADMET
- 4-coumaric acid methyl ester
Drug discovery is a multifaceted and interdisciplinary endeavor that pursues a sequential process, begins with the discovery of a target and lead, followed by lead optimization and preclinical studies to define and verify the suitability of such lead agents through a number of predetermined guidelines for kick-starting clinical development [1, 2]. The high cost and time-consuming nature of these processes against an ailment demand a novel kind of cost-effective and less time-consuming, intensive in-silico approach [2, 3]. In-silico techniques employ bioinformatics tools to identify drug targets, to explore target protein structure for prediction of possible active sites to dock molecules with targets, rank ligands based on their binding affinities (i.e., energies), and optimize lead molecules [4]. Proficiency in calculating precise, significant, and diverse conformations of ligands within the target site make the molecular docking the most desired drug designing tool [5]. Hence, this tool can be used to understand the various interactions between phytochemicals and breast cancer targets or biomarkers. Despite the availability of modern tools and increasing expertise to identify a new anticancer molecule or lead, the potential of natural products (medicinal plants) and their compounds remains immense and undiscovered.
Many FDA-approved drugs such as paclitaxel and vinblastine (anticancer drugs), and quinine and artemisinin (antimalarial drugs) are plant-based. Therefore, combining the potential of phytomedicine with modern technology can help to develop more effective and economical drugs [6]. Phytochemicals from medicinal plants not only serve as drugs but also provide a lead for the development of new drugs. Punica granatum L., commonly known as pomegranate, is one such medicinal plant with a known reservoir of active biomolecules such as a series of compounds known as ellagitannins, and specifically punicalagin, the unique largest molecular weight polyphenol known to date [7]. A plethora of research reports describe the vasculo protective [8], antidiabetic, antioxidant [7], antiproliferative [9], antitumor [9, 10], anti-inflammatory [11], neuroprotective [12], and antimicrobial [13] properties of pomegranate along with its potential for treating asthma [14]. Previously, Usha et al. (2014) [15] have identified anticancer targets from pomegranate using an in-silico approach.
Breast cancer is the most common type of cancer worldwide, which affects 2.1
million women annually. In the year 2018, the World Health Organization estimated
that 627,000 deaths in women were caused due to breast cancer accounting for
nearly 15% of all cancer-related mortality among women
(https://www.who.int/cancer/detection/breastcancer/en/) [16]. The current study aims at identifying a lead molecule for breast cancer
from a natural source like pomegranate. Aromatase is an enzyme that converts C19
androgen to C18 estrogen that plays a vital role in breast cancer, which was
reported to be present in high concentrations in breast tumors [17]. Several
ellagitannins-derived compounds isolated from pomegranate were previously
reported to have potential anti-aromatase activity and inhibit
testosterone-induced breast cancer cell proliferation [18]. However, there is a
dearth of knowledge about the molecular basis and mechanism(s) of these compounds
against breast cancer. The aim of the present study is to design a drug-like
entity for breast cancer from Punica granatum L. phytochemicals. To be
specific, our study involves a gas chromatography-mass spectrometry
(GC-MS)/MS-based metabolite profiling of hydromethanolic extract of
Punica granatum L. leaves (HMPGL), to identify a lead candidate, which
can act as an antagonist and further verification against the ligand-binding
domains (LBDs) of the three different isoforms of human estrogen receptor (ER),
namely, ER alpha (ER
The leaves of Punica granatum L. plant were collected from Gandhi
Krishi Vignan Kendra (GKVK), Bengaluru, Karnataka, India. After authentication by
the plant taxonomist, a voucher specimen (BOT/mLAC/BMPG001) has been submitted to
Botany Department, Maharani Lakshmi Ammanni College for Women, Bengaluru,
Karnataka, India. The leaves were shade dried, grounded, and the powder was
subjected to soxhlation using 70% methanol as a solvent system. The extract was
lyophilized using freeze-drying technology under –55
The yield of hydromethanolic leaf extract was calculated using the following
equation: Yield (g/100 g of dry plant material) = (W1
Qualitative phytochemical analysis of HMPGL, for the detection of alkaloids, carbohydrates, flavonoids, glycosides, phenols, saponins, steroids, tannins [19], fatty acids, coumarins and resins [20] was carried out as per the standard procedures.
DPPH radical scavenging assay was used to determine the antioxidant activity of
HMPGL. Sample aliquots of different concentrations were incubated with 1.8 mL of
DPPH for 30 min at room temperature and the absorbance was measured at 540 nm
[19]. The ability of the HMPGL to scavenge DPPH radical was calculated using the
standard formula: DPPH free radical scavenging activity (%) = (Absorbance of
control (DPPH)–Absorbance of DPPH radical + HMPGL)/(Absorbance of control
(DPPH))
The total phenolic content of HMPGL was estimated using the Folin-Ciocalteu reagent as previously reported by Goyal et al. 2010 [19].
The gas chromatogram-mass spectrometer (Agilent Technologies 7890B GC and Triple
Quadrupole mass spectrometer 7000D series) was used for the analysis, having a
blend silica column, bundled with 5% biphenyl 95% dimethylpolysiloxane
(Elite-5MS), merged with a capillary column (30 m
The GC-MS/MS results were obtained to find the most probable lead molecules present in HMPGL. The metabolites thus obtained, if were available in PubChem, then their structures were downloaded from the PubChem compound database (https://www.ncbi.nlm.nih.gov/pccompound) [21] in 3D conformation and .sdf format. The structures of few metabolites, which were not available in PubChem, were obtained from NIST Library and sketched using ChemSketch. The standard protocols of BIOVIA Discovery Studio v3.5 (DS) software (Accelrys Software Inc., USA, 2012) was used for ligand preparation. This standard program was widely employed to prepare all the lead-like compounds to fix and resolve all different chemical properties such as different protonation states, ionization states, isomers, tautomers adding hydrogen, removing duplicates, and fixing bad valencies. This step is crucial because the receptor-ligand interactions have different protonation states; isomers and tautomers typically have different 3D geometries and binding characteristics.
This study is mainly centered on drug target proteins related to breast cancer;
therefore, a list of principal biomarkers and prognostic biomarkers that were
immunohistochemically tested in breast cancer tissue, are considered for the
study [22]. A network of these proteins was constructed using a string database
version 11.0 (https://string-db.org/) [23] and analyzed using Cytoscape, and the
protein with a medium number of edges was used as a protein target. Basic
sequence and structure analysis of the ER was conducted. The protein structures
of the different isoforms of ER structure such as ER
The 145 compounds identified through GC-MS/MS analysis were then subjected to a dual-step virtual screening process, in which, first, the compounds were screened for their pharmacokinetic activities, such as ADMET, which were predicted using ADMET Descriptors protocol in DS v3.5.
ADME studies are widely employed in drug discovery to optimize the properties to convert leads into drug molecules or medication. The lead compounds are mostly identified through virtual high-throughput screening approaches or by virtual screening. Drug discovery statistics reveal that around 50% of the drugs fail in the course of clinical trials due to nonstandard effectiveness (efficacy), which has low bioavailability due to poor intestinal absorption and undesirable metabolic stability [25]. The failure of drug-like candidates in the later stages of drug development proves very expensive. Therefore, to scale back the value and clinical failures of recent drug-like molecules, the lead compounds were screened within the initial stages for ADMET. Secondly, the ADMET screened compounds were checked for Lipinski’s rule of five (RO5) violations to ensure that the compounds have drug-likeness properties [5]. Finally, the remaining compounds were taken for further process.
The docking study was performed by identifying the binding site of drug-target
protein using the LibDock algorithm available in Discovery Studio 3.5. It uses
protein binding site features to direct docking. It finds polar and apolar probes
by placing a grid around the ligand 20 Å by 20 Å by 20 Å and extra
space of 5 Å in each direction [26]. The site volume and binding site sphere
will vary for all the three drug targets and were as follows; for ER
The molecular dynamic simulation (MDS) was ideally carried out to inspect the
stability and rationality of the binding patterns between the probable ligands
and the specific target protein. The finest interaction hits obtained from
receptor-ligand docking were used for MDS. In this experiment, top pose and elite
compound interaction with individual drug targets were simulated using the
GROMACS version 2016 package [3, 7]. 3ERD-2-propenoic acid, 3-(4-hydroxyphenyl)-,
methyl ester, 3OLS-2-propenoic acid, 3-(4-hydroxyphenyl)-, methyl ester and
2GPU-2-propenoic acid, 3-(4-hydroxyphenyl)-, methyl ester complex topologies were
prepared separately for the protein and the ligand. For the ligand molecular
topology, the coordinate files were generated by the PRODRG 2.5 server, followed
by solvating the receptor-ligand complex in the dodecahedral box with a minimal
distance of 1.0 nm. The whole protein-ligand complex and aqueous system are
maintained at neutral conditions using Na
The % yield of the HMPGL was found to be 1.44%. Qualitative phytochemical
analysis revealed the absence of resins and the presence of alkaloids,
carbohydrates, coumarins, flavonoids, glycosides, saponins, steroids, tannins,
fatty acids, phenols and terpenoids. The IC
Fig. 1 indicates the GC-MS/MS chromatogram of the HMPGL. After comparing mass spectra of the components with the NIST Library, 145 phytocompounds were identified and listed in Table 1. Out of the 145 compounds screened, 102 and 42 compounds (Table 2, Ref. [27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67]) were identified to have more than 80% match with NIST library, respectively. Among the screened compounds, 19 were identified as novel metabolites. The most prevailing compounds among the 145 metabolites are 9,12,15-octadecatrienoic acid, (Z,Z,Z)- (linolenic acid), 1,2,3-benzenetriol(pyrogallol), n-hexadecanoic acid (palmitic acid), maltol, 5-hydroxymethylfurfural, 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl-, undecanol-5 and 3-furaldehyde. These phytocompounds are different fatty acids, terpenes, heterocyclic compounds, flavonoids, pyrrolidines, sesquiterpenoids and phenols. The mass spectrum corresponding to the structure of 2-propenoic acid, 3-(4 hydroxyphenyl)-, methyl ester (also known as 4-coumaric acid methyl ester), which exhibits the highest binding affinity with the breast cancer receptors considered in the current investigation, was represented in Fig. 2.
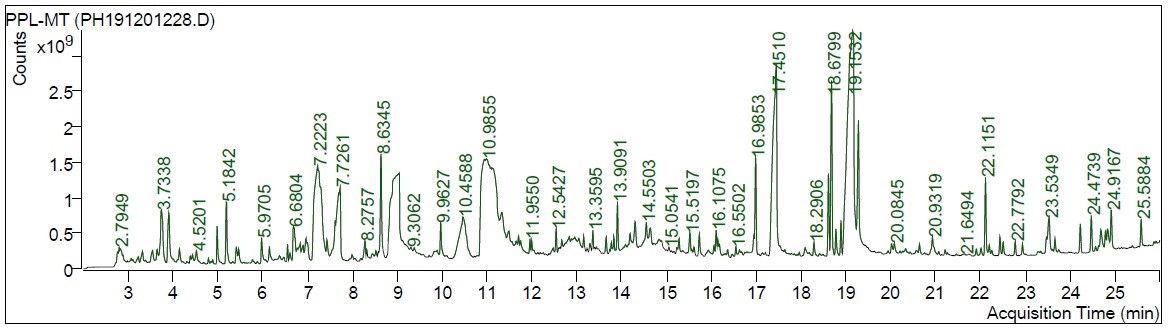 Fig. 1.
Fig. 1.GC-MS/MS chromatogram of the hydromethanolic extract of Punica granatum L. leaves.
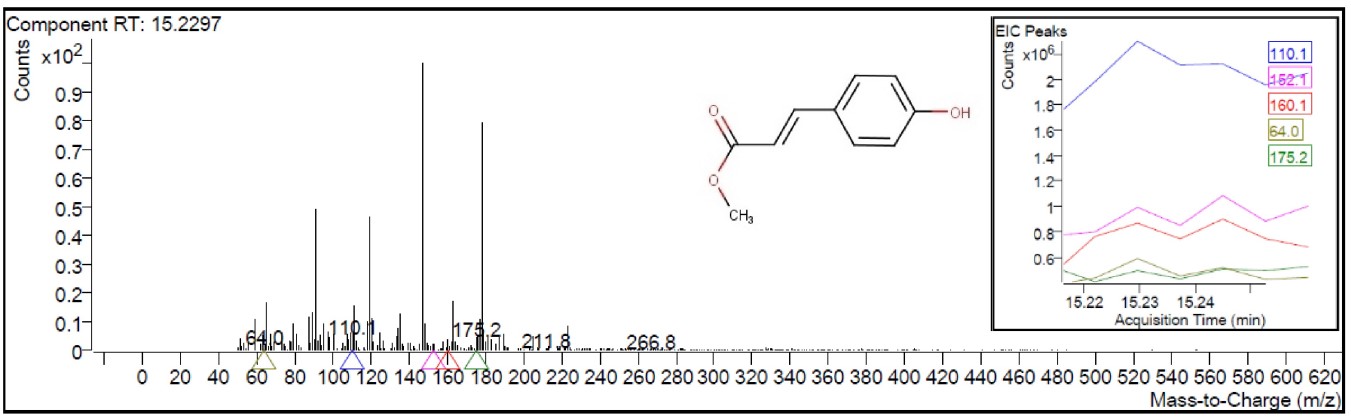 Fig. 2.
Fig. 2.Mass spectrum of the component (2-propenoic acid,
3-(4-hydroxyphenyl)-, methyl ester) at retention time (RT) 15.2297 (represented
in the insert) with a y-axis scale from 0 to 0.9
| S. No | Retention time | Compound name | Chemical formula | Area | MW (g/mol) |
| 1 | 11.6496 | (3-Nitrophenyl) methanol, n-propyl ether | C |
728600034 | 195.21 |
| 2 | 12.1305 | (4H)4a,5,6,7,8,8a-Hexahydrobenzopyran-5-one-3-carboxamide,2-(2-hydroxypentyl)-8a-methoxy-4a-methyl | C |
28662212 | 325.40 |
| 3 | 24.6876 | Alpha-Tocospiro A | C |
1087514521 | 462.70 |
| 4 | 24.8403 | Alpha-Tocospiro B | C |
909893093 | 462.00 |
| 5 | 25.9167 | Gamma-Tocopherol | C |
77931713 | 416.70 |
| 6 | 6.9628 | [1,2,3,4]Tetrazolo[1,5-b][1,2,4]triazine,5,6,7,8-tetrahydro- | C |
1406037645 | 126.12 |
| 7 | 12.5427 | 1,1,4,5,6-Pentamethyl-2,3-dihydro-1H-indene | C |
470619079 | 188.31 |
| 8 | 11.4893 | 1,2,3,6-Tetrahydropyridine, 1-methyl-5-phenyl- | C |
2272849467 | 173.25 |
| 9 | 10.9855 | 1,2,3-Benzenetriol | C |
29449616456 | 126.11 |
| 10* | 9.8863 | 1,8-Dioxaspiro[4.5]decan-2-one, 4-(2-aminothiazol-4-yl)-7,7-dimethyl- | C |
392180530 | 282.00 |
| 11* | 3.3827 | 1H-Cyclopropa[3,4]benz[1,2-e]azulene- 4a,5,7b,9,9a(1aH)-pentol, 3-[(acetyloxy)methyl]-1b,4,5,7a,8a9-hexahydro-1,1,6,8-tetramethyl-,5,9,9a-triacetate, [1aR-(1a.alpha., 1b.beta.,4a.beta.,5.beta.,7a.alpha.,7b.alpha.,8.alpha.,9.beta.,9a.alpha) | C |
50122079 | 534.59 |
| 12 | 8.3139 | 1H-Imidazole, 1-methyl- | C |
58733639 | 82.10 |
| 13 | 12.9549 | 1H-Inden-1-one, 2,3-dihydro-3,3,4,6-tetramethyl- | C |
920997462 | 188.26 |
| 14 | 11.7031 | 1H-Inden-1-one, 2,3-dihydro-3,3,5,6-tetramethyl | C |
548026939 | 188.26 |
| 15 | 16.1457 | 2-(3-Isopropyl-4-methyl-pent-3-en-1-ynyl)-2-methyl-cyclobutanone | C |
383367099 | 204.31 |
| 16* | 12.8862 | 2-(3-Isopropyl-4-methyl-pent-3-en-1-ynyl)-2-methyl-pent-3-en-1-ynyl-2-methylcyclobutanone | C |
264669072 | 204.00 |
| 17 | 12.8328 | 2(4H)-Benzofuranone, 5,6,7,7a-tetrahydro-4,4,7a-trimethyl-,(R)- | C |
828417676 | 180.24 |
| 18 | 3.5354 | 2(5H)-Furanone | C |
532754993 | 84.07 |
| 4.4285 | 287429887 | ||||
| 19 | 3.3140 | 2,2’-Bioxirane | C |
455025294 | 86.09 |
| 20 | 6.4666 | 2,4(1H,3H)-Pyrimidinedione, 5-hydroxy- | C |
90374515 | 128.09 |
| 21* | 2.7949 | 2,4,6,8,10-Tetradecapentaenoic acid, 9a- decahydro-4a,7b-dihydroxy-3-(hydroxymethyl)- cyclopropa[3,4]benz[1,2-e]azulen-9-yl ester, (1a.alpha.,1b.beta.,4a.beta.,7a.alpha.,7b.alpha, 8.alpha, 9.beta,9a.alpha.) | C |
1729589562 | 606.00 |
| 22 | 5.1842 | 2,4-Dihydroxy-2,5-dimethyl-3(2H)-furan-3-one | C |
1800447816 | 144.12 |
| 23 | 12.474 | 2,4-Di-tert-butylphenol | C |
162473760 | 206.32 |
| 24 | 13.1534 | 2,5-Dimethoxy-4-ethylamphetamine | C |
258752481 | 223.31 |
| 25 | 6.6040 | 2,5-Furandicarboxaldehyde | C |
194264167 | 124.09 |
| 26* | 3.0926 | 2-[4-Chloro-2-nitrophenyl]-1-(2-diethyaminoethyl)-3-formyl-1H-indole | C |
55662163 | 399 |
| 27 | 4.5201 | 2-Amino-4-methyl-oxazole | C |
518087636 | 98.10 |
| 28 | 14.1915 | 2-Cyclohexen-1-one, 4-(3-hydroxy-1-butenyl)-3,5,5-trimethyl- | C |
560078241 | 208.30 |
| 29 | 15.6113 | 2-Dodecen-1-yl(-)succinic anhydride | C |
133093417 | 266.38 |
| 30 | 4.9781 | 2-Furancarboxaldehyde, 5-methyl- | C |
898805469 | 110.11 |
| 31 | 6.9017 | 2-Furancarboxylic acid | C |
661021535 | 112.08 |
| 32 | 3.9018 | 2-Furanmethanol | C |
2300435655 | 98.10 |
| 33 | 6.1460 | 2-Heptanol, 5-ethyl- | C |
358696294 | 144.25 |
| 34 | 4.7873 | 2-Hexene, 4-methyl-, (E)- | C |
92798753 | 98.19 |
| 35 | 9.3062 | 2H-Pyran, 2-(bromomethyl)tetrahydro- | C |
39730800 | 179.05 |
| 36 | 5.4056 | 2H-Pyran-2,6(3H)-dione | C |
511684838 | 112.08 |
| 37 | 6.3674 | 2-Methylthio-2,3-dimethylbutane | C |
191303503 | 132.27 |
| 38 | 15.0541 | 2-Propanone, 1-hydroxy-3-(4-hydroxy-3-methoxyphenyl)- | C |
76991990 | 196.20 |
| 39 | 15.2297 | 2-Propenoic acid, 3-(4-hydroxyphenyl)-, methyl ester or 4- Coumaric acid methyl ester | C |
79623370 | 178.18 |
| 40 | 11.7489 | 3,3-Dimethyl-4-phenyl-4-penten-2-one | C |
815120462 | 188.26 |
| 41 | 13.2984 | 3a,7-Methano-3aH-cyclopentacyclooctene, decahyydro-1,1,7-trimethyl-,[3aS-(3a.alpha.,7.alpha.,9a.beta.)]- | C |
99198977 | 191.00 |
| 42 | 8.1918 | 3-Amino-N-(pyridin-4-yl)propanamide | C |
46243181 | 165.19 |
| 43* | 4.8789 | 3-Aminopyrazine 1-oxide | C |
69030002 | 111.00 |
| 44 | 14.5503 | 3-Chloropropionic acid, heptadecyl ester | C |
971108451 | 347.00 |
| 45 | 3.7338 | 3-Furaldehyde | C |
2958433873 | 96.08 |
| 6.6804 | 1614869390 | ||||
| 46 | 3.6422 | 3-Furanmethanol | C |
505585085 | 98.10 |
| 47 | 14.8327 | 3-Furoic acid, benzyldimethylsilyl ester | C |
203867247 | 260.36 |
| 48 | 15.9319 | 3H-Cyclodeca[b]furan-2-one, 4,9-dihydroxy-6-methyl-3,10,dimethylene-3a,4,7,8,9,10,11,11a-octahydro- | C |
257866769 | 264.32 |
| 49 | 16.1075 | 3-Hexadecyne | C |
586051279 | 222.41 |
| 50 | 16.3670 | 3-Octadecyne | C |
162514947 | 250.50 |
| 51 | 12.1840 | 4-(2,6,6-Trimethylcyclohexa-1,3-dienyl)but-3-en-2-one | C |
19815134 | 190.28 |
| 52 | 14.2984 | 4,4,5,8-Tetramethylchroman-2-ol | C |
1110933501 | 206.28 |
| 53 | 7.7261 | 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl- | C |
7618503933 | 144.12 |
| 54 | 8.2757 | 4H-Pyran-4-one, 3,5-dihydroxy-2-methyl- | C |
434272719 | 142.11 |
| 55 | 7.4284 | 4H-Pyran-4-one, 5-hydroxy-2-methyl- | C |
802533562 | 126.11 |
| 56 | 9.9627 | 4-Hydroxy-2-methylacetophenone | C |
1075532343 | 150.17 |
| 57 | 17.8479 | 5-(2-Morpholino-1-thiophen-2-yl-vinyl)-1,2,4-thiadiazole | C |
39520603 | 279.40 |
| 58 | 15.7335 | 5,5,8a-Trimethyl-3,5,6,7,8,8a-hexahydro-2H-chromene | C |
458070336 | 180.29 |
| 59* | 25.39 | 5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one, chloro-1,1a,1b,2,3,4,4a,7a,7b,8,9,9a- tetramethyl-, [1aR-(1a.aplha.,7b.alpha.,8.alpha.9.beta.,9a.alpha)] | C |
53025185 | 489 |
| 60* | 18.4891 | 5H-Cyclopropa[3,4]benz[1,2-e]azulen-5-one,9,9a-bis(acetyloxy)-3-[(acetoxy)methyl]-1,1a,1b,2,3,4,4a,7a,7b,8,9,9a-dodecahydro-2,3,4a,7b-tetrahydroxy-1,,6,8-tetramethyl-,[1ar-(1a.alpha.,1b.beya,2.alpha.,3.alpha.,4a.beta.,7a.alpha.,7b.alpha.,8.alpha.,9.beta.,9a.alpha) | C |
62085503 | 446.00 |
| 61 | 9.0390 | 5-Hydroxymethylfurfural | C |
12338767206 | 126.11 |
| 62 | 13.6572 | 5-Isopropenyl-2-methylcyclopent-1-enecarboxaldehyde | C |
344352899 | 150.22 |
| 63 | 15.5197 | 6-Hydroxy-4,4,7a-trimethyl-5,6,7,7a-tetrahydrobenzofuran-2(4H)-one | C |
607605365 | 196.24 |
| 64 | 18.4128 | 7,8,9,10-Tetrahydro-6(5H)-phenanthridinone | C |
83259442 | 199.25 |
| 65 | 25.8709 | 7,8-Epoxylanostan-11-ol, 3-acetoxy- | C |
53150204 | 502.80 |
| 66 | 16.5502 | 7-Heptadecyne, 17-chloro- | C |
206699451 | 270.90 |
| 67 | 20.5654 | 8,11,14-Eicosatrienoic acid, methyl ester,(Z,Z,Z)- | C |
114885329 | 320.50 |
| 68 | 19.1532 | 9,12,15-Octadecatrienoic acid, (Z,Z,Z)- | C |
30390789406 | 278.40 |
| 69 | 16.4739 | 9,12,15-Octadecatrienoic acid, 2,3-dihydroxypropyl, ester (Z,Z,Z)- | C |
80048369 | 352.50 |
| 23.5395 | 2227344617 | ||||
| 70 | 22.5120 | 9,12,15-Octadecatrienoic acid, ethyl ester, (Z,Z,Z)- | C |
364409437 | 306.50 |
| 71 | 18.6799 | 9,12,15-Octadecatrienoic acid, methyl ester,(Z,Z,Z) | C |
5071324434 | 292 |
| 72 | 18.6112 | 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | C |
2018420993 | 294.50 |
| 73 | 18.0922 | 9-Hexadecenoic acid | C |
510568704 | 254.41 |
| 74* | 23.2907 | Acetic acid, 17-acetoxy-3-hydroxyimino-4,4,13-hexadecahydrocyclopenta[a]phenanthren-10-ylmetyl ester | C |
90416430 | 433.00 |
| 75 | 11.3290 | Azetidine, 1,1’-methylenebis[2-methyl- | C |
4498160447 | 154.25 |
| 76 | 11.9550 | Benzaldehyde, 4-ethyl- | C |
327861429 | 134.17 |
| 77 | 8.6345 | Benzofuran, 2,3-dihydro- | C |
2678521082 | 120.15 |
| 78 | 16.7869 | Benzoic acid, 3,4,5-trihydroxy-, methyl ester | C |
168553503 | 184.15 |
| 79 | 12.6649 | Benzoic acid, 4-ethoxy-, ethyl ester | C |
183455032 | 194.23 |
| 80 | 9.3749 | Benzoic acid, 4-methyl- | C |
28589154 | 136.15 |
| 81 | 5.9705 | Benzyl alcohol | C |
821031061 | 108.14 |
| 82 | 7.9857 | beta.-1,5-Dibenzoyl-2-deoxy-ribofuranose | C |
372597103 | 342.3 |
| 83 | 13.8251 | Bicyclo[2.2.1]hept-2-ene, 1,7,7-trimethyl- | C |
275857190 | 136.23 |
| 84 | 4.1384 | But-1-ene-3-yne, 1-ethoxy- | C |
568495293 | 96.13 |
| 85 | 13.4358 | Butyrovanillone | C |
22430637 | 194.22 |
| 86* | 24.2296/25.5884 | Carbonic acid, eicosyl vinyl ester | C |
702086983/675042558 | 368.60 |
| 87 | 8.436 | Catechol | C |
104187122 | 110.11 |
| 88 | 23.3517 | cis-5,8,11,14-Eicosatetraenoic acid, picolinyl ester | C |
75303340 | 395.60 |
| 89* | 8.52 | Cyclobutane, 1,2:3,4-di-O-ethylboranediyl- | C |
117519035 | 195.82 |
| 90 | 5.5735 | Cyclohexane, 1,3,5-trimethyl-2-octadecyl- | C |
43383586 | 126.10 |
| 91* | 16.1762 | Cyclopropane, 1 heptaonyl-3-methylene-2-pentyl- | C |
301623110 | 378.7 |
| 92* | 17.138 | Cyclopropanepctanoic acid, 2-[(-pentylcyclopropyl)methyl]-,methyl ester, trans, trans- | C |
39520603 | 322.00 |
| 93* | 5.7644 | Cyclopropylamine, N-isobutylidene- | C |
133377747 | 111.18 |
| 94* | 13.0465 | Dihydroxanthin | C |
492844334 | 280.00 |
| 95 | 14.0846 | Doconexent | C |
146857459 | 328.5 |
| 96* | 21.2677 | Doconexent, TBDMS derivative | C |
31973570 | 442.70 |
| 97 | 11.9931 | E-11,13-Tetradecadien-1-ol | C |
430872421 | 210.36 |
| 98 | 20.9319 | Eicosanoic acid | C |
795269485 | 312.50 |
| 99 | 4.3827 | Ethanone, 1-(2-furanyl)- | C |
165889275 | 110.11 |
| 100 | 20.3364 | Ethyl 5,8,11,14,17-icosapentaenoate | C |
252266022 | 330.50 |
| 101* | 24.5731/24.7945 | Furan, 2,5-bis(3,4-dimethoxyphenyl)tetrahydro-3,4-dimethyl-,[2R-(2.alpha.,3.beta.,4.beta.,5.alpha.)] | C |
101227511/485260974 | 372.00 |
| 102 | 4.3064 | Furan-2-ylmethyl palmitate | C |
36491320 | 336.50 |
| 103 | 21.4662 | Heneicosanoic acid, methyl ester | C |
34798280 | 340.60 |
| 104 | 22.0158/22.7792 | Heptacosane | C |
291521130/296780030 | 380.70 |
| 105 | 22.199 | Hexadecanoic acid, 1-(hydroxymethyl)-1,2-ethanediyl ester | C |
317213707 | 568.90 |
| 106 | 17.9624 | Hexadecanoic acid, 15-methyl-, methyl ester | C |
82387516 | 284.50 |
| 107* | 22.1151 | Hexadecanoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester | C |
2065095285 | 330.00 |
| 108 | 16.9853 | Hexadecanoic acid, methyl ester | C |
3020358912 | 270.50 |
| 109 | 19.9395 | Indane-4-carbonitrile, 2,2,5,7-tetramethyl-1-oxo | C |
71510002 | 213.27 |
| 110 | 9.7108 | Indole | C |
20622699 | 117.15 |
| 111 | 22.9395 | Licarin A | C |
224689115 | 326.40 |
| 112 | 7.2223 | Maltol | C |
14939408876 | 126.11 |
| 113 | 15.3671 | Mannofuranoside, 1-allyl-2,3-5,6-tetra-O-acetyl- | C |
25787993 | 372.40 |
| 114 | 13.7717 | Megastigmatrienone | C |
240529958 | 190.00 |
| 115* | 16.0617 | Methanone, (2,6-dimethyl-4-morpholyl)(9H-xanthen-9-yl)- | C |
238398621 | 323.40 |
| 116 | 5.3293 | Methanone, [4-(2-furfurylthio)-3-nitrophenyl](morpholino) | C |
76240602 | 348.40 |
| 117 | 23.7487 | Methyl 18-methylicosanoate | C |
148572467 | 340.60 |
| 118 | 20.6418 | Methyl 18-methylnonadecanoate | C |
237808761 | 326.60 |
| 119 | 18.8937/22.2525 | Methyl stearate | C |
831716164/166816879 | 298.50 |
| 120 | 5.459 | N-Butyl-tert-butylamine | C |
514116590 | 129.24 |
| 121 | 17.451 | n-Hexadecanoic acid | C |
16562951600 | 256.42 |
| 122 | 16.6266 | Nootkaton-11,12-epoxide | C |
174277673 | 234.33 |
| 123 | 17.6418/21.0769 | Octadecanal, 2-bromo- | C |
305383125/46483241 | 347.4 |
| 124 | 21.2143 | Octadecane, 3-ethyl-5-(2-ethylbutyl)- | C |
94740807 | 364.00 |
| 125 | 5.8789/21.909 | Octadecanedioic acid | C |
34874865/234355861 | 314.5 |
| 126 | 19.283 | Octadecanoic acid | C |
3813012247 | 284.5 |
| 127 | 23.6647 | Octadecanoic acid, 2,3-dihydroxypropyl ester | C |
433327098 | 358.6 |
| 128 | 20.4357 | Oxiranedodecanoic acid, 3-octyl-, cis- | C |
15785811 | 354.6 |
| 129 | 22.4357 | Phthalic acid, octyl tridec-2-yn-1-yl ester | C |
486477919 | 456.7 |
| 130 | 18.7792 | Phytol | C |
776137812 | 296.50 |
| 131 | 20.7563 | Pregnane-7,8,9,11,20-pentaol-18-oic acid,7,11-diacetate-18,20-lactone | C |
49740067 | 478.50 |
| 132 | 3.2224 | Propane, 2-fluoro- | C |
193906460 | 62.09 |
| 133 | 6.5506 | Pyridazine | C |
251310771 | 80.09 |
| 134 | 24.4739 | Squalene | C |
923530021 | 410.70 |
| 135 | 24.9167 | Sulfurous acid, hexyl pentadecyl ester | C |
1241617690 | 376.60 |
| 136 | 13.9091/13.3595 | syn-Tricyclo[5.1.0.0(2,4)]oct-5-ene, 3,3,5,6,8,8-hexamethyl- | C |
1025801501/321999626 | 190.32 |
| 137 | 15.2831/18.2906 | Tetradecanoic acid | C |
324806663/394717404 | 228.37 |
| 138 | 20.0234 | Tetradecanoic acid, 2-hydroxy- | C |
269881507 | 244.37 |
| 139 | 6.833 | Thymine | C |
1117152651 | 126.11 |
| 140 | 14.6419 | Tibolone | C |
783232577 | 312.40 |
| 141 | 3.0621 | Trifluoromethyltrimethylsilane | C |
112646880 | 142.19 |
| 142 | 16.3136 | Undecanoic acid, 2-nonyl-, methyl ester | C |
109936717 | 326.60 |
| 143 | 10.4588 | Undecanol-5 | C |
5922534581 | 172.31 |
| 144 | 20.2067/21.6494 | Ursodeoxycholic acid | C |
34678472/97533782 | 396.60 |
| 145 | 20.0845 | Z-(13,14-Epoxy)tetradec-11-en-1-ol acetate | C |
320849197 | 268.39 |
| S. No | Compound | Bioactivity and references |
| 1 | 2-Furanmethanol or Furfuryl alcohol | Flavoring agent [27] and antioxidant [28] |
| 2 | 2-Furancarboxaldehyde, 5-methyl- | Food additive and used for fragrance [29] |
| 3 | 2,4-Dihydroxy-2,5-dimethyl-3(2H)-furan-3-one | Flavoring agent [30] |
| 4 | 2H-Pyran-2,6(3H)-dione | Antiallergic [31], analgesic, mild sedative, soporific, fungicide, fungistatic, antihypoxic, spasmolytic, and muscle relaxant activities [32] |
| 5 | Benzyl alcohol | Antimicrobial activity [33] |
| 6 | 2-Furancarboxylic acid | NR* |
| 7 | Maltol | Antitumor [34] and antinephrotoxicity activities [35]; alleviates hepatic fibrosis [36] |
| 8 | 4H-Pyran-4-one, 5-hydroxy-2-methyl- | NR* |
| 9 | 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl- | Alleviates male reproductive toxicity [37]; stimulates the autonomic nerve activity [38]; antiproliferative and pro-apoptotic [39]; antibiotic activities [40] |
| 10 | 4H-Pyran-4-one, 3,5-dihydroxy-2-methyl- or 5-Hydroxymaltol | Nutrient [41] |
| 11 | Benzofuran, 2,3-dihydro- | Antiarrhythmic, spasmolytic, and antiviral activities [42] |
| 12 | 5-Hydroxymethylfurfural | Antiinflammatory, antioxidant and antiproliferative activities [43] |
| 13 | 1,2,3-Benzenetriol or Pyrogallol | Antitumor, cytotoxic and antiproliferative activities [44] |
| 14 | Syn-tricyclo(5.1.0.0(2,4))oct-5-ene, 3,3,5,6,8,8-hexamethyl- | NR* |
| 15 | Megastigmatrienone | Flavoring agent [45] |
| 16 | 2-(3-Isopropyl-4-methyl-pent-3-en-1-ynyl)-2-methyl-cyclobutanone | NR* |
| 17 | n-Hexadecanoic acid or Palmitic acid | Antifungal [46] and antioxidant activities [47] |
| 18 | 9,12-Octadecadienoic acid (Z,Z)- or Linoleic acid | Antimicrobial activity [47] |
| 19 | 9,12,15-Octadecatrienoic acid, (Z,Z,Z)- or Alpha-linolenic acid | Regulates butyrylcholinesterase [48], antioxidant and antimicrobial [47]; antiinflammatory activities, antiacne, antiandrogenic, antiarthritic, antibacterial and anticandidal, anticancer, anticoronary, antieczemic, antihistaminic, hepatoprotective, hypocholesterolemic, insectifuge, nematicide, 5-alpha reductase inhibitor and cancer preventive activities [40] |
| 20 | Octadecanoic acid | Antimicrobial [40] |
| 21 | 3-Furaldehyde | Inhibits polyphenol oxidase 2, phenolase, cresolase and tyrosinase [49] |
| 22 | 1H-Inden-1-one, 2,3-dihydro-3,3,5,6-tetramethyl- | Antifungal activity [50] |
| 23 | 1,1,4,5,6-Pentamethyl-2,3-dihydro-1H-indene | NR* |
| 24 | Benzoic acid, 4-ethoxy-, ethyl ester | Antimicrobial activity [40] |
| 25 | 4,4,5,8-Tetramethylchroman-2-ol | Possible treatment against oligospermy and oliguria [45] |
| 26 | 3-Hexadecyne | Antiandrogenic agent [51] |
| 27 | Hexadecanoic acid, methyl ester or Methyl palmitate | Prevents Kupffer cell activation [52]; antiinflammatory [53, 54] and antifibrotic activities [53] |
| 28 | 9-Hexadecenoic acid | Regulates lipogenesis, desaturation, and β-oxidation in bovine adipocytes [55] |
| 29 | Phytol | Antioxidant [47], anticancer, antiinflammatory, antidiuretic, antimicrobial activities [40]; Fragrance [56] Antinociceptive activities [57] |
| 30 | Methyl stearate | Nutrient, membrane stabilizer and energy source [58] |
| 31 | Eicosanoic acid/Arachidic acid | Nutrient, membrane stabilizer and energy source [59] |
| 32 | Phthalic acid, octyl tridec-2-yn-1-yl ester | Antiplatelet activity [60] |
| 33 | Heptacosane | NR* |
| 34 | Squalene | Antitumor and anticancer effects [breast, colon, lung and ovarian cancer], hypocholesterolemic activity, reduces skin damage caused by UV radiation, cardioprotective effect and , detoxifying agent [61] |
| 35 | Alpha-Tocospiro A | NR* |
| 36 | Alpha-Tocospiro B | Cytotoxic [62] and α-Glucosidase inhibiting activities [63] |
| 37 | Sulfurous acid, hexyl pentadecyl ester | NR* |
| 38 | Carbonic acid, eicosyl vinyl ester | NR* |
| 39 | 4-Hydroxy-2-methylacetophenone | Acaricidal activity [64] |
| 40 | 2,5-Furandicarboxaldehyde | Insulin receptor partial antagonist [65] |
| 41 | 4H-Pyran-4-one, 5-hydroxy-2-(hydroxymethyl)-methyl- or Kojic acid | Skin-lightening agent [inhibits tyrosinase] antioxidant, antidiabetic, anticancer, antiinflammatory, antimicrobial, antiproliferative, antiparasitic, antiviral, antitumor, antispeck, pesticidal,insecticidal, radio protective properties [66] |
| 42 | Hexadecanoic acid, 2-hydroxy-1- (hydroxymethyl)ethyl ester | Food additive [67] |
| Or 2-Palmitoylglycerol | ||
| NR*, no activity reported. | ||
The protein network of text mined and experimental interactions; and interaction
score with 0.9 confidence, consisted of 81 number of nodes and 234 edges with a
P-value of
With the advent of new technology and computational tools, there is a massive
increase in the deposition of protein structures in PDB. This consequently
creates an increased level of difficulty in selecting an optimal PDB entry for
docking. X-ray crystallographic structures of ER
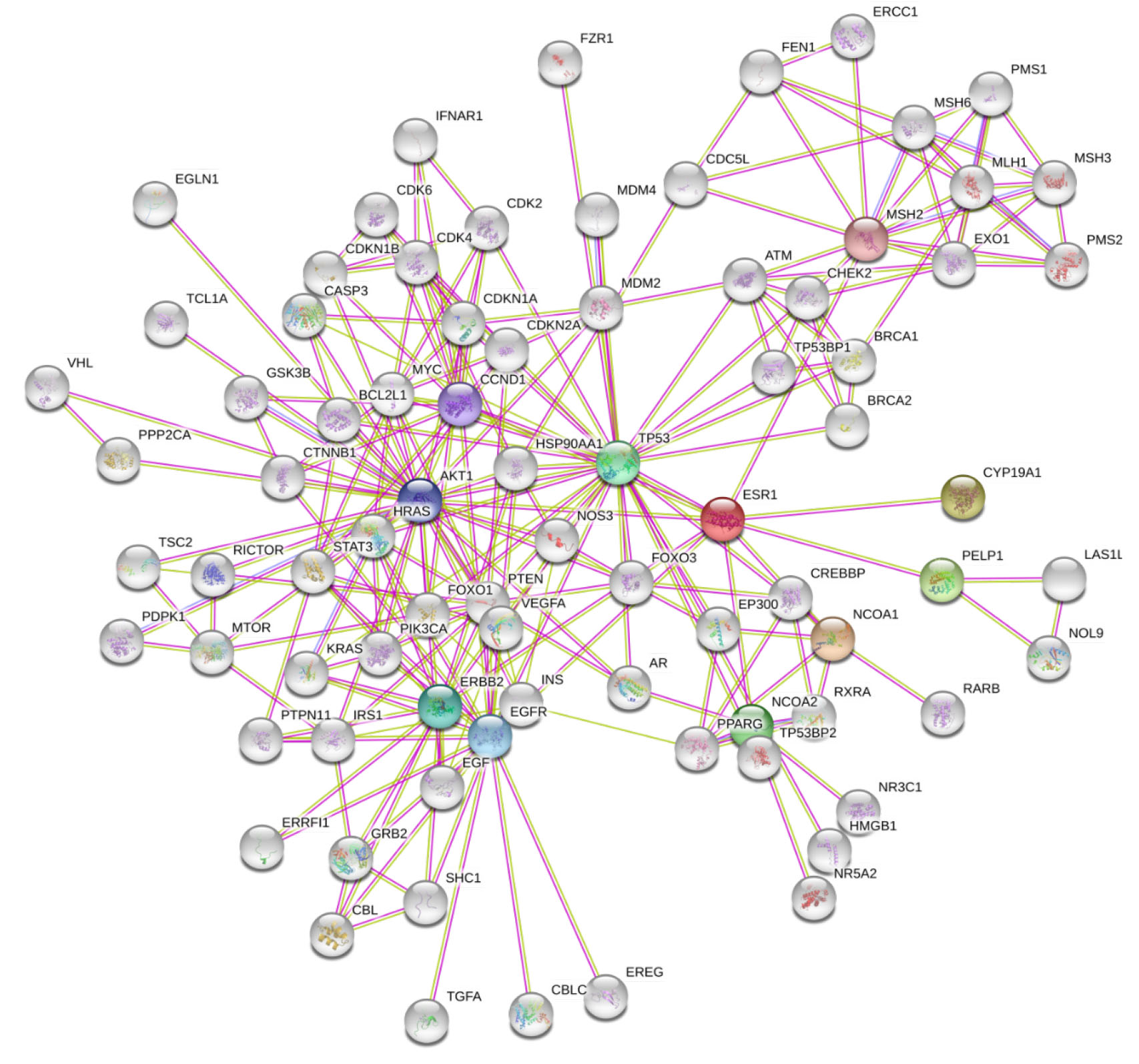 Fig. 3.
Fig. 3.Protein interaction network of principal and prognostic biomarkers of breast cancer.
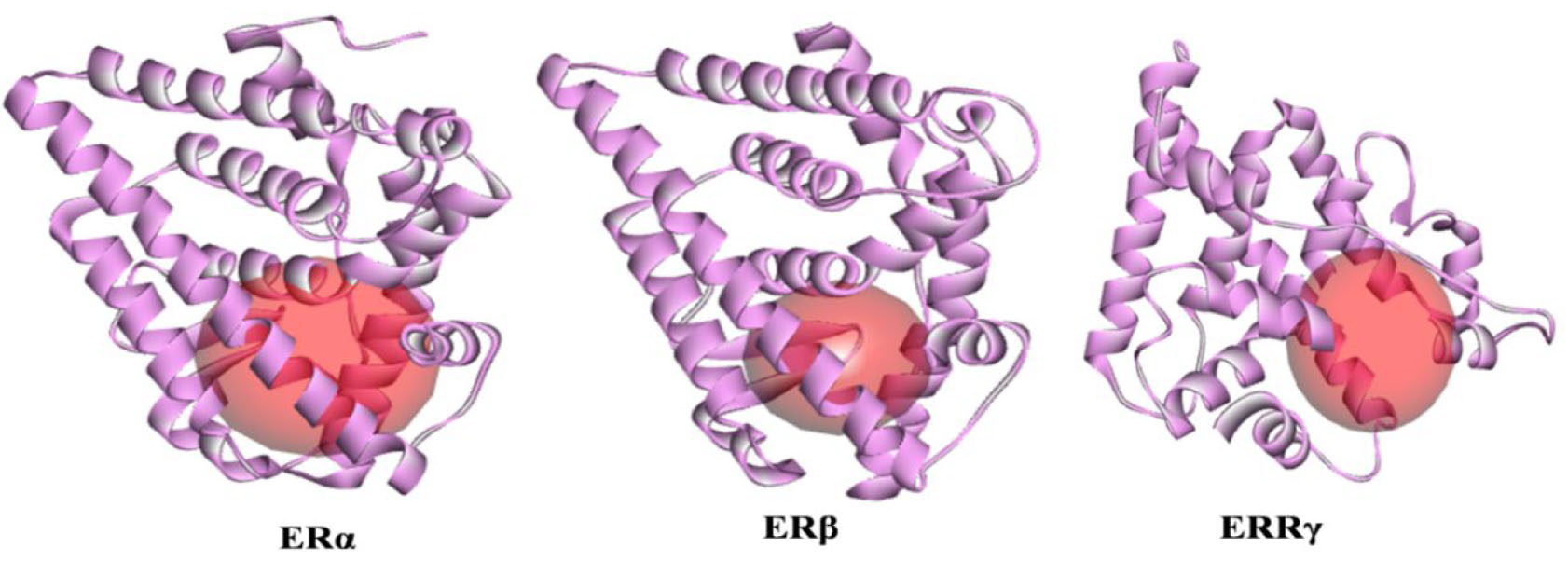 Fig. 4.
Fig. 4.Active site representation of isomers of human estrogen receptor.
Efficacy and toxicity are the pivotal determinants of successful drug
development. Therefore, all the GC-MS/MS characterized metabolites were subjected
to the virtual screening process thoroughly. Initial screening with Absorption,
Distribution, Metabolism, Excretion and Toxicity (ADMET) drastically narrowed
down the count of molecules (Figs. 5,6) followed by RO5 violation AlogP
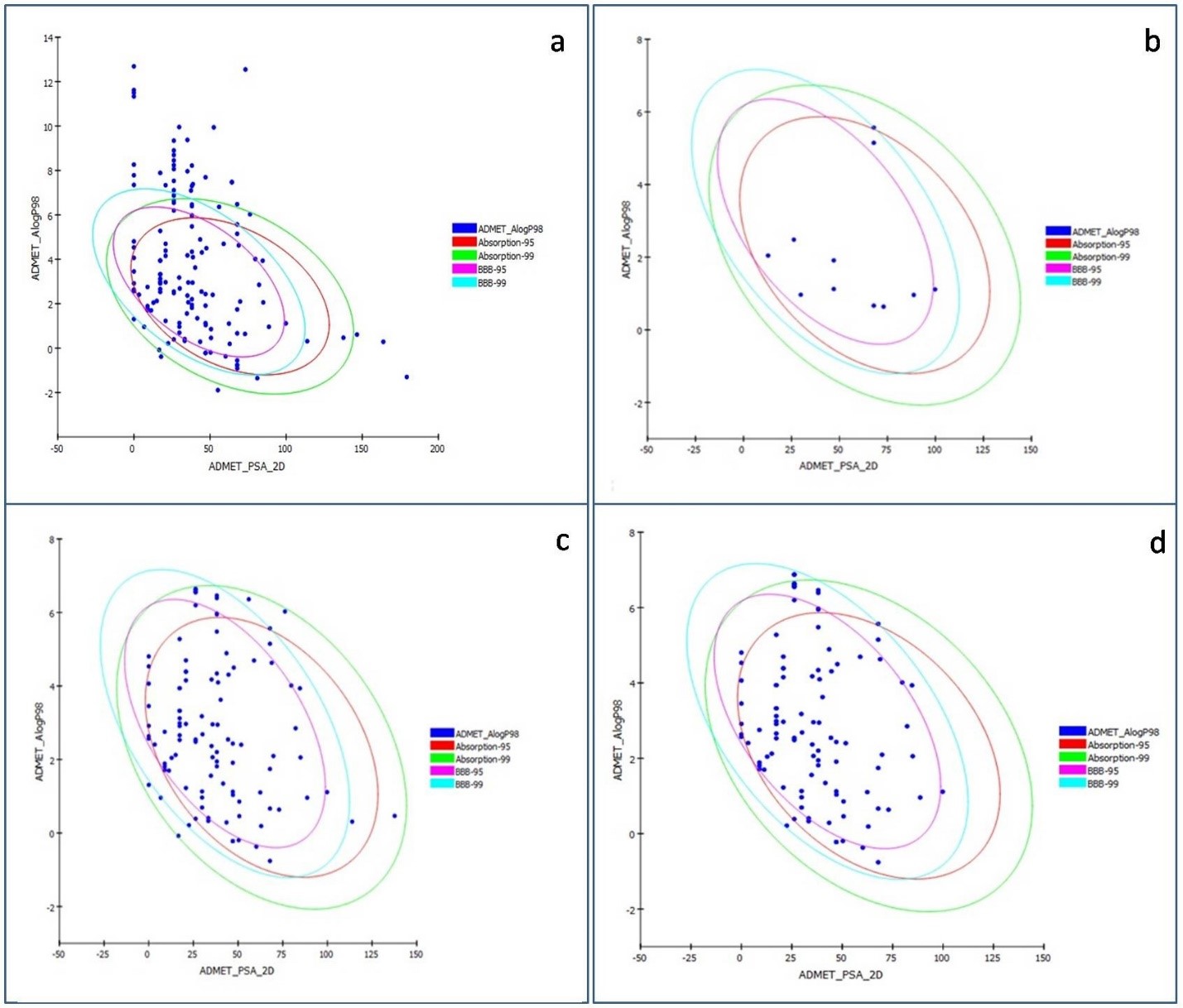 Fig. 5.
Fig. 5.The graph of ADMET 2D polar surface area (PSA_2D) vs. ALogP of 145 phytochemicals of Punica granatum L. representing the confidence limit ellipses of 95% and 99% corresponding to the intestinal absorption models and blood-brain barrier (BBB). (a,b) represent molecules before and after ADMET screening. (c) The ellipses define regions where well-absorbed Human Intestinal Absorption (HIA) compounds are expected to be found after oral administration. (d) The graph represents compounds with low and medium blood-brain barrier (BBB) penetration.
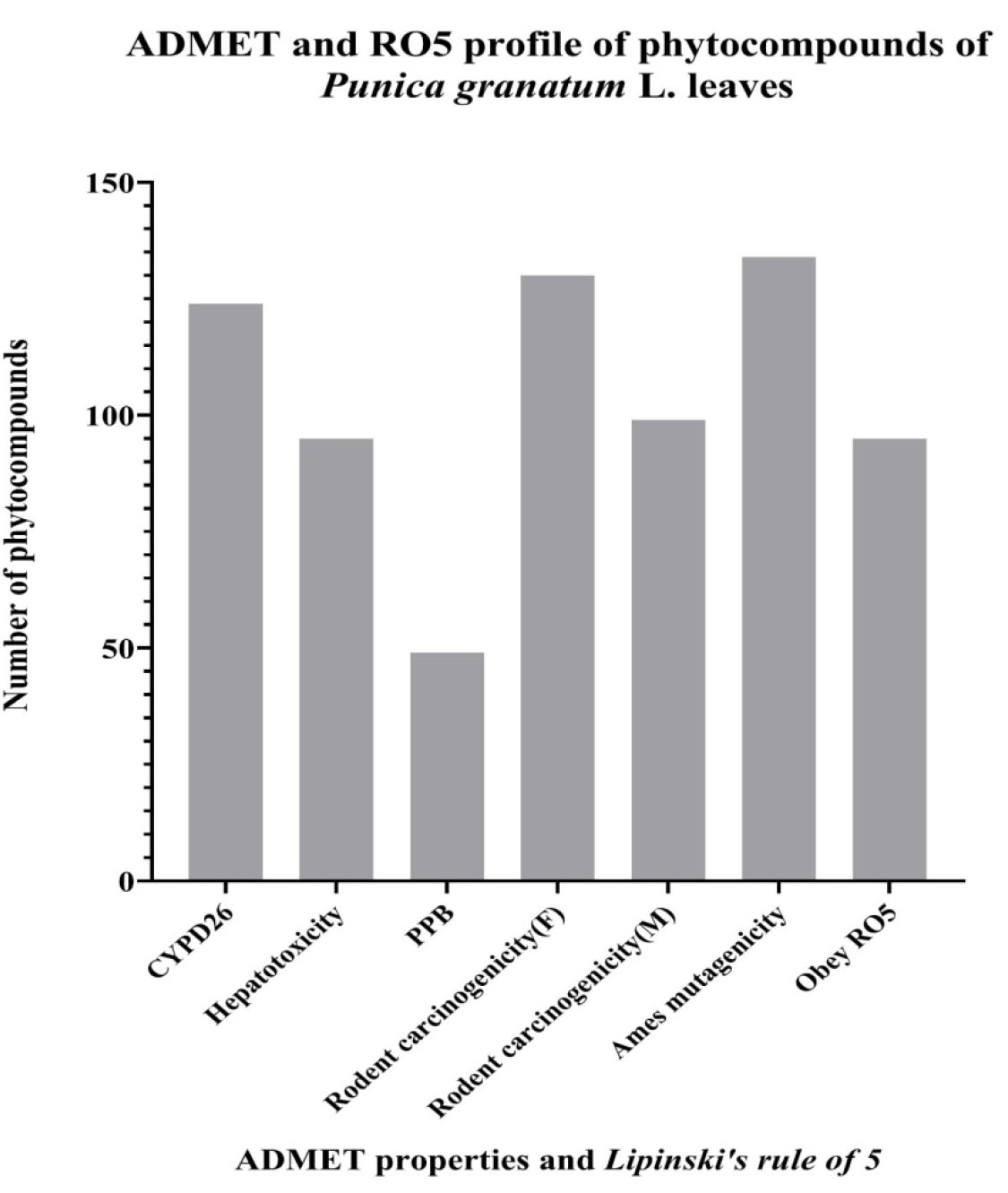 Fig. 6.
Fig. 6.Number of phytocompounds of hydromethanolic extract of Punica granatum L. leaves predicted to be CYPD26 inhibitors, non hepatotoxic, no plasma protein binding (PPB) affinity, non-carcinogens in Female (F) and Male (M) non-mutagens and obey RO5.
Detailed tabulations of the screened compounds, which completely satisfied the ADMET properties and drug-likeness, are given in Table 3. It is evident from the table that the standard drug, tamoxifen, an anti-estrogen that is widely used in the clinic to treat ER-positive breast tumors) violated both RO5 and exhibits hepatotoxicity and high affinity to plasma binding proteins. 2-Propenoic acid, 3-(4-hydroxyphenyl)-, methyl ester, a natural compound, followed RO5 and did not exhibit any toxicity in ADMET prediction suggesting its efficacy and nontoxic nature. Some of the natural compounds of HMPGL also showed hepatotoxicity and affinity to plasma proteins.
| Compounds ID | RO5 | Solubility level | BBB level | Absorption | CYPD26 | Hepatotoxic | PPB | NTP | Ames mutagen |
| Female & male rat | |||||||||
| 7428 | Y | 4 | 3 | 0 | F | F | F | NC | NM |
| 14334 | Y | 3 | 3 | 0 | F | F | F | NC | NM |
| 85447 | Y | 4 | 1 | 0 | F | F | F | NC | NM |
| 92203 | Y | 4 | 2 | 0 | F | F | F | NC | NM |
| 586459 | Y | 4 | 3 | 0 | F | F | F | NC | NM |
| 587806 | Y | 3 | 3 | 0 | F | F | F | NC | NM |
| 6432173 | Y | 3 | 1 | 0 | F | F | F | NC | NM |
| 2733526 | N | 1 | 0 | 1 | T | T | T | NC | NM |
| Tamoxifen | |||||||||
| (Standard drug) | |||||||||
| RO5 (Lipinski’s rule of five): Y, Yes (Follow RO5); N, No (Don’t follow RO5). Solubility level: (1) No; (2) Very low but possible; (3) Good; (4) Optimal. BBB (Blood Brain barrier): (0) Very high penetrant; (1) High Medium; (2) Medium; (3) Low. Human Intestinal Absorption: (0) Good; (1) Moderate. CYP2D6: F, False (Non-binding); T, True (Binding). Hepatoxicity: T, True (Toxic); F, False (Nontoxic). PPB (Plasma protein binding): F, False (Non-binding); NC, Non-carcinogen; NM, Non-mutagen. | |||||||||
The screened compounds and the standard drug tamoxifen were docked with three
different ER structures. The docked poses were examined based on the LibDock
score (Kcal/mol) and various types of interactions in hydrogen/hydrophobic
interaction analyses. The structural conformations of 4-coumaric acid methyl
ester (92203) were the most favorable for the binding cavity of all the three
receptors, especially hydroxyl group (-OH) exhibits a major structure-activity
relationship, where the removal of (-OH) in the para position of the structure is
directly proportional to dock score values. Thus, this position is considered
crucial for the binding of this phytochemical to these receptors. Other compounds
like benzoic acid, 3,4,5-trihydroxy-, methyl ester (7428),
2-propanone,1-hydroxy-3-(4-hydroxy-3-methoxyphenyl) (586459),
(4H)4a,5,6,7,8,8a-hexahydrobenzopyran-5-one-3-carboxamide,2-(2-hydroxypentyl)-8a-methoxy-4a-methyl
(587806) and tamoxifen (2733526) also showed favorable binding, but the standard
drug tamoxifen violated the RO5 and demonstrated toxicity. The structure of
4-coumaric acid methyl ester showed high complementarity to
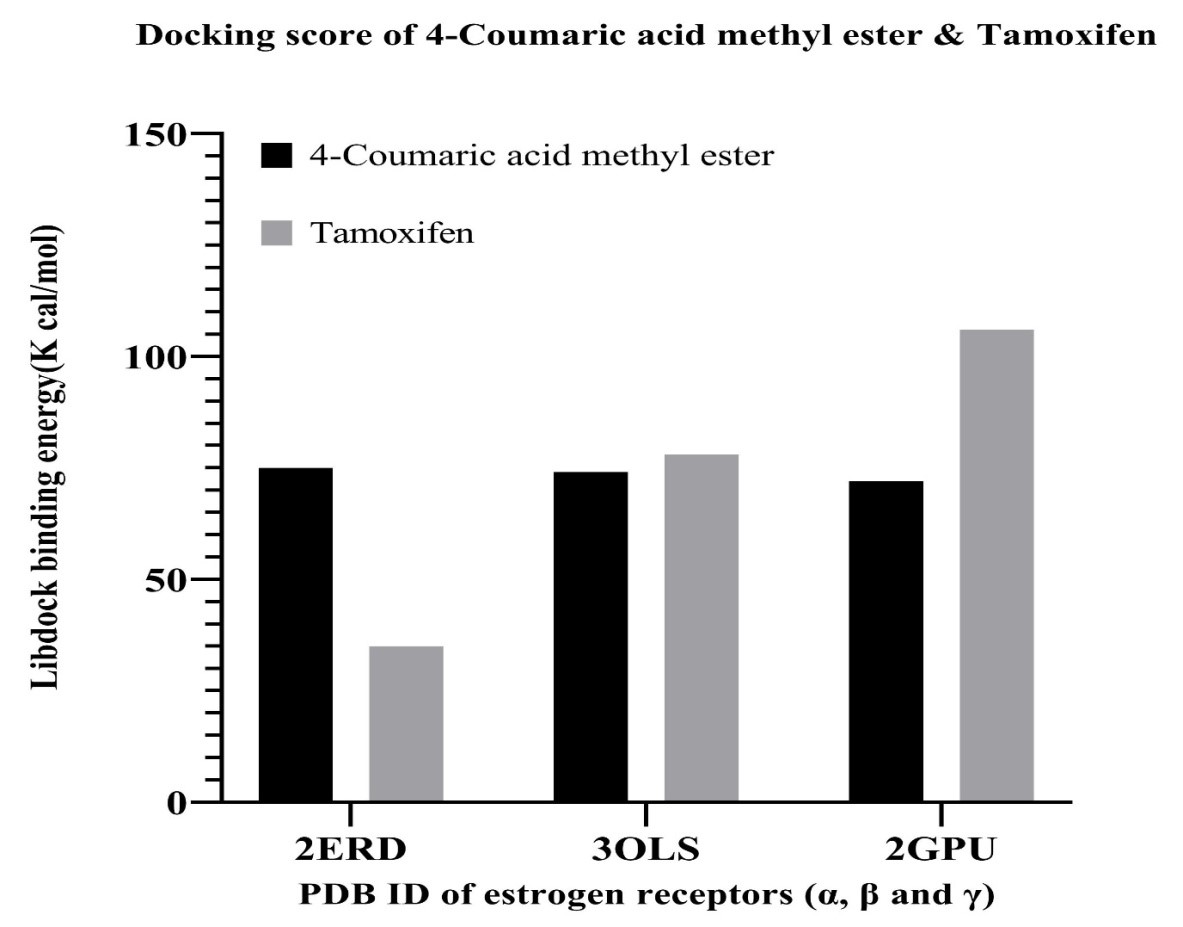 Fig. 7.
Fig. 7.
The binding energy of 4-coumaric acid methyl ester present in
hydromethanolic extract of Punica granatum L. leaves and tamoxifen with
three isoforms of estrogen receptor (3ERD, 3OLS and 2GPU corresponds to the PDB
ID of the LBDs of ER
The binding pattern and chemical interactions of nontoxic natural 4-coumaric
acid methyl ester present in HMPGL with good docking score, with the three
isoforms of ER, were demonstrated in Fig. 8. The
A time-dependent MD simulation at 50 ns was conducted using GROMACS 2016 to investigate the flexibility and overall stability of docked complexes. MD simulation were carried for the best candidate molecule, 4-coumaric acid methyl ester, with three drug target receptors and time-dependent parameters, were analyzed as described by Dhivya et al., 2018 [3]. The root mean square deviation (RMSD), the radius of gyration (Rg) and hydrogen bond (H-bond) interaction graphs were generated by using Xmgrace software in a Linux environment (Fig. 9).
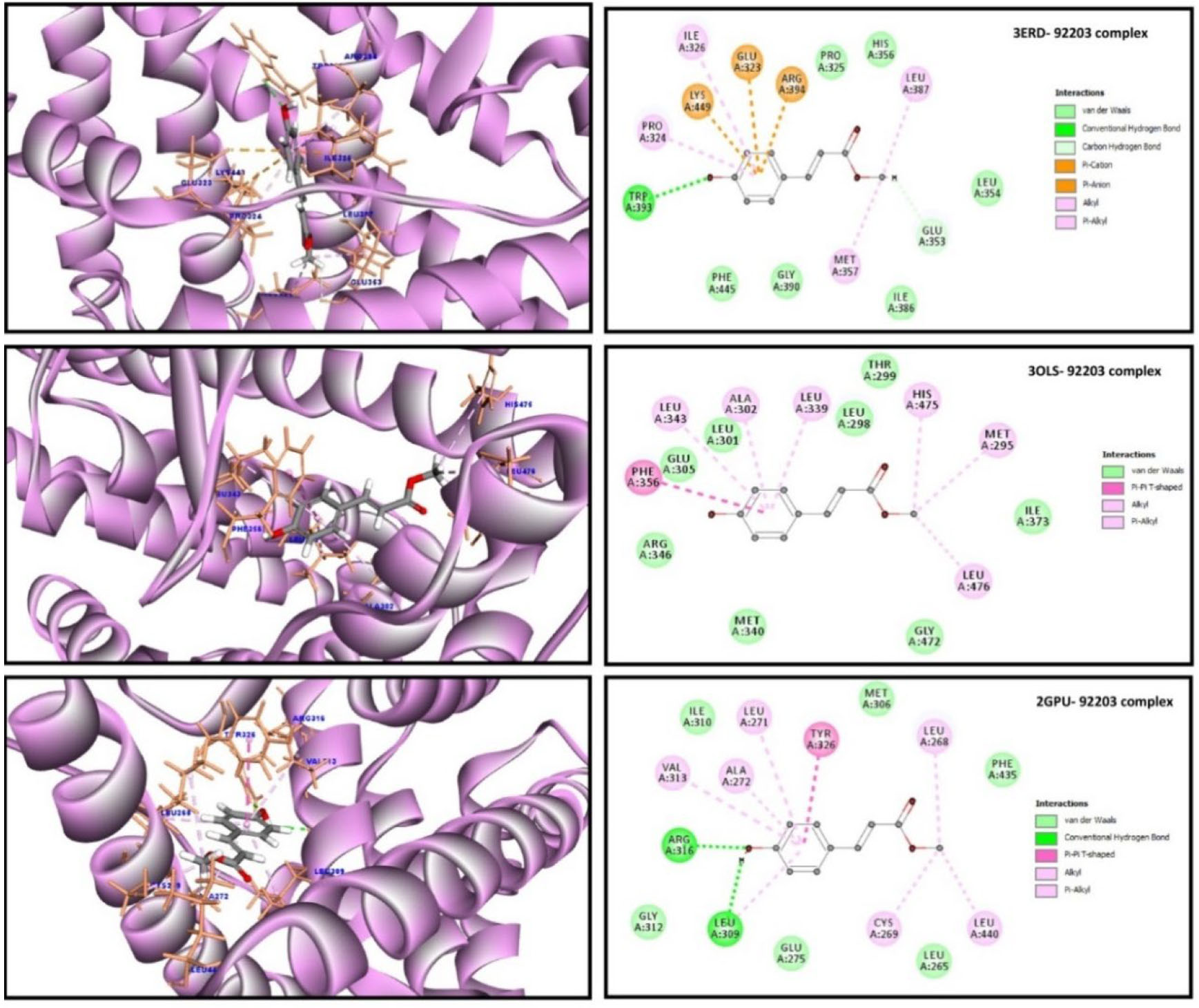 Fig. 8.
Fig. 8.Both 3D and 2D docked representation 4-Coumaric acid methyl
ester with three different estrogen receptors’ (drug targets) active sites.
4-Coumaric acid methyl ester docked with 3ERD (the ligand-binding domain of
ER
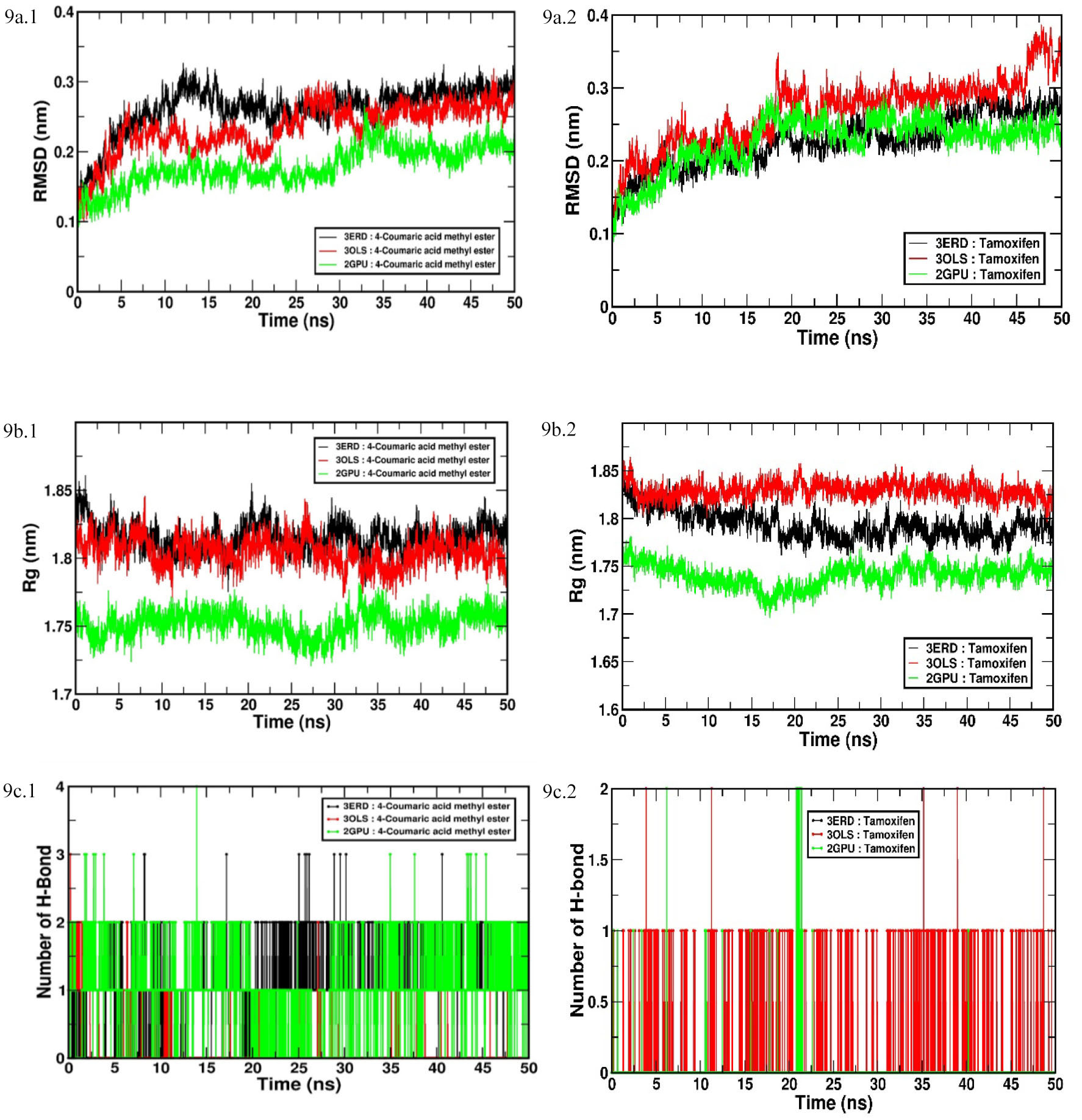 Fig. 9.
Fig. 9.Time-dependent MD and simulation of 4-coumaric acid methyl ester
and tamoxifen with three different ERs’ (drug targets) active sites (LBD of
ER
As is evident in Fig. 9a.1, the increasing trend of RMSD was observed for all
the six complexes, 3ERD/ER
Breast cancer is regarded as one of the major burdens observed in women. Consequently, there is an upsurge in efforts for the discovery of new drugs to combat this disease. Plants are widely regarded as a reservoir of various types of bioactive metabolites with various therapeutic and pharmacological potentials [68]. Due to their diverse nature, several plant-based molecules are used as drugs and are in the process of discovery routes [69]. Most of the research studies reported to date involved the characterization of peel, seed and bark by GC-MS and very few studies on leaves of Punica granatum L. using the NMR technique. However, no studies have been reported on leaves by GCMS analysis. Notably, in the present study, we attempted to characterize the possible phytochemicals exclusively by GCMS in Punica granatum L. leaves [70, 71]. The present study identified 145 phytoconstituents from HMPGL by GC-MS profiling. Similar to our report, 5-hydroxymethylfurfural was reported in high concentrations in ethyl acetate extract of the fruit peel of Punica granatum by Barathikannan et al. (2016) [71]. These phytochemicals are known to contribute to the diverse medicinal properties of the plant, as previously reported [69]. A majority of these phytocompounds were reported to have antioxidant, antiinflammatory, anticancer, and antimicrobial activities, and used as food additives (Table 1). Due to the presence of alkaloids, coumarins, flavonoids, glycosides, phenols, saponins, tannins and terpenoids, HMPGL has better DPPH radical scavenging activity. Bekir et al. (2013) [72] had previously reported the antioxidant potential of Punica granatum L. leaves extracted with different solvents based on their polarity.
Using the in-silico approach, the pharmacokinetics and toxicity studies of compounds are investigated before evaluating their biological activity [73, 74]. Poor pharmacokinetic profile and toxicity are the main reasons for last stage failures in drug discovery. Therefore, in the present study, all the identified 145 phytochemicals from the HMPGL were initially screened for ADMET and Lipinski’s RO5, and only eight molecules were identified to be drug-like nontoxic molecules. Among the 138 phytoconstituents, a majority of them were identified to be fatty acids and terpenes. Due to the presence of the long hydrophobic -acyl chains and terpenes, the fatty acids exhibited low solubility and high affinity to plasma binding proteins indicating low efficacy. They will be highly toxic as they can penetrate BBB, bind to CYPD26 and exhibit hepatotoxicity.
Docking studies were carried out with 51 metabolites (eight drug-like nontoxic
molecules + 42 mentioned in Table 1). Those 42 metabolites were also used for
docking studies as most of them exhibited hepatotoxicity, and it is one very
common toxic property noted in many FDA-approved drugs like tamoxifen. One of the
most important steps in developing a new lead molecule is target selection.
Systematic analysis of protein interaction networks (disease networks) and
identification of hub protein enhances the understanding of the molecular basis
of the disease. They also help in determining the key node as a potential target
protein in the drug discovery process. Several hub proteins have been identified
to be not suitable as drug targets as their inhibition may affect crucial
activities of the cell; However, ER, a middle degree node and with high cluster
coefficient, is an optimal drug target for breast cancer treatment [75, 76]. Our
binding pocket analysis revealed that ER
MD simulations of the docked complexes from docking studies help refine docking
and enhance the accuracy of the binding affinity predictions [3, 68]. Post
docking MD simulations at 50ns reflected the time-dependent behaviour of the
docked complexes. The ligand in the ERR
To conclude, the current research work was an attempt to draw insight into the
structural, functional, and dynamical aspects of phytocompounds of Punica
granatum L. In the process, the GC-MS/MS metabolite profiling revealed
the presence of 145 compounds. These can be deployed to discover novel drugs
against various cancers, as Punica granatum L. is reported to have
anticancer properties. Estrogen receptor plays a crucial role in cellular
proliferation and differentiation of breast cancer cells, which was identified as
a novel target for breast cancer by network pharmacology. 96% of the
phytoconstituents exhibited toxicity in the virtual ADMET screen, and 35% did
not exhibit drug-likeness. In-silico, molecular docking was performed
against three isoforms of ER and compared with the standard drug tamoxifen.
4-Coumaric acid methyl ester, a nontoxic natural, demonstrated the highest
affinity with core residues of 3ERD (ER
SKM and KRS conceived the idea. TU performed in-vitro and computational studies and drafted the manuscript. DS assisted in in-silico experimentations. AKG helped in the experimental procedure and partially drafted the manuscript. HSY helped in the computational facility and wrote a part of the manuscript. DB helped in GC-MS/MS analysis and thoroughly revised the manuscript. SKM arranged the funds and supervised the whole study, edited, and upgraded the final version of the manuscript. All the authors analyzed, discussed the results, and approved the version to be submitted.
Not applicable.
The authors acknowledge Neoscience Labs Private Limited, Chennai, India for allowing to utilize their GC-MS facility and their technical support. DBT-BIF computational facility and BiSEP facility at MLACW was used to carry out the research work. Dhivya also expresses her sincere thanks for the fellowship provided by the DBT-BIF facility at MLACW by Govt of India. The authors are grateful to Dr. V.R. Devraj, Professor, Bangalore Central University and Dr. C.S. Karigar, Professor, Bangalore University, India, for their valuable suggestions. This publication was supported by the Deanship of Scientific Research at Prince Sattam Bin Abdulaziz University, Al-kharj, Saudi Arabia.
This research was partially supported by a minor research grant (MLACW-MRP-057) from MLACW, Bengaluru, India.
The authors declare no conflict of interest.
The pomegranate extract samples are available with the authors.