


Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Biotechnology, School of Applied Sciences, REVA University, Rukmini Knowledge Park, Bangalore-560064, Karnataka, India
2 Molsys Scientific (Molsys Pvt. Ltd) Yelahanka, Bangalore, Karnataka, India
3 Department of Biotechnology, Bodoland University, Kokrajhar- 783370, Assam, India
4 Department of Plant Pathology, University of Agricultural Sciences, Bengaluru- 560065, India
5 Department of Biochemistry, Maharani Lakshmi Ammani College for Women, Bengaluru-560012, Karnataka, India
6 Department of Biotechnology, Maharani Lakshmi Ammani College for Women, Bengaluru-560012, Karnataka, India
Abstract
Soil constitutes a major component of the agro-ecosystem. Unrestrained uses of chemical pesticides and increased human activities have contributed to unprecedented changes in soil microflora affecting productivity. Modern microbiomics has proven to be an indispensable tool to understand the adaptations underlying complex soil microbial communities and their beneficial applications. In this review, we seek to emphasize the scope of microbiomics in enhancing soil productivity by providing an overview of the various sequencing platforms considering key parameters such as the accuracy, read lengths, reads per run, time involved and weighing out their pros and cons. The advances in modern ultra-high-throughput microbiomics platforms in combination with cloud-based analytics for in-depth exploration of soil-microbe associations can help achieve sustainable soil management contributing to better plant yield and productivity.
Keywords
- Microbiomics
- Soil
- Metagenomics
- DNA
- Sequencing
- Review
The agro-ecosystem has been affected due to the unrestrained use of chemical pesticides and increased human activities that have contributed to unprecedented changes in soil microbial diversity, as a result of which agriculture productivity has declined. Several reports have shown the positive effects of adopting organic farming practices that include reduced loss of nutrients, lower global warming potential, lower energy requirements and enhance soil biodiversity (1–3). While other farming practices such as organic farming have proven to address these difficulties, more research is required to determine factors that enhance diversity of the soil microbiome. The microbiome constitutes a composite mixture of a wide variety of microorganisms living either symbiotically or asymbiotically (4, 5). The association of these diverse microorganisms present in a particular place can be different in dissimilar conditions. In depth understanding of the agro-ecosystem considering the soil microbiome and the host-microbiome interactions can be key factors in enhancing the soil productivity (6).
In exploring the soil microbiome, traditional microbiological methods come with several limitations and fall short particularly with non-cultivable microorganisms that make up the vast majority of the microflora. Several reports have shown the use of Next Generation Sequencing (NGS) techniques that have addressed such limitations through metagenomic approaches (8–11). NGS methods have greatly impacted microbiomics and in due course of time, sequencing technologies have evolved dramatically. With the advent of new techniques in NGS and analysis techniques and the rapidly declining costs, NGS has emerged as a gold standard in ascertaining the presence and role of key regulators in soil productivity (12). With emerging advanced methods such as Metatranscriptomics, Metaproteomics and Metabolomics, Soil microbiome analysis provides an approach to study regulation of the soil’s nutrient cycle by way of which sustainability in soil could be achieved (13). fig1 depicts the process and applications involved in metagenomics.
In the field of environmental microbiology and microbial ecology, metagenomics can help study genomes of uncultivable microbes such as ubiquitous groups and methanogenic Archaea using the techniques in metagenomics (14).
Metagenomics field and the advances in metagenomics have revolutionized the domain of soil microbiology. Bacterial communities can be described in the natural environment with high resolution. Ofaim et al. (2017) (15), worked on a broad framework which could analyse the functions of the microbial community through analysis based on metagenomics approach. From metagenome analysis, gene catalogs which are very specific to the environment are correlated into metabolic-network representation. The predominant taxa are assigned the metabolic functions according to the established conventions in ecology. The objective is to link taxonomic group with the function. Then prediction is done with respect to the effect of the predominant taxonomic group on the performances of the overall community. This is achieved by simulation of edges which are removed or added which represent the taxa associated functions. The network developed was implemented in the metagenomics data of rhizobacteria of wheat and cucumber as well as the microbiome away from the root zone. The ancestor algorithm used to assign taxonomy was MEGAN. There were considerable differences between the treatments, root vs soil for the environment dependent effects. In simulated roots, the specific plant exudate metabolism for metabolites such as flavonoids, organic acids, could be correlated with distinct taxonomic groups. This concludes that community structure is to a considerable extent decided by the metabolic functions of root associated microbes. In case of simulation studies involving pair of taxonomic groups at the order level, prediction could be done regarding the production of complementary metabolites. A key taxonomic group in the soil environment was found to be Actinomycetales along with other groups such as Burkholderiales, Rhizobiales, Xanthomonadales. Pseudomonadales, Sphingomonadales. With the framework designed, the correlation between the structure of the microbial community, the functional role and production of metabolites could be unfolded by integrating the metagenomic and metabolomics data obtained through high-throughput techniques. Once the metabolism of microbial community is explored, the manipulation and optimization of their functions can enhance the plant productivity.
Land use changes have profound influence on properties of ecosystem and functions, as the conversion of native habitats can lead to the loss of ecological functions (16). Metagenomics have also opened the possibility of exploring the variation in soil microbial community according to the land use. Goss-Souza et al. (17) reported the effects of land used as a result of forest conversion to grassland and no tillage land on microbial communities. In their study, further to DNA isolation and sample preparation, and employing MiSeq Reagent Kit V2, the indices such as Shannon's alpha and Whittaker's global beta diversity indices were used for estimating the diversities in functional and economic potential across land use. While results from the study revealed no significant changes in the alpha diversity, the beta diversity was found to be minimum in grasslands, and was comparable between forest and no tillage system. The sequence similarity was 46.6% for Proteobacteria whereas, 20.9% was reported for Actinobacteria. The Acidobacteria representation was 9.5%, followed by Firmicutes with 4.1%.
The influence of soil functional potential was also explored in the study. In long term grasslands, there was significant change in the relative abundance observed in 12 categories. The increase in potential function was observed with respect to carbohydrates, rate of respiration, extent of virulence, metabolism of sulphur, regulation and cell signaling. Increased rate of motility, chemotaxis, iron acquisition and metabolism were also observed. There was a decrease in the synthesis of amino acids, fatty acids, lipids and isoprenoids, cofactors and vitamins. There was a reduction in the production of aromatic compounds as well in the rate of photosynthesis. In case of forest conversion to no tillage land, increase in amino acid content, concentration of nucleosides and nucleotides were observed along with a reduction in virulence and rate of photosynthesis. Similar study using metagenomic analysis was carried out to investigate the diversity in taxonomy as well as the soil microbiota present in Chilean vineyards and the neighboring native forests (18). In this study, further to DNA isolation from soil samples of native forest and the area converted to organic vineyard, shotgun metagenomic approach was employed. Results revealed comparable microbial species from both habitats. The predominant bacterial group included Bradyrhizobium and Candidatus solibacter. Gibberella comprised the most abundant fungal species in both habitats. This result led to the hypothesis that the forest area may be acting as a buffering area for microbes ensuring similar taxonomic diversity in the converted forest area. Metabolic diversity showed significant differences. Forest soils had enriched levels of genes associated with amino acids, fatty acids, nucleotide metabolism and secondary metabolism. Soils of vineyards had abundance of genes related to miscellaneous functions. The results from these studies suggest that the maintenance of the vineyard in an organic way, which is ecofriendly, may ensure greater diversity in the microbial community and functions in the ecosystem of natural habitats. The use of new gene-targeted assembler MegaGTA, has made it possible to assemble gene sequences from ultra-large metagenomic datasets. Usage of this tool can avoid false positive contigs. An efficient Hidden Markov Model can be built by incorporating the multiplicity of k-mers (19).
Metagenomic approaches can be used to investigate the soil microbes associated with fruit quality and products derived from the agricultural crops. High-throughput sequencing was employed to investigate the microbial communities with Cannonau grapes, which is an important variety of Sardinia and the must prepared from it. Following Illumina guidelines, bacterial and fungal DNA libraries were prepared for each DNA sample. QIIME pipeline scripts were used for analysis of the fungal and bacterial population (20). VSEARCH 1.1.8. software version was used in processing the reads from bacteria (21). To assign the microbial taxonomy of the representative sequences, RDP Bayesian Classifier was used against the fungal database UNITE.
The sequencing studies reported the presence of soil bacteria from various orders on berries such as Pasteurellales, Bacteroidales, Lactobacillales and Rhodospirillales, most of which were bacteria causing wine fermentation. The predominant fungal taxa were found to be Dothioraceae, Pleosporaceae, and Saccharomycodaceae. Vinification processes were carried out at a wine cellar under controlled conditions. Yeast was not used as starter culture. More than 50% of the bacteria associated with the berries were present in the musts. This work indicated that soil, climatic conditions and farm management practices play a crucial role in deciding the microbial community in an agroecosystem. The results proved that microorganisms on the berries persist during the fermentation process. Hence, along with the plant, entire holobiont which includes all the symbionts and the host, should be considered for accurate wine genotyping. Microbiomic data may aid in developing improved approaches for both to increase the yield of grapes and improve wine quality (22).
Metagenomic techniques can also be employed for studying the diversity of viral population. Viral metagenomics is a powerful approach for obtaining genetic information from viruses as the technique does not demand the isolation of virus. There are a number of bottlenecks in viral metagenomics and one of them being a high proportion of reads don’t find match with known sequences in published viromes. Thereby the efficiency of the study can be enhanced by isolating and characterizing individual viruses from soil samples (23).
The soil microbial diversity varies significantly with the soil depth as the environmental conditions in the soil in terms of carbon and other nutrients fluctuate across the soil profile (24). The microorganisms present in the subsoil are distinct from the ones present in top soil as the nutrient status and carbon content in the subsoil is comparatively lower than top soil. The microbial diversity is characterized by the communities adapted to limited nutrient status as reported by Hartmann et al., 2009 (25). Before the advent of metagenomic techniques to unravel the microbial diversity across the soil depth, methods like phospholipid fatty acid analysis were employed. Fierer et al. (2003) (26), analysed the microbial diversity of mollisol soil across a soil depth of 2 m from a location adjacent to Santa Barbara, USA. The study revealed that there was a proportional increase in the diversity of Gram-positive bacteria as well as Actinomycetes population with the increase in soil depth whereas the reverse trend was reported for Protozoa, fungi and Gram-negative bacteria. The changes in the soil microbial diversity was attributed to the reduction in the availability of carbon across the soil depth which was in consensus with the findings reported by Hartmann et al. (2009) (25). Other methods to decipher the microbial diversity included quantitative PCR targeting 16S rRNA and 18S rRNA for bacteria and fungi respectively. The study on the abundance of genes involved in N-cycling for the soil samples collected from three different regions in China where paddy was cultivated across the soil depth of 0-100cm reported that except Ammonia oxidizing archaea which were prominent in deeper layers of 20-40 cm, other genes involved in nitrogen cycling such as nitrogen fixation, nitrite reduction and ammonia oxidizing bacteria were more prevalent in top soils. The ammonia oxidizing archaea were prominent in paddy soils compared to bacteria. The top layer of 10 cm had maximum abundance of bacteria and fungi compared to deeper layers.
Metagenomics approaches using next generation sequencing techniques facilitated in acquiring deeper insights into the genus and species level microbial diversity existing in various ecosystems. Uroz et al.(2013), explored the microbial diversity and functional aspects of soil samples collected from a spruce plantation (27). The objective of the study was to correlate the variations in the nutrient status of organic and mineral horizons of the soil profile on the functionality and diversity of the microbial population. The techniques employed for sequencing were pyrosequencing and Illumina platforms. The nutrient rich horizon containing organic matter was dominated by bacteria and Ascomycota whereas archaea was predominant in the mineral horizon down the soil depth. Glycoside hydrolases sequences were significantly higher in organic horizon while glycoside transferases dominated in the mineral horizon emphasizing the role of nutrients status across the soil profile on the functionality of the microbial population. This was revealed through functional analysis and MG-RAST. The results revealed the dominance of Proteobacteria, Bacteroidetes and Verrumicrobia in the organic horizon at the top whereas the mineral horizon below revealed the abundance of Firmicutes and Chloroflexi. At the genus level, Burkholderia was more prominent in organic horizon and Candidatus solibacter in the mineral horizon (27).
Human population is increasing at an alarming pace. Increased food production is the need of the hour. The improved varieties with good yield also demand the application of inorganic fertilizers. Proper exploitation of beneficial soil microorganisms can considerably decrease the use of inorganic fertilizer in agriculture. Metagenomic studies can culminate in modification and better utilization of soil microbes for promoting plant growth. Based on the responses of plants to the soil microbial community obtained through metagenomic approaches, the genotypes favoring beneficial microbial interactions can be selected. Such genotypes can be incorporated in the breeding programme (28).
RNA based metagenomic studies called as metatranscriptomics focuses on microorganisms which are metabolically active. In this technique, environmental RNA is reverse transcribed to a cDNA and thereafter sequenced using Illumina Hi Seq (29). In future, plant selection could be done on the basis of soil metagenomic approaches.
Jin et al. , (2017) (30), through 16srRNA sequencing on Ion Torrent Platform investigated the microbiota present in the rhizosphere of foxtail millet and rhizoplane biome community and their functions. In this study, the abundant microbes in the rhizosphere were reported to be Acidobacteria, Actinobacteria, Proteobacteria, Bacteroidetes and Firmicutes. Appreciable reduction was reported in the alpha diversity of the bacteria. Total 16,109 operational taxonomic units (OTU) were identified in the study out of which 187 were found common between rhizosphere and rhizoplane. Microhabitat followed by geographic locations may have had an influence on the bacterial community in the rhizosphere of foxtail millet. Pathway involving tryptophan metabolism was enhanced in the rhizoplane microbiome when compared with rhizosphere. Rhizoplane microbiome showed the ability to degrade xenobiotics along with prominent catabolism pathways. The study concluded that rhizoplane microbes could be used to isolate enzymes useful for biodegradation of xenobiotic compounds.
Metagenomic approach is a highly effective approach to study the effect of invasive plant species on the microbial diversity of that area. A comprehensive investigation was carried out by Kamutando et al., (2017) (31) on Acacia dealbata, an invasive tree across South African grasslands. The study was focused on the influence of factors such as soil nutrition and biogeography on the microbial community present in the rhizosphere. Illumina MiSeq was used to generate data from 16S rRNA genes and (ITS) regions using shotgun metagenomics approach. Microorganisms in the rhizosphere were compared with bulk soil with respect to taxonomic diversity and metabolic capacity. Sequencing data revealed a significant difference in microbial communities of bulk and rhizosphere soil. The abundant bacterial and fungal classes were found to be Alphaproteobacteria and Gammaproteobacteria & Pezizomycetes and Agaricomycetes, respectively in the rhizosphere. The specific microbial community found in rhizosphere could be the result of modification of soil nutrient status by Acacia dealbata. The core bacterial and fungal taxa irrespective of the soil and geographical attributes were found to be the ones associated with N and P cycling. The remaining community members were categorized as plant pathogens. The finding stated that there was an overrepresentation of functional genes concerned with plant growth promotion in the rhizosphere. Therefore, the results suggest microbial communities present in the soil rhizosphere could play a crucial role in the establishment of the invasive crops like Acacia. In another study, a metagenomics approach was employed to investigate the Arbuscular Mycorrhizal fungus (AMF) diversity in agricultural lands (32). The symbiotic association of AMF benefits by providing better nutrition and tolerance to environmental stress. Advanced metagenomic studies can throw light on the taxonomic diversity and identification of key genes involved in symbiotic association and plant growth. By promoting the native AMF diversity, better crop health can be ensured (33).
Soil metagenomics has proven to be a highly efficient technique to study functional genes and microbial diversity. In another study, the metagenome of southern maritime Antarctic soil was analyzed using a 454 pyrosequencing platform. From soil genomic DNA metagenomics library was constructed and sequenced. Seventy seven percent (77%) of functional genes were related to structure, metabolism of carbohydrate, processing and modification of DNA and RNA. The predominant genera in Antarctic terrestrial environments were found to be Proteobacteria, Actinobacteria (34).
Various agricultural practices can also influence the bacterial communities. Crop residue management influence the specific taxa even with the same organic matter present in the field. This was revealed through 16s-metagenomics and IndVal index analysis (35). Prasannakumar et al (2020) reported metagenome sequencing of finger millet and associated microbial consortia (35). They have identified 1029 species (includes obligate endophytes) of microbiota using the whole genome metagenome sequencing approach of GPU-28 (GPU) and Uduru mallige (UM) finger millet varieties, consisting unique 385 and 357 species, respectively. Actinobacteria were found to be more abundant in GPU as compared to UM (36). Another comparative metagenomics study by Prasannakumar et al. reveals antagonistic activity of certain bacteria against Magnaporthe oryzae strain MG01 (Unpublished data).
The community structure of microbes may be dependent on two factors which are soil and host genotype. Soil metagenomic studies on two crop species, wheat and cucumber, very distinct in lineage revealed the predominant bacterial genera as Pseudomonas and Cell vibrio respectively. BLASTX algorithm1 was used for assigning annotations for the taxonomic and functional aspects. Non-redundant NCBI protein database was used for mapping the Open Reading Frames. In cucumber, genes involved in pectin and mannan synthesis were associated with root colonization. In case of wheat, the genes abundantly expressed were involved in assembly of flagella, bacterial chemotaxis, C4-dicarboxylates transport, denitrification process, glutathione synthesis and protein export (37).
Diversity of microbial species may be influenced by the physicochemical properties of the soil and diversity of plants. Pyrosequencing followed by clustering of OTU and taxonomic classification using the Evolutionary Placement Algorithm (EPA) showed remarkable changes in the Fusarium community with monoculture and polyculture agricultural practice. F. tricinctum and F. oxysporum were found to be the predominant species (38).
Gene profiling using 16S rRNA combined with shotgun metagenome were employed to analyse the microbial communities with both wild and domesticated barley. The predominantly present bacterial families were found to be Comamonadaceae, Flavobacteriaceae, and Rhizobiaceae in the rhizosphere. Microbiotas in the rhizosphere of barley were enriched with traits associated with mobilization of nutrients, pathogenesis, secretion and interactions with phage. The findings of this study led to the conclusion that a combined effect of microbe-microbe and host-microbe interactions may have an influence on the microbial diversity at the root-soil interface (39).
Population Genomics Analysis of particular genotypes of Rhizobium leguminosarum bv. viciae also support the fact that host selection is primary in deciding the microbiota in the rhizosphere. Rhizobial biota of Vicia sativa, Pisum sativum, Vicia faba and Lens culinaris were studied using R. leguminosarum bv. viciae 3841 as a reference. The sequences were highly conserved in all subpopulations. An appreciable fraction of 16-22% of reads did not map to the genome used as a reference as these microbes were very specific to that particular soil population. A single nucleotide polymorphism (SNP) pattern in the nod gene cluster was very specific for the host plant. These findings support the fact that the specific rhizobial genotypes are preferred by the host plant (40).
Barcoded pyrosequencing method was used to identify fungal populations in the soil rhizosphere of Ulleungdo and Dokdo, two volcanic islands. Ulleungdo samples explored 768 OTU while it was 640 and 382 from the Dongdo and Seodo samples, respectively. Ulleungdo samples had Basidiomycota as the dominant phylum, whereas Ascomycota was the predominant phylum in the samples from Dokdo. Ulleungdo samples showed an abundance of Ectomycorrhizal fungi. The conclusion of the study was that the species richness and diversity in vascular plants of Ulleungdo and Dokdo had an impact on ectomycorrhizal fungal abundance present in the rhizospheres (41).
Metagenomic studies in plants of alpine soil have highlighted the influence of physiotype of the plant on the microbial diversity in the rhizosphere. The production of HCN and intensity depended on microhabitat associated with the plant along with the soil type. The predominant group was HCN producing Pseudomonas species. These microbes helped in mineralization of soil through HCN production, solubilization of inorganic phosphate and iron complexation with siderophores (42).
Metagenomic approaches have also thrown light on the specific plant-microbe interactions in polluted soils. For sequencing RNA, a Shotgun metagenomics and targeted amplicon sequencing was employed to investigate the impact of certain plants such as horseradish, black nightshade and Tobacco along with NPK fertilization on microbial community. The rhizosphere community showed appreciable increase in abundance of copiotrophic taxa, α, β and µ-proteobacteria or Bacteroidetes than the bulk soil. Functional genes for mechanisms like signal transduction, plant defense, transport of amino acid and metabolism were overrepresented in these copiotrophic organisms. The effect of mineral fertilization was not significant compared to the plant type on microbial community (43).
Eco-functional genes performing important ecosystem functions can be assessed using techniques like targeted Illumina sequencing. Evaluation of diversity of actinomycetes genus Frankia across three soils of Illinois, Colarado and Wisconsin was carried out. The study covered the soils vegetated with host and non-host plants. Nitrogenase reductase gene fragments, i.e., nifH were subjected to targeted Illumina sequencing. Paired-end reads were analysed using a modified QIIME pipeline. Similarity in the sequences of all the three soils with Frankia strains belonging to Alnus as well as Elaeagnus host infection groups were observed (44).
A study employed Shotgun metagenomic approach to explore the microbial diversity in soybean. Both the rhizosphere and bulk soil were analysed. There was a clear-cut distinction in the community as influenced by the selective pressure of the host. The predominant and exclusive class in rhizosphere was found to be Gammaproteobacteria, Solibacteres. The two most abundant orders in Gammaproteobacteria were Enterobacteriales and Pseudomonadales having beneficial effects on the growth of soybean. These findings indicate that the rhizosphere community may select considering the functions of microbiota linked to metabolism of nutrients which may promote growth in the host plant (45).
Rhizosphere bacteria enhance crop productivity by suppressing pathogenic bacteria in soil. The role of soil microbes and various mechanisms associated with disease control is not fully understood. A study considering the characterization of 33,000 bacterial and archeal species employed a combination of metagenomics using a PhyloChip for microbes in the rhizosphere and functional analysis that is culture-dependent. The phylum Actinobacteria Proteobacteria, and Firmicutes were the predominant groups involved in disease suppression. In the γ-Proteobacteria group, disease suppression was attributed to the regulation by nonribosomal peptide synthetases. The results of the study suggested that plants have the ability to exploit the microbes that are beneficial for disease suppression caused by root pathogens (46).
Suhaimi et al., (47) employed 16srRNA sequencing method using an Illumina MiSeq platform to explore the difference in the microbial diversity of banana plants infected with bacterial wilt. The plants showing symptoms of disease along with the non-symptomatic plants were studied. 17 bacterial phyla were detected in non-symptomatic plants whereas symptomatic plants revealed nine phyla, Cyanobacteria and Proteobacteria being the two predominant phyla in both the plants. At the generic level, both the samples revealed the presence of Ralstonia, Sphingomonas, Methylobacterium, Flavobacterium, and Pseudomonas. Ralstonia comprised 59% of the entire genera in symptomatic plants whereas in non-symptomatic plant, it was only 36%. In total, 102 bacterial genera could be assigned to non-symptomatic plants. It was concluded that species richness and abundance of microbiota were associated with the banana plants that were non-symptomatic when compared to the banana plants showing symptoms of wilt disease. The study led to the conclusion that the increase in diversity of the microbes present as endophytes in banana plants which are non-symptomatic could be the reason for the suppression of pathogens which reflected in delayed expression of disease or prevented the disease.
With the advancement of sequencing technologies, while the costs of sequencing have significantly declined in recent years, longer read lengths have been achieved. However, the challenge still persists wherein there is a tradeoff considering the read depth and read lengths, particularly with studies involving soil biomes a greater depth or more the number of reads is highly desirable. The read depth is important in order to cover some least significant features to facilitate differentiating the samples. The challenge considering the desired depth, quality and lengths is further dependent on the costs of sequencing (48). Table 1 provides an overview of the various sequencing platforms considering a number of parameters such as the accuracy, read lengths, reads per run, time involved and weighing out their pros and cons. The comparative overview may assist a researcher in considering the most suited platform for a soil study considering their pros and cons.
| Sequencing Platform | Accuracy (%) | Read Length | Reads per Run | Time per Run | Pros | Cons |
|---|---|---|---|---|---|---|
| 454 GS FLX |
99.9 |
500-1000 bp | 1 million | 24 h | Long read length |
High cost per base; Homopolymer errors; Low throughput |
| Illumina HiSeq |
99.9 |
2 x 150 bp | 5 billion | 1 – 3.5 days | High accuracy; High throughput | Shorter read lengths; High computation costs; High concentrations of DNA |
| Illumina HiSeq |
99.9 |
2 x 250 bp | 5 billion | 7 h - 6 days | High accuracy; High throughput | Shorter read lengths; High computation costs; High concentrations of DNA |
| Ion Torrent PGM | 98.3 |
600 bp | 5 million |
2 - 3 h | Relatively longer reads; Accuracy | High cost per Mb |
| Ion Torrent S5 | 99 | 600 bp | 10 million |
2.5 - 4 h | Relatively long reads; Accuracy | High cost per Mb |
| PacBio |
99.9 | 15 Kb | 500 K |
2 - 3 h per cell | Longest read length; No amplification errors | Low outputs, Higher costs per Mb; Error rates |
| Oxford Nanopore |
85 | 2 Mb | 1 million | 1 min - 48 h | Long Read length; Data streamed real-time | Low read quality; Low device cost |
Plant-microbe interaction may result in beneficial effect or detrimental considering growth and yield (49). Ravinath et al., (2019) explored the VAM fungal association with Punica granatum in organic plot in tumkur region of southern India (50). Prior reports have shown that microbial diversity is influenced by several environmental factors such as moisture content, temperature and clay content. Abundance of microbial community is also influenced by seasonal variation of environmental factors. Agriculture systems may be affected by factors such as suppression of diseases, promotion of plant growth, organic matter decomposition, nutrient cycling, nitrogen fixation and bioremediation and also anthropogenic action on soil ecosystem (51). Studies have supported the fact that extreme environmental conditions from Antarctic regions (52) and also regions faced with water restriction, lack of nutrient availability, freeze-thawing cycles during summer may be the reason for presence of lower taxa. The evidence collected is based on culture-based studies and cloned libraries. Metagenomic studies have enabled the assessment of soil samples from these extreme regions in more comprehensive way and give an insight into the geochemical functional analysis of certain genes (53). In another study it is shown that the Fusarium communities varies among different plant species in rhizosphere soil due to changes in the soil physicochemical characteristics. The targeted metagenomic analysis of Fusarium community revealed the clustering of operational taxonomic units (OTUs) revealed the presence of two predominant phylogenetic lineages namely F. tricinctum and F. oxysporum. The studies support the findings that soil physicochemical characteristics influenced the two predominant phylogenetic and thereby confirms the influence of plant species, soil physicochemical characteristics on abundance of Fusarium communities (38). Another study reports the diversity of fungal populations in arid and semi-arid ecosystems in Baja California and Mexico between seasons was reported to be different due to significant differences in clay content and moisture in case of topsoil. Comparative analysis between the two samples revealed that the fungal community structure varied widely in top soil than burrow soil and Ascomycota and Basidiomycota species were found to be dominated in soil samples (54). Prasannakumar et al., (2020) have reported Metagenome sequencing of finger millet associated microbial consortia (35). 1029 species (includes obligate endophytes) of microbiota have been identified using the whole genome metagenome sequencing approach of GPU-28 (GPU) and UM finger millet varieties, consisting unique 385 and 357 species, respectively. Actinobacteria were found to be more abundant in GPU as compared to UM (36).
Antibiotics released in the soil poses a threat to all microbial communities by affecting their structural and functional diversity. Emergence and rapid spread of resistance to antibiotics by bacteria by harboring antibiotic resistance genes (ARG) also hampers microbial community structure, enzyme activity, mineral cycling and nitrogen cycling. Besides, the rapid transfer of ARG among the microbial communities, and spread to human and animals poses a major challenge (55). Although there are also several reports of bacteria degrading antibiotics in the soil, the impact of antibiotics on the soil microbiome functions is not fully understood. Interestingly, a study reported the discovery of novel antibiotics namely malacidins, a class of calcium dependent antibiotics through soil metagenomics (56).
Cloud-based data analysis has facilitated quick, accurate and comprehensive analysis of metagenomics sequencing data. Several web-servers and commercial platforms are available. A typical overview of the various steps involved in metagenome analysis and the various available platforms are depicted in fig1. Most services are freely accessible and provide analysis of both from both long amplicon and whole-genome metagenomes. An overview of the popular web platforms has been depicted in Table 2. GAIA, an integrated open source metagenomics suite (version 2.0. https://metagenomics.sequentiabiotech.com/ accessed on 01 February 2019) is a tool which facilitates quick and comprehensive analysis of metagenomics sequencing data. MG-RAST (Metagenomics Analysis Server 4.0.3. https://www.mg-rast.org/ accessed on 01 February 2019) metagenomics web server and API version 4.0.3. is a popular web platform which provides free and comprehensive analysis of metagenomics sequencing data from both long amplicon and whole-genome metagenomes. Web publication has analyzed upto 366,341 metagenomes containing 1,355 billion sequences and has facilitated up to 186.76 Tbp for 27,705 registered users. One Codex (https://app.onecodex.com/ accessed on 01 February 2019) is a popular web server for metagenomics sequencing data analysis which provides access to standard databases including its own one codex database. The One Codex database provides a comprehensive collection of 53,193 bacterial, 27,020 viral, 1,724 fungal, 1,756 archaeal, and 168 protozoan genomes (83,863 including host) and targeted Loci presenting 247,647 records (31,633 unique species) covering 5S, 16S, 23S, gyrB, rpoB, 18S, 28S, and ITS genes. Kaiju (http://kaiju.binf.ku.dk/ accessed on 01 February 2019) is another popular webserver that facilitates taxonomic classification based on protein-level hits. It also presents an easy to use tool to query sequences without taxonomic classifications against custom protein/nucleotide databases. Other commercial applications include Oxford Nanopore’s cloud-based analysis platform, EPI2ME (https://epi2me.nanoporetech.com/ accessed on 01 February 2019), which provides data analysis in real time. Alongside various advanced technologies that also utilize the cloud extensively, various methods such as Metatranscriptomics, Metaproteomics and Metabolomics are being used in soil microbiome exploration. Figure 2 depicts an overview of such methods. Table 3 gives a general idea of the suitable usage of high-throughput sequencing approach such as high-throughput targeted-amplicon sequencing, shotgun metagenomics sequencing, metagenomics shotgun sequencing, targeted-amplicon sequencing for the identification and characterization of novel microbial community in microbiomics study.
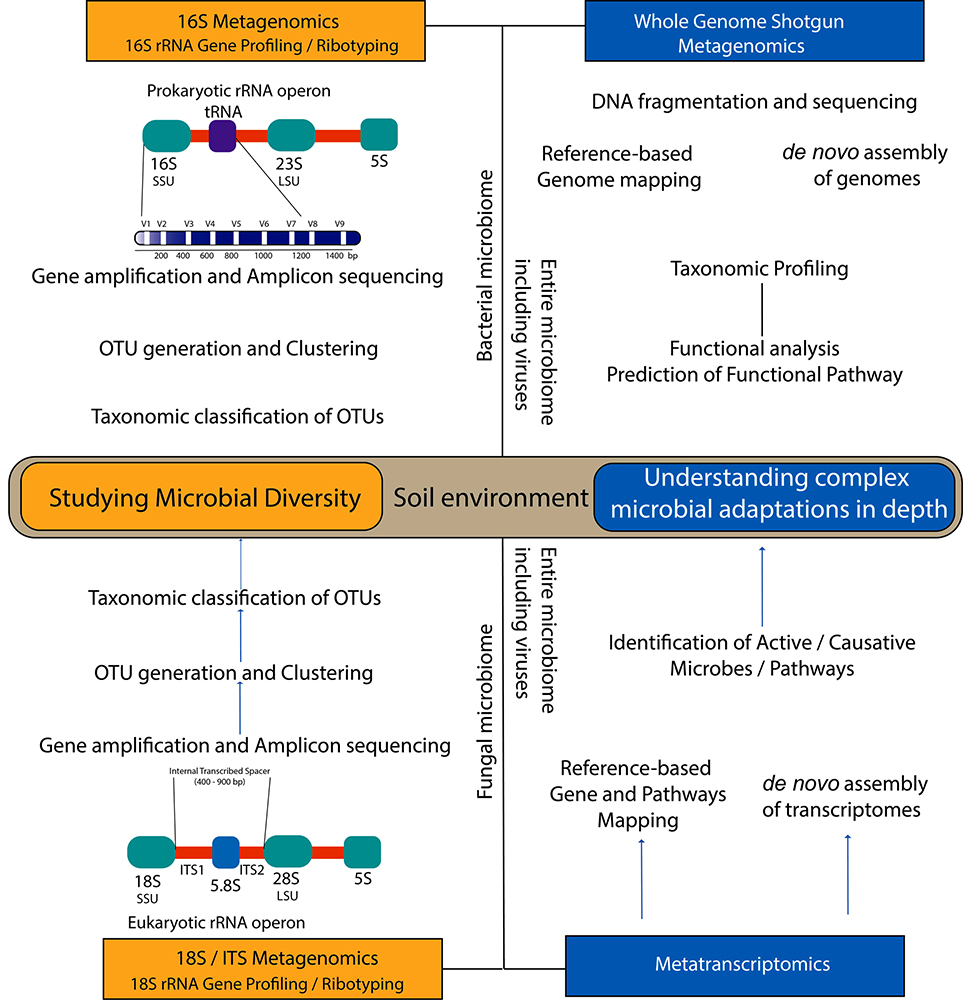 Figure 1
Figure 1Schema of the process and applications involved in metagenomics/microbiomics.
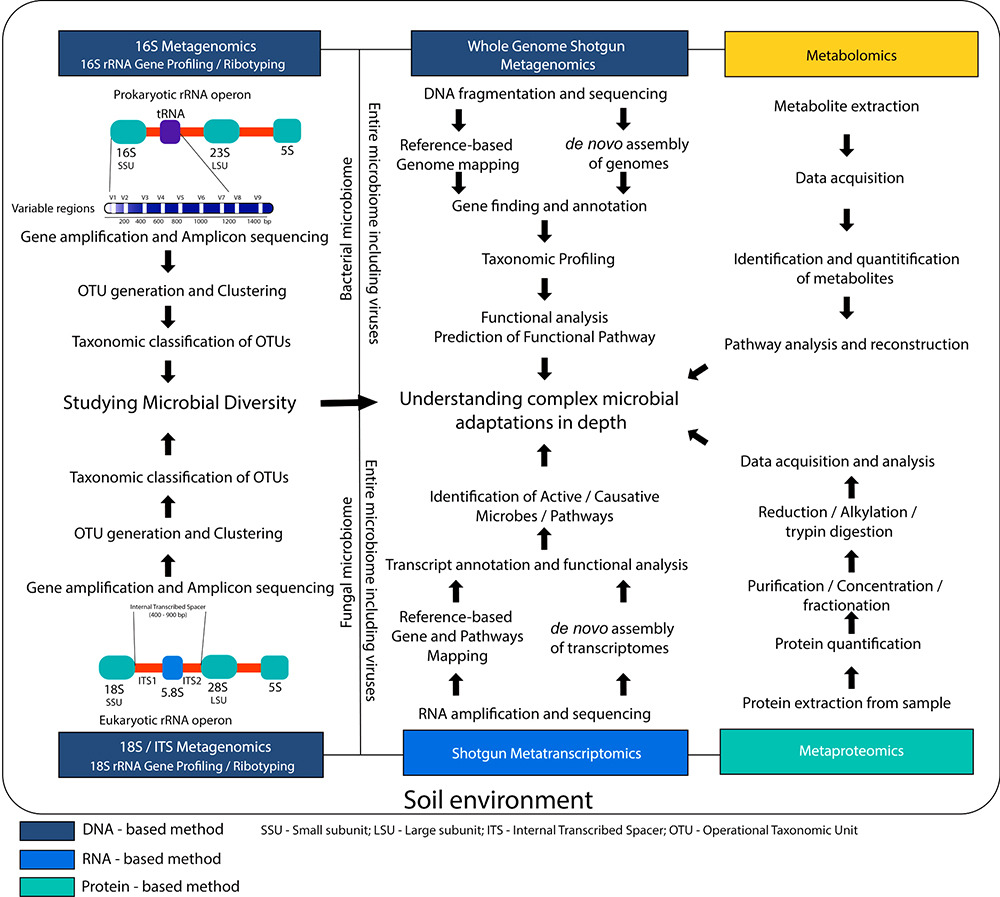 Figure 2
Figure 2Overview of various methods employed in microbiome exploration.
| Server | Application | Platforms |
Databases | Analyses | Output |
|---|---|---|---|---|---|
| GAIA | Web Service | I, ON, IT, R | NCBI (sequences containing 5.8s rRNA genes, 16s rRNA genes, 18s rRNA genes, 28s rRNA genes, ITS1 region and ITS2 region) | QC, Trimming, BWA Alignment, Lowest Common Ancestor Algorithm, Identity Thresholds for OTU Identification, Alpha and Beta Diversity Calculation using phyloseq, Differential Abundance Analysis using DESeq2 | QC, Alpha and Beta Diversity; |
| MG-RAST | Web Service, API | All | SEED, GenBank, RefSeq, IMG/M, Uniprot, eggNOGG, KEGG, PATRIC, greengenes, SILVA, RDP | Sequence Statistics; |
Assignment Tables; |
| One Codex | Web Service, API | I, ON, IT, R, PB, S | RefSeq; NCBI; |
Closed reference: matching the reads to reference; |
Alignment; Sample classification at different levels, |
| Kaiju | Web Service, Source Code | I, R | RefSeqGenomes; proteins from completely assembled (Bacteria, Archaea Viruses) |
Translate nucleotides to protein, Sorting with MEM or Greedy algorithms, Assign taxonomy based on longest or highest scoring match. | .out file with classification evidence |
| Study purpose | Suggested sequencing approach |
|---|---|
| Identification and characterization of an explicit collection of microbiota (exclusive of viruses) in sample(s) | HTTA approach |
| Identification and characterization of the entire microbial DNA in sample(s) | MSS approach |
| Functional profiling | TAS approach |
| Identification and characterization of new/novel microbial | TAS relies on highly curated databases of identified microbes |
Metagenomic studies play a crucial role in exploring the abundance and diversity of microbial communities associated with the commercially important crop plants and a promising avenue for the same. Various NGS platforms and easily available bio-analytical algorithms utilised in metagenomics would facilitate the process of unraveling the complex host-microbe interaction, the associated metabolic pathways which are inevitable for enhanced plant productivity, sequence data to molecular structure and functional possessions. From the ecological perspective, such studies also help in explaining the specific environmental adaptations of microbial communities.
The studies reported in this review along with others highlight the abundance of microbial species in the ecosystem, which indicate that better soil management practices such as the use of biofertilizers could enhance the efficacy of the soil. Phylum phytophthora was found to be the most abundant phyla across the globe (Table 4). Furthermore, with the advent of futuristic approaches such as microbiomics, the host response to the microbial community could be better understood by way of which, plants favoring the beneficial interactions could be chosen for further breeding.
| Geographical location | Phyla Abundance | Reference |
|---|---|---|
| Europe, Argentina, Japan, South Korea Hawaii, Côte d’Ivoire | Phytophthora species | 56-59 |
| China | Acidobacteria, Actinobacteria, Proteobacteria, Bacteroidetes and Firmicutes | 29 |
| South African grasslands | Alphaproteobacteria, Gammaproteobacteria, Pezizomycetes and |
30 |
| India | Actinobacteria | 35 |
| Minnesota, USA | Fusarium community | 37 |
| Ulleungdo and Dokdo | Ectomycorrhizal fungal | 40 |
| Illinois, Colarado and Wisconsin | Frankia | 43 |
| Brazil | Gammaproteobacteria, Solibacteres | 44 |
| Tumkur region of southern India | VAM fungal association | 49 |
| Baja California and Mexico | Ascomycota and Basidiomycota | 53 |
Furthermore, the rapid pace at which metagenomic/sequencing libraries can be prepared with lower cost per base makes these methods desirable for studies demanding deeper sequencing of microbial communities, which can provide comprehensive information on the identity and host-microbe interactions. Several limitations of NGS technologies particularly with the read lengths and error rates can be overcome by using hybrid assemblies. Genomic data obtained from microbiomic studies could contribute to the existing data collated in databases such as one under the program of Microbial Systems in the Biosphere (MSB). Such databases can complement the efforts of plant biologists various beneficial microbe associated traits in crop plants. Some of the challenges associated with such studies are the data management aspects that include storage, processing, distribution, curation and security. Aside of exploration of uncharacterized microbes, analytical modelling through multistep simulations through bioinformatics, efficient data storage and high-performance processing systems such as Cloud-based systems in combination with high throughput sequencing technologies can be achieved. Such cloud-based platforms can not only facilitate superior data management but also improve screening and identification of causative organisms across complex microbiomes and improve the discovery of biocatalytic genes leading to better crop yield and productivity.
DBT-Bioinformatics Facility (BIF) facility, Government of India and Biotechnology Skill Enhancement Program (BiSEP) by Government of Karnataka, at Maharani Lakshmi Ammanni College for Women (mLAC), Bengaluru, India for providing the facility.
HTTA
High-throughput targeted-amplicon sequencing
Shotgun metagenomics sequencing
Metagenomics shotgun sequencing
Targeted-amplicon sequencing
Illumina
Roche454
Oxford Nanopore
SOLiD
PacBio
Ion Torrent