






 , Wenqi Gao 3,*
, Wenqi Gao 3,*1 Department of Pharmacy, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
2 Department of Hematology, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University and Technology, 430015 Wuhan, Hubei, China
3 Institute of Maternal and Child Health, Wuhan Children’s Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University and Technology, 430015 Wuhan, Hubei, China
4 Department of Radiation and Medical Oncology, Zhongnan Hospital, Wuhan University, 430071 Wuhan, Hubei, China
† These authors contributed equally.
Abstract
Purpose: The purpose of our present study was to, for the
first time, identify key genes associated with postpartum depression (PPD) and
discovery the potential molecular mechanisms of this condition. Methods:
First, microarray expression profiles GSE45603 dataset were acquired from the
Gene Expression Omnibus (GEO) in National Center for Biotechnology Information
(NCBI). The weighted gene co-expression network analysis (WGCNA) was performed to
identify the top three modules from differentially expressed genes (DEGs).
Furthermore, cross-validated differential gene expression analysis of the top
three modules and DEGs was used to identify the hub genes. Gene set enrichment
analysis (GSEA) was conducted to identify the potential functions of the hub
genes. We conducted a Receiver Operator Characteristic (ROC) curve to verify the
diagnostic efficiencies of the hub genes. Lastly, GSE44132 dataset was used to
search the association between the methylation profiles of the hub genes and
susceptibility to PPD. Results: Altogether, 8979 genes were identified
as DEGs for WGCNA analysis. The turquoise, yellow, and green functional modules
were the most significant modules related to PPD development after WGCNA
analysis. The enrichment analysis results of the Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway demonstrated that hub genes in the three modules were
mainly enriched in the neurotrophin signaling pathway, chemokine signaling
pathway, Fc
Keywords
- WGCNA
- Postpartum depression
- Potential diagnostic biomarkers
Postpartum depression (PPD) is one of the most prominent mood disorders affecting 15% of parturient women [1]. Its clinical symptoms typically occur within two weeks after delivery, with patients reporting persistent and severe moodiness, including negative emotions (e.g., anxiety, sadness, hopelessness and/or worthlessness), low energy, and social withdrawal [2]. PPD may result in both short-term and long-term negative effects on neonatal and infant development, cognition, emotion, and behavior. Previous studies have shown that maternal depression is a risk factor for higher rates of premature and low-birth-weight babies, infant malnutrition and stunting, and infant diarrhoeal, which can lead to mothers’ reluctance to care for their children and impair normal mother-child relationships. And it’s becoming a major public health problem [3].
PPD is the result of the combined influence of multiple factors. Previous studies have reported hypotheses regarding the physiopathologic mechanisms of PPD, such as the abnormal changes of hormones, neurotransmitters, inflammatory factors [4], among others. The molecular pathways implicated in the PPD was involved in genetic inheritance [5], HPA axis [6], serotonin transporter [7], and GABA receptor [8]. However, there is no exact evidence to support any of the above conclusions, and the physiopathologic mechanisms of PPD remain largely obscure.
Despite the severity of PPD on adverse infant and maternal outcomes, early diagnosis would help establish mechanisms for the prevention and early cure of PPD. Therefore, it is necessary to identify reliable diagnostic biomarkers of PPD. In practical work, it is difficult to acquire brain tissue for clinical testing. Peripheral blood mononuclear cells (PBMCs) share mRNA expression patterns in brain tissues [9]. In both PBMCs and the prefrontal cortex, the expression levels of many genes and biological processes are similar [10]. On the one hand, mounting evidence has indicated that gene expression changes in PBMCs may intervene molecular alterations in the region of brain [11]. On the other hand, gene expression of peripheral lymphocytes may be affected by the central nervous system (CNS) via neurotransmitters, cytokines, and hormones [12]. Therefore, PBMCs can serve as an effective tissue to search the gene expression in PPD.
With the development of high-throughput expression microarray technology, gene expression profiling has become a powerful tool for in-depth study of the pathogenesis and diagnostic biomarkers of mental disorders at the genomic level [13]. For example, microarrays have been used to detect differences in gene expression characteristics between healthy controls and PPD patients [14]. Many microarray-based studies have focused only on screening for differentially expressed genes (DEGs) without identifying their links. Genes with similar expression patterns may be functionally related. Weighted gene co-expression Network analysis (WGCNA) algorithm can divide genes into different modules according to the similarity of gene co-expression in multiple samples. This produces a set of genes that function similarly, allowing selection of disease-related biomarkers and pathways. In the present study, we, for the first time, used WGCNA to construct a co-expression network of genes and determined key modules and hub genes associated with PPD.
The design of this study was exhibited in Supplementary Fig. 1. We downloaded the mRNA microarray expression profile dataset GSE45603 from GEO, a free and publicly available database. This dataset was used to screen DEGs, then construct co-expression networks using WGCNA, and finally to identify hub genes associated with PPD.
There were 210 samples from PBMCs in the GSE45603 dataset, from which 21 samples were finally selected, containing 5 healthy control samples (GSM1110484, GSM1110493, GSM1110498, GSM1110515, GSM1110539) and 16 PPD samples (GSM1110374, GSM1110378, GSM1110380, GSM1110400, GSM1110403, GSM1110409, GSM1110412, GSM1110424, GSM1110433, GSM1110447, GSM1110461, GSM1110477, GSM1110505, GSM1110506, GSM1110518, GSM1110547), the samples of preconception, 1st, 2nd, and 3rd trimester of pregnancy and postpartum were not included in present study.
GEO2R was used to analyze DEGs between PPD and healthy control samples,
p
Gene annotation with functions involving cellular components (CC), molecular
functions (MF), and biological pathway (BP) were analyzed by Gene ontology (GO)
[15, 16]. The key MF, BP, CC and pathways of statistically significant DEGs were
visualized (p
The results from the STRING (version 11.5, https://www.string-db.org/) [17] were
analyzed and structured using Cytoscape software (Cytoscape version 3.9.0,
https://cytoscape.org/). The MCODE plugin was employed to identify modules of the
PPI network. Two modules were selected (score
| Gene symbol | log |
p-value | Score |
| UBA52 | 0.56 | 1.21 |
72 |
| HNRNPA2B1 | –0.847 | 6.80 |
43 |
| IL10 | –0.783 | 8.90 |
39 |
| NHP2 | 0.581 | 2.52 |
35 |
| CAT | –1.04 | 1.77 |
34 |
| CDC5L | –0.517 | 3.39 |
34 |
| PTGES3 | –0.748 | 3.87 |
34 |
| RPL23 | –0.847 | 4.84 |
34 |
| SIRT1 | –0.512 | 3.18 |
34 |
| RPS3A | –0.725 | 2.28 |
33 |
| CCT3 | 0.503 | 3.58 |
32 |
| RPS28 | –0.787 | 2.82 |
32 |
| ALDH18A1 | 0.797 | 7.78 |
31 |
| ATM | –0.821 | 4.56 |
31 |
| PABPC1 | –1.2 | 7.00 |
31 |
| ACTR3 | –0.907 | 8.00 |
30 |
| RPL13A | 0.538 | 2.57 |
30 |
| RPL37A | –0.511 | 4.49 |
30 |
| FBL | 0.741 | 2.34 |
29 |
| RPL10A | 0.538 | 5.93 |
29 |
| RPS15A | –0.607 | 1.36 |
29 |
| SPI1 | –0.73 | 2.77 |
29 |
| CCL5 | 0.536 | 2.14 |
28 |
| RPS29 | 0.515 | 3.66 |
28 |
| CCR7 | 0.582 | 2.04 |
27 |
| HSPE1 | –1.13 | 3.65 |
27 |
| KPNB1 | –0.516 | 2.26 |
27 |
| RPS27 | –0.551 | 2.97 |
27 |
| RAD51 | –0.501 | 1.97 |
26 |
| RPS26 | 1.28 | 2.79 |
26 |
There were 8979 DEGs after analysis by GEO2R. WGCNA (Version 1.6.0, https://git.bioconductor.org/packages/GmicR) was performed using 8979 DEGs [20]. The adjacency matrix was transformed into topological overlap matrix (TOM) by one-step method, and the minimum value was 30. Hierarchical clustering was used to generate hierarchical clustering tree [21, 22]. The co-expressed gene modules were generated by hierarchical clustering tree with different colors, and the module structure was displayed by topological overlapping matrix.
Correlations between modular characteristic genes (MEs) and clinical features were calculated to identify relevant modules. Gene significance (GS) was measured as the log10 transformation of the p-value (GS = lg p) of the linear regression between gene expression and clinical information. Modular significance (MS) was the average GS of all genes in a module. Overall, of all the selected modules, the module with the absolute first rank in MS was considered to be relevant for clinical features. In our study, the genes in key modules were imported into Cytoscape and screened out according to degree. Key modules and two genes were overlapped using Venn diagrams.
The distribution of all DEGs in GSE45603 was identified, and the functional similarity between proteins was evaluated by GOSemSim package (version 2.18.1, http://bioconductor.org/packages/release/bioc/html/GOSemSim.html) [23]. Then, the methylation of the hub gene was detected. Genome-wide models of prenatal DNA methylation analysis data (GSE44132) were also downloaded. This dataset includes 55 samples. Among these, 25 samples without depression (GSM1079474, GSM1079478, GSM1079480, GSM1079481, GSM1079482, GSM1079483, GSM1079484, GSM1079485, GSM1079486, GSM1079488, GSM1079491, GSM1079492, GSM1079493, GSM1079494, GSM1079495, GSM1079497, GSM1079498, GSM1079499, GSM1079501, GSM1079503, GSM1079504, GSM1079505, GSM1079507, GSM1079510, GSM1079528) and 11 samples with PPD were selected (GSM1079475, GSM1079476, GSM1079477, GSM1079479, GSM1079487, GSM1079489, GSM1079490, GSM1079496, GSM1079500, GSM1079502, GSM1079506). The raw data were analyzed using “minfi” package (version 1.38.0, http://www.bioconductor.org/packages/release/bioc/html/minfi.html) in R and were converted into a score, referred to as a beta value.
Receiver operating characteristic (ROC) analysis combines sensitivity and specificity to comprehensively evaluate diagnostic accuracy or discriminant effect. The “pROC” package (version 1.68.1, http://www.bioconductor.org/packages/release/bioc/html/ROC.html) [24] was applied to evaluate the diagnostic value of hub genes and to screen out the diagnostic biomarkers of PPD.
All statistical analysis were measured as mean
There was a total of 21 PBMCs samples in this study, which included 5 healthy
control and 16 PPD samples. There were 8979 DEGs. Then, the cutoff criteria were
adjusted p value
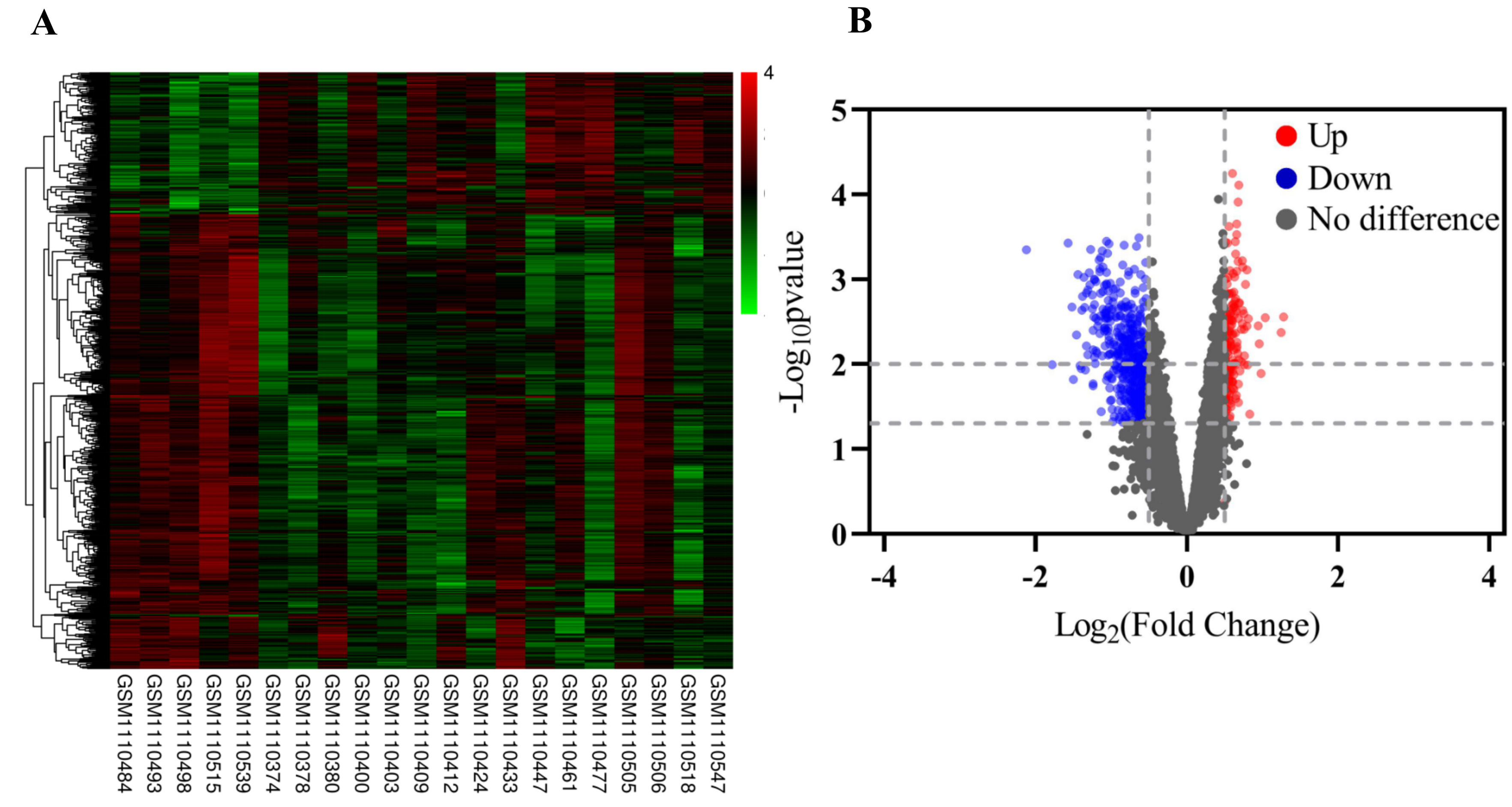 Fig. 1.
Fig. 1.The DEGs from GSE43056. (A) Heat map of differentially expressed genes. (B) Volcano plot of gene expression. Red means upregulated DEGs; green means downregulated DEGs; black means no difference.
GO function annotation and KEGG pathway enrichment analysis were conducted to obtain more comprehensive knowledge of the selected DEGs. As for BP, the upregulated DEGs were mainly implicated in the regulation of translation, rRNA processing, regulation of immune response, and SRP-dependent cotranslational protein targeting the membrane. Mitochondrion, T cell receptor complex, ribosome, mitochondrial inner membrane, and cytosolic large ribosomal subunits were involved in the CC. Changes in MF were mainly enriched in the structural constituents of ribosomes, methyltransferase activity, poly (A) RNA binding, protein binding, and mRNA binding (Supplementary Fig. 2A). The downregulated DEGs were mainly responsible for reciprocal meiotic recombination, antigen processing, and presentation of exogenous peptide antigen via MHC class I, TAP-independent, positive regulation of long-term synaptic potentiation, positive regulation of macroautophagy, and negative regulation of gene expression in BP. Cytoplasmic, cytosolic, phagocytic vesicle membrane, NADPH oxidase complex, and focal adhesion in CC. Protein binding, four-way junction DNA binding, superoxide-generating NADPH oxidase activator activity, superoxide-generating NADPH oxidase activity, and enzyme binding in MF (Supplementary Fig. 2B). The enrichment analysis results of KEGG pathway demonstrated that upregulated DEGs were mainly involved in ribosome enrichment, biosynthesis of amino acids, oxidative phosphorylation, ribosome biogenesis in eukaryotes, and hematopoietic cell lineage (Supplementary Fig. 2C). The downregulated DEGs were mainly enriched in phagosomes, osteoclast differentiation, antigen processing and presentation, glucagon signaling pathway, and Forkhead box O3 (FoxO) signaling pathway (Supplementary Fig. 2D).
The PPI network consisted of 571 nodes and 2213 edges, and module A (score =
17.263, including 20 nodes and 164 edges; Supplementary Fig. 3A), and B
(score = 8.5, including 37 nodes and 135 edges; Supplementary Fig. 3B)
with score
Firstly, we checked the data quality in GSE45603. All samples were taken for subsequent analysis (Supplementary Fig. 5). DEGs containing 8979 genes was analyzed using WGCNA, and modules containing highly related genes were identified. Based on the approximate scale-free topology criterion, soft threshold power was 14 (scale-free R2 = 0.86) was optimized (Supplementary Fig. 6A and Supplementary Fig. 6B). There were 12 modules (Fig. 2A), 1921 genes in turquoise module, 704 genes in yellow module, and 447 genes in green module. The 2736 genes that could not be included in any module were placed into the gray module and identified as non-co-expressed.
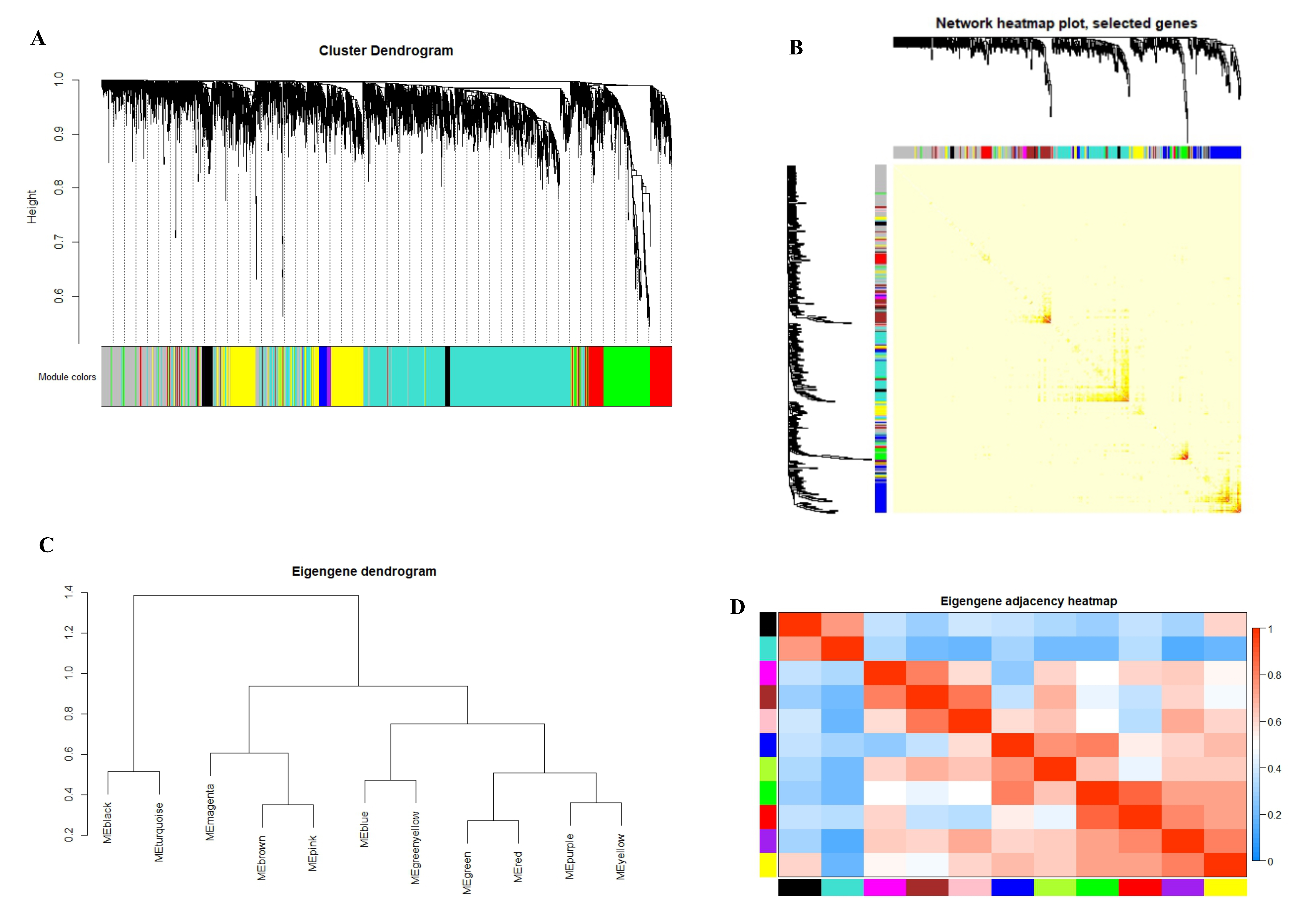 Fig. 2.
Fig. 2.Construction of co-expression modules by the WGCNA. (A) The cluster dendrogram of genes in GSE45603. Branches of the cluster dendrogram of the most connected genes gave rise to eleven gene co-expression modules. Every gene represents a line in the hierarchical cluster. The distance between two genes is shown as the height on the y-axis. (B) Interaction relationship analysis of 400 selected co-expression genes. Different colors of the horizontal axis and vertical axis represent different modules. The brightness of yellow in the middle represents the degree of connectivity of different modules. There was no significant difference in interactions among different modules, indicating a high degree of independence among these modules. (C) Hierarchical clustering of module hub genes that summarize the modules yielded in the clustering analysis. (D) Heatmap plot of the adjacencies in the hub gene network.
The 400 genes were selected to draw a network heat map (Fig. 2B). Our results so far showed that the turquoise, yellow, green, and blue modules were independently verified and showed a high degree of independence between modules and the relative independence of the genes expressed in each module. The 11 modules were divided into two main clusters: one consists of two modules (turquoise, black), while the other consists of nine modules (magenta, brown, pink, blue, greenish-yellow, green, red, purple and yellow modules; Fig. 2C). Finally, heat maps were drawn according to the connectivity of characteristic genes to visualize the results (Fig. 2D).
The turquoise, yellow, and green modules were visualized by Cytoscape, and the top 30 genes were screened out by sorting node degree candidate genes for further analysis (Fig. 3A–C). Cross-validation of the data from these three modules and DEGs revealed 3 genes (HNRNPA2B1, IL10, and RAD51) in both DEGs and the green module, 4 genes (UBA52, NHP2, RPL13A, FBL) in both DEGs and the turquoise module, and 1 gene (SPI1) in both DEGs and the yellow module (Supplementary Fig. 7A). These 8 genes were observed based on the average functional similarity (Supplementary Fig. 7B).
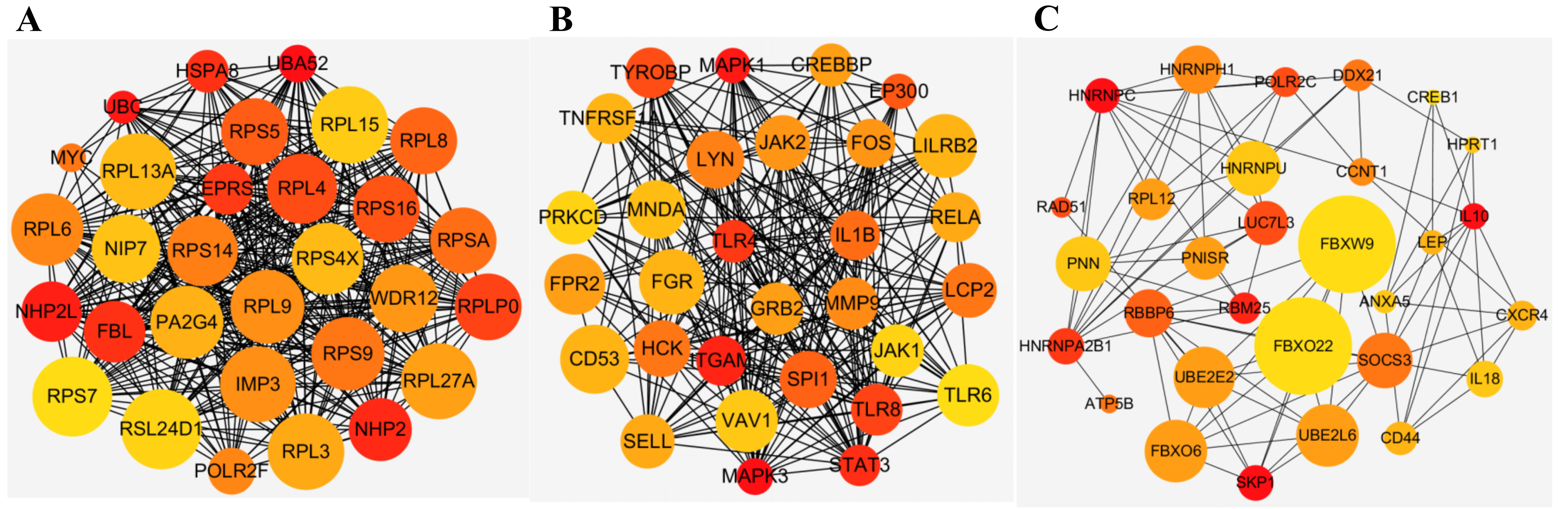 Fig. 3.
Fig. 3.Top 30 genes in (A) turquoise module, (B) yellow module, and (C) green module. The size of the node represents the clustering coefficient. The color of the node indicates its degree, the bigger the node, the greater the number of connections it has, blue, yellow and orange color represent big, middle and small degrees respectively. (A) Hub genes in turquoise module. (B) Hub genes in yellow module. (C) Hub genes in green module.
The diagnostic efficiencies of 8 hub genes were evaluated by ROC curve analysis (Fig. 4). The AUC of ROC curves were 0.902, 0.790, 0.797, and 0.808 for HNRNPA2B1, interleukin 10 (IL-10), RAD51, and ubiquitin A-52 residue ribosomal protein fusion product 1 (UBA52), respectively (Fig. 4A). The AUC of ROC curves were 0.827, 0.818, 0.865, and 0.763 for NHP2, ribosomal protein L13a (RPL13A), fibrillarin (FBL), and Spi-1 proto-oncogene (SPl1), respectively (Fig. 4B). AUC values within the range of 0.700–0.900 were considered to have moderate accuracy. Thus, HNRNPA2B1, IL10, RAD51, UBA52, NHP2, RPL13A, FBL, and SPI1 were the hub genes with higher diagnostic value.
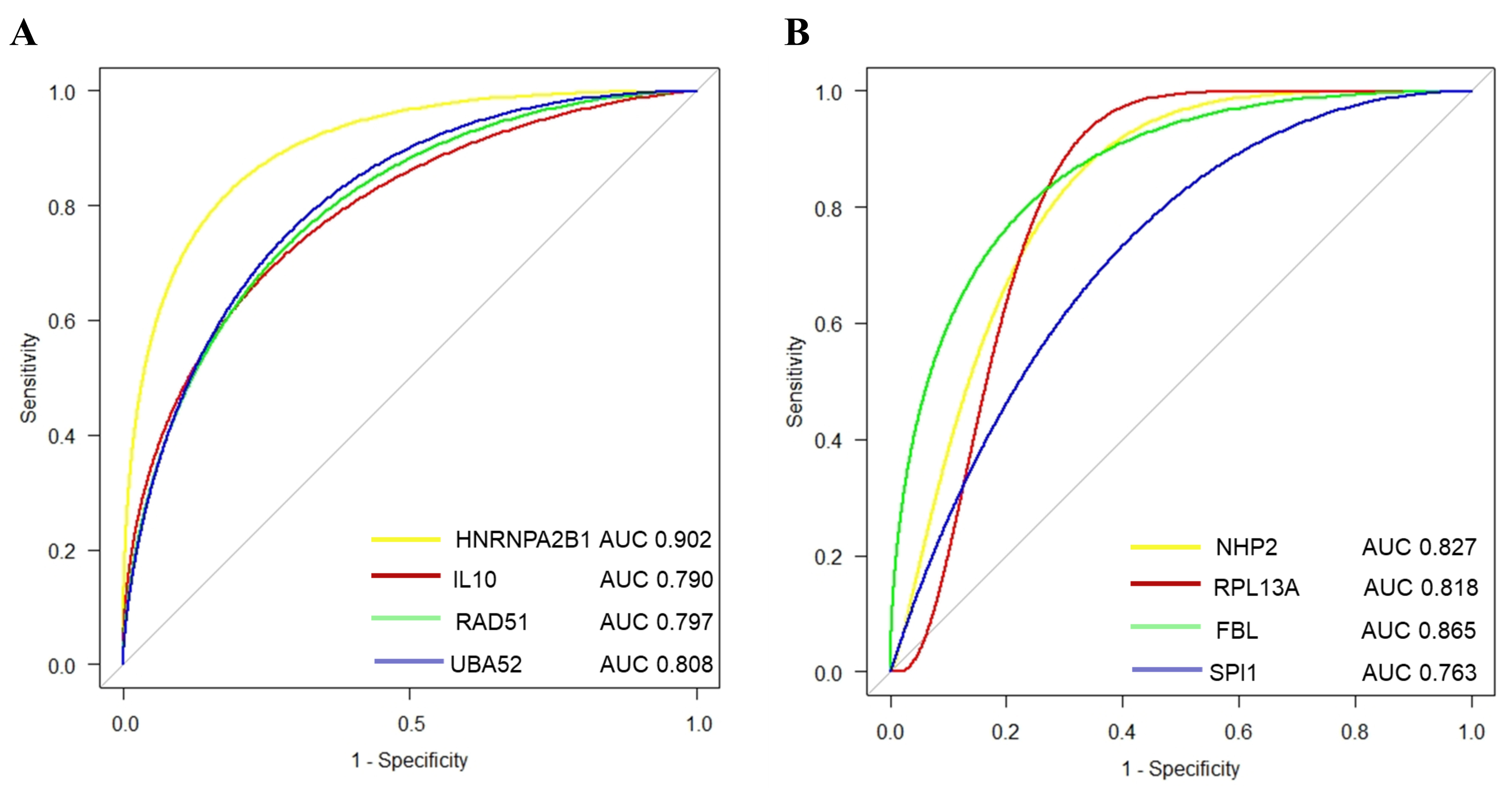 Fig. 4.
Fig. 4.Validation of diagnostic value for the hub genes in PPD. (A,B) ROC curve of the hub genes for PPD diagnosis.
Using GSE44132 dataset, we investigated the relationship between methylation levels of 8 genes and PPD susceptibility, and explored the possible mechanism of PPD occurrence. Data from 36 whole blood samples (11 PPD samples and 25 healthy controls) passed quality control indicators and were analyzed. In the GSE45603 dataset, HNRNPA2B1 was down-regulated, and RPL13A and UBA52 were significantly up-regulated in the PPD group, compared with the control group (Fig. 5A). It’s worth noting that, highly differentiated methylation sites were found in HNRNPA2B1, RPL13A, and UBA52 genes: cg19062098 (p-value = 0.036), cg18208268 (p-value = 0.039), and cg25699533 (p = 0.012) (Fig. 5B).
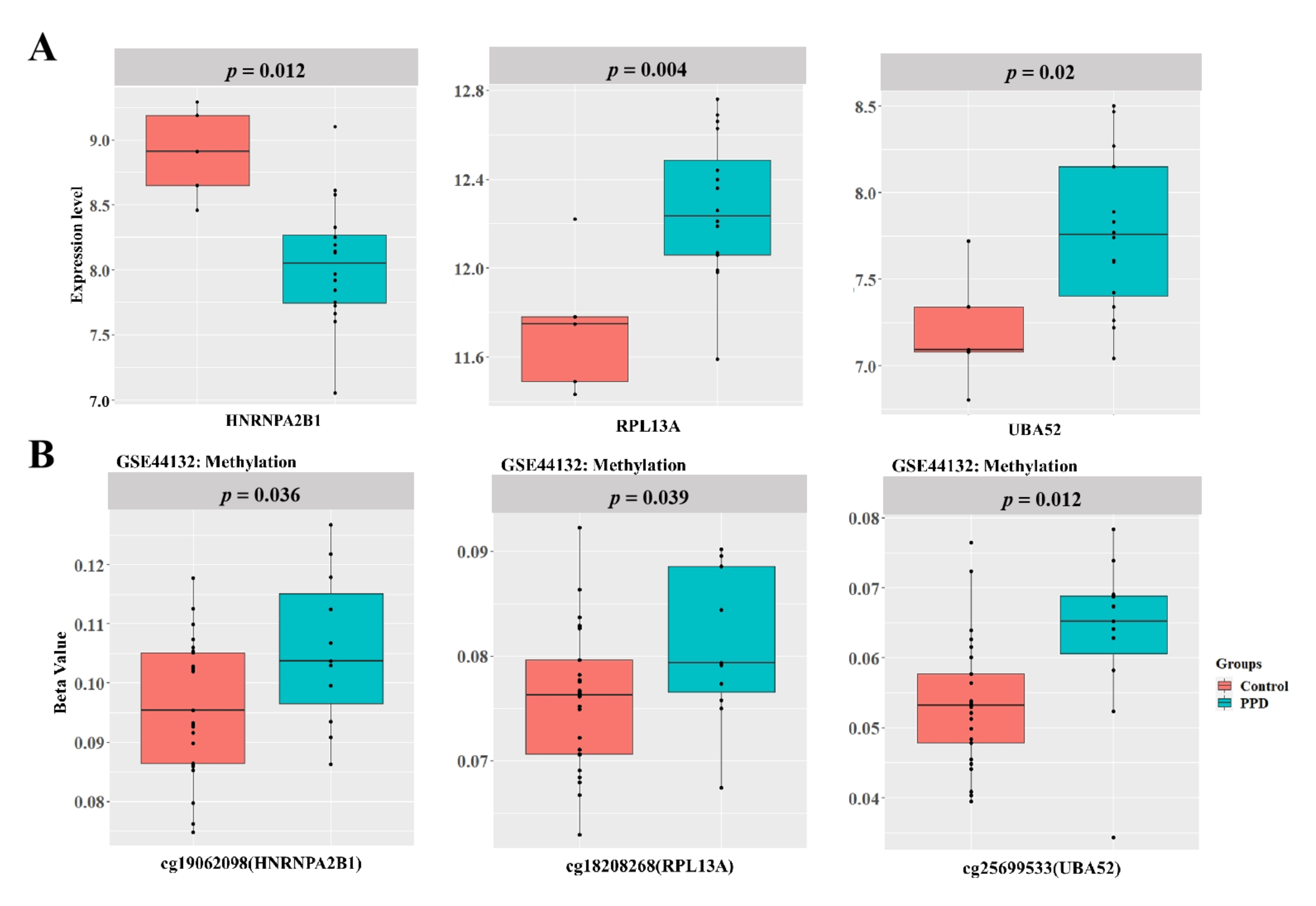 Fig. 5.
Fig. 5.The expression and methylation levels of HNRNPA2B1, RPL13A and UBA52. (A) The expression levels of the three genes (HNRNPA2B1, RPL13A and UBA52) were correlated with PPD (based on microarray data of GSE44132). (B) The methylation levels of HNRNPA2B1, RPL13A and UBA52 (based on microarray data of GSE44132).
Because HNRNPA2B1, RPL13A, and UBA52 had highly significantly differentiated methylated positions, we selected the three genes for follow-up studies. To identify the potential functions of these hub genes, GSEA was conducted to identify enriched biological processes in the samples. Geneset enrichment analysis (GSEA) for gene sets related to HNRNPA2B1, RPL13A and UBA52 expressions. Our results found that, compared with healthy control, HNRNPA2B1 was down-regulated and RPL13A and UBA52 were up-regulated in the PPD samples, respectively (Fig. 5A), thus we considered that lowly expression of HNRNPA2B1 and highly expressions of RPL13A and UBA52 were involved in which pathways. Results suggested that HNRNPA2B1 was lowly expressed in the gene set of “OLFACTORY_TRANSDUCTION”, “BUTANOATE_METABOLISM”, and “MELANOMA” (Fig. 6A). RPL13A and UBA52 were highly expressed in the gene sets of “AMINOACYL_TRNA_BIOSYNTHESIS”, “LYSINE_DEGRADATION” and “BUTANOATE_METABOLISM” (Fig. 6B,C).
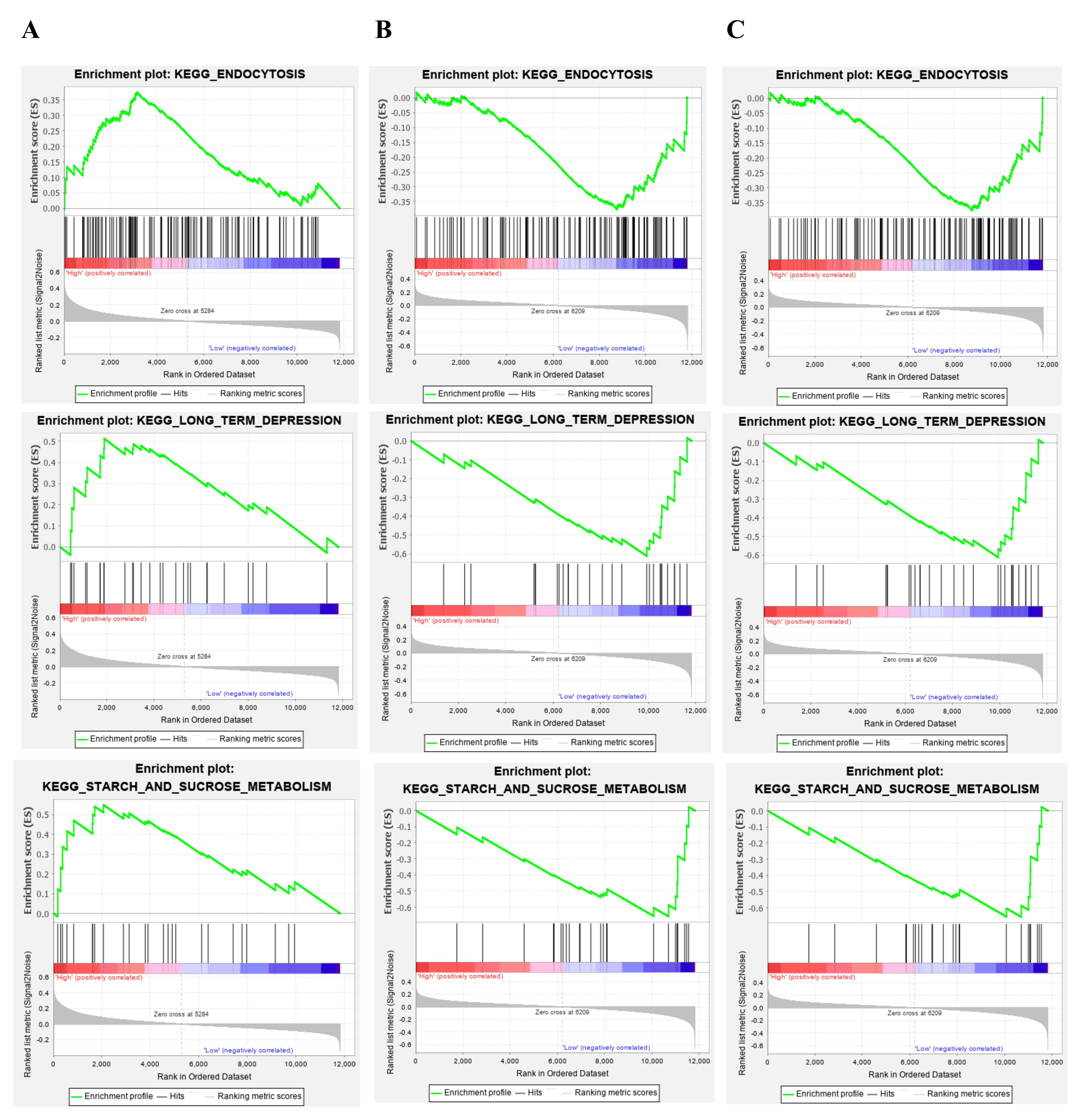 Fig. 6.
Fig. 6.Gene set enrichment analysis (GSEA) for gene sets related to HNRNPA2B1, RPL13A and UBA52 expressions. (A) HNRNPA2B1 was lowly expressed in the gene set of “OLFACTORY_TRANSDUCTION”, “BUTANOATE_METABOLISM”, and “MELANOMA”. (B) RPL13A was highly expressed respectively in the gene sets of “AMINOACYL_TRNA_BIOSYNTHESIS”, “LYSINE_DEGRADATION” and “BUTANOATE_METABOLISM”. (C) UBA52 was highly expressed in the gene sets of “AMINOACYL_TRNA_BIOSYNTHESIS”, “LYSINE_DEGRADATION” and “BUTANOATE_METABOLISM”.
PPD is a major public health concern, affecting 10 to 15 percent of mothers
worldwide [25]. Investigations are being performed to develop a non-invasive and
quantitative clinical test to support PPD diagnosis, nevertheless, no specific or
sensitive biomarkers in whole blood are currently available for the diagnosis of
PPD [26, 27]. Thus, we performed several bioinformatic methods to identify the
exact pathological mechanisms and effective diagnostic biomarkers of PPD. In the
first part of our study, we determined the hub genes and pathways associated with
PPD within whole blood samples. A total of strictly screened 673 DEGs
(
Recently, it has been widely accepted that endocrine [28], immune response [29], and genetic and epigenetic factors [30], are all involved in the pathogenesis of PPD. Our study found that genes in the three modules identified by WGCNA (turquoise, green, and yellow modules) were mainly enriched in innate immune response, signal transduction, intracellular signal transduction, inflammatory response, and viral processes. The green module was enriched in the regulation of transcription (DNA-templated), gene expression, response to ionizing radiation, and RNA processing.
According to the cross-validation of data from the three modules and DEGs, eight
genes acted as high degree genes. Among these eight genes, we were interested in
the HnRNPA2B1, Fibrillarin (FBL), and Spi-1 proto-oncogene (SPI1). HnRNPA2B1, an
RNA-binding protein and one member of the heterogeneous nuclear ribonucleoprotein
family, initiates and amplifies the innate immune response. HnRNPA2B1
dimerization is required for nucleocytoplasmic translocation and initiation of
IFN-
There were two previous studies to find biomarkers of PPD based on GSE45603 dataset. Lauren et al. [51] reported that the objective of this study was to independently replicate their previously published prediction model of postpartum depression and women without a history of psychiatric disorders, and to further investigate the DNA methylation status of postpartum depression biomarkers associated with changes in pregnancy hormone levels and the timing of major hormonal changes. Maria et al. [52] focused on whether abnormal patterns of circulating levels of cytokines and chemokines may offer a suitable biomarker for disease development and/or therapeutic response. The objective of the present study and the previous two study were both searching for biomarkers of PPD. However, there were several differences among these studies. Our present study only observed the postpartum mental state and healthy controls. Lauren et al. collected different gestation process samples and used their own model to identify biomarker, their key point was changes in hormone levels. The emphasis of Maria’ study was chemokines levels. Our present study did not classify genes, we put all DEGs into WGCNA and obtained hub genes, and the functions of genes were involved in various aspects of cell.
There were several limitations to our present study. First, in order to comprehensively determine dysfunctions related to PPD, both brain tissue and blood samples would need to be integrated. However, our present study did not perform an analysis of brain tissue. Second, only one microarray was included in the computerized analysis, and likely leading to one-sided results and a high false-positive rate. Third, our present study only performed data mining and data analysis and did not conduct any experiments to validate the results. Finally, there were only 5 healthy control samples and 16 PPD samples selected for analysis; thus, it is necessary to increase the sample size to validate the accuracy of the results in a follow-up study.
Based on our knowledge, we for the first time used the system biology-based
WGCNA method to predict several potential biological pathways and diagnostic
biomarkers involved in PPD. WGCNA and co-expression network analysis identified
key biological processes and signaling pathways, especially the neurotrophin
signaling pathway, chemokine signaling pathway, Fc
GW designed this study. GW and XH provided administrative support. DZ and CW collected the data, writed the manuscript. LJ, DA, YY, TJ, and YC analyzed the data. All the authors approved the final version of manuscript.
Not applicable.
Not applicable.
This study was supported by grants from the Wuhan Clinical Medical Research Project (WX20C15).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://www.fbscience.com/Landmark/articles/10.52586/5006.
PPD, Postpartum depression; PBMCs, Peripheral blood mononuclear cells; HNRNPA2B1, Heterogeneous nuclear ribonucleoprotein A2/B1; NHP2, NHP2 ribonucleoprotein; RAD51, RAD51 recombinase; CXCL1, C-X-C motif chemokine 1; GSEA, Gene set enrichment analysis; FBL, Fibrillarin; SPI1, Spi-1 proto-oncogene.