




Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Biotechnology, Indian Institute of Technology Roorkee, Roorkee – 247667, Uttarakhand, India
Abstract
Chemokines are small regulatory proteins that play a crucial role in the coordinated migration of cell populations to the site of infection/inflammation by binding to their cognate receptors. In principle, chemokine receptors, which are serpentine G protein-coupled receptors (GPCRs), mediate the series of downstream intracellular signaling events that occur inside the cells to resolve the pathogenicity. Intracellular signaling pathways regulated by the kinase protein sub-families are the center of attention for chemokine derived functional responses. Kinases potentially influence cell migration, cell growth, transcriptional activation, and other essential molecular events. The regulation and flow of the signals by the kinases are different for each physiological and pathological event and are tightly regulated by the nature and pairing of chemokine(s) with its receptor(s). For example, phosphoinositide 3-kinase (PI3K) is activated during the initial steps of the chemokine induced signaling cascade to regulate chemotaxis, transcription, and cell survival. G protein-coupled receptor kinase (GRKs) plays a crucial role in the desensitization and internalization of the chemokine receptors. The regulation of chemokine receptor is also governed by kinases like protein kinase A (PKA), protein kinase C (PKC), mitogen-activated protein kinases / extracellular signal-regulated kinases (MAPK/ERK), etc. It was also established that tyrosine-protein kinases (TECs) such as ITK and RLK play a significant role in chemokine signaling in T lymphocytes. On a similar note, many others like janus kinases (JAKs), Protein kinase B (PKB), PKC, etc. are also studied in chemokine mediated disease models. The present review elucidates the role of different kinases involved in the chemokine/chemokine receptor mediated signaling cascade during various pathophysiological processes.
Keywords
- Chemokines
- Chemokine receptors
- Kinases
- G protein coupled receptors
- Molecular signaling
- Leukocyte trafficking
- Review
Chemokines are small (8-10 kDa) signaling proteins that regulate cell trafficking. They play many important roles in multiple physiopathological activities like hematopoiesis, wound healing, organogenesis, inflammation, angiogenesis, tumorigenesis by regulated cell trafficking (1-5). Approximately 50 chemokines have been reported in humans to date. Depending upon the location of the cysteine residue at N-terminal, they are classified into two major (CXC and CC), and two minor (CX3C and C) families. Chemokines bind to cell surface glycosaminoglycans and G-protein coupled receptors (GPCRs) to perform their biological functions (5-8).
All the members of chemokine sub-groups essentially form a similar monomeric structure. It comprises of a disordered N-terminal, followed by an extended N-loop, three-stranded β-sheets forming the core of protein, and a helix at the C-terminal (Figure 1A) (9). However the oligomerization or dimerization of CXC and CC type chemokines vary significantly. In case of CXC chemokines such as CXCL8/IL8 (Interleukin-8), the intermolecular interactions between the C-terminal helix to the β-strand and the H-bonding between the β1-β1’ sheets mediate the dimerization (Figure 1B). Whereas, For CC chemokines such as CCL2, the extended N-loop forms the dimerization interface without involving the C-terminal helix (Figure 1C). Indeed, chemokines such as CXCL7 forms higher order oligomers such as tetramers, where they use both the CC and CXC type dimer interfaces (Figure 1D).
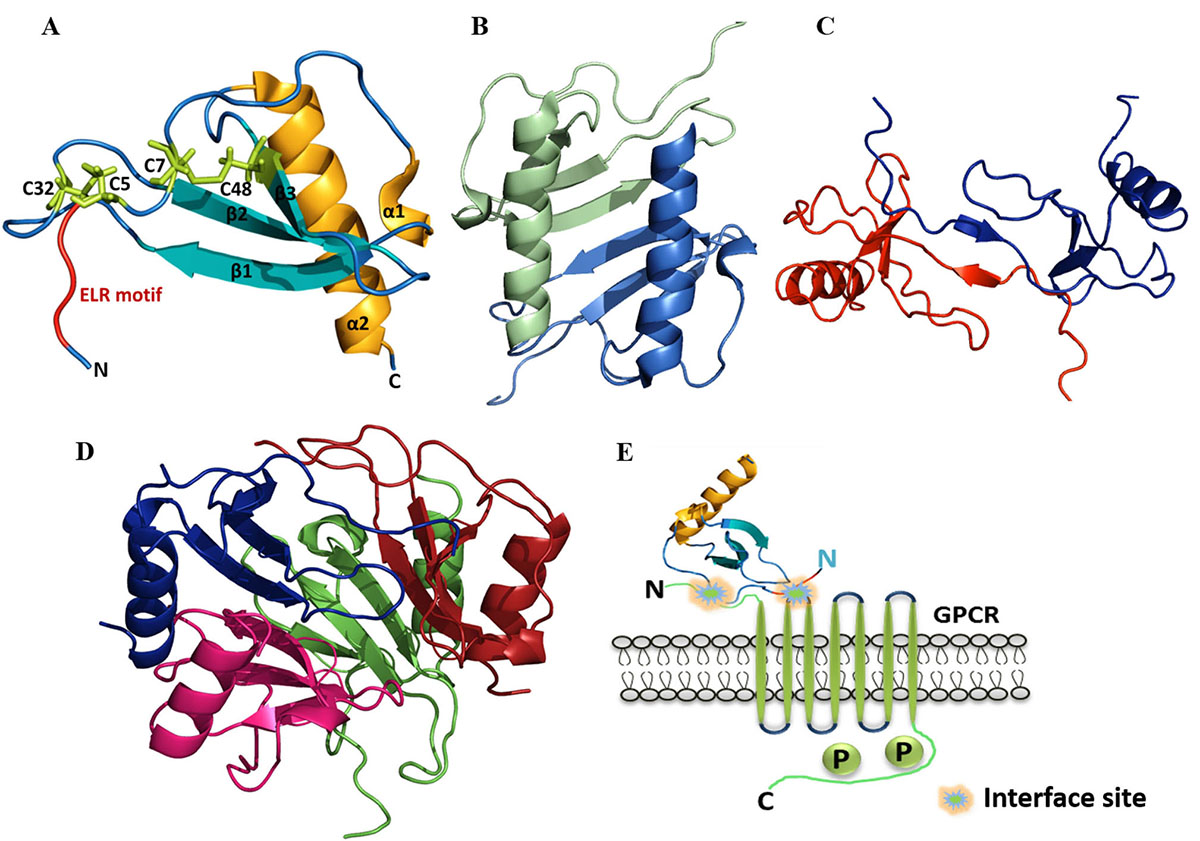 Figure 1
Figure 1A) Structural components in the monomeric structure of ELR-CXC (IL8) chemokine (PDB ID: 5D14). B) Dimeric structure of Human CXCL8 (PDB ID: 1IL8). C) Dimeric structure of Human CCL2 (PDB ID: 1DOM). D) Tetrameric structure of CXCL7 (PDB ID: 1NAP). E) Schematic representation of the interaction between monomeric chemokine and GPCR.
G protein-coupled receptors (GPCRs) are the prevalent family of cell surface receptors which evolved to sense and communicate wide range of extracellular signals (10). GPCR based signaling is involved in various biochemical cascades such as immune, endocrine, cardiovascular, nervous, etc. Their existence on the plasma membrane makes GPCRs easily accessible to the drug molecules (agonist and antagonist) for the stimulation and blockade of the physiological processes. It is well manifested that defects in the receptor signaling triggered by genetic mutation or by overexpression of the receptors lead to various pathological conditions such as cancer, atherosclerosis, dwarfism, etc. (11, 12). The abundant presence of GPCRs on eukaryotic cells and its involvement in the human diseases accentuate the significance of studying the GPCRs. It is well elucidated that affinity of chemokines towards GPCR binding and activation significantly vary with their oligomerization (13). In general, the N-terminal domain of the chemokine binds to receptors at its N-terminal loop and first extracellular loop as shown in Figure 1E.
Binding of the chemokine to its cognate heptahelical GPCR induces a number of cellular responses like migration, adhesion, and different events of chemotaxis including changes in the shape of the cells, actin cytoskeleton reorganization, integrin expression, and activation, etc. (14, 15). Moreover, a particular chemokine can lead to a different response on the basis of the receptor to which it binds, and also the cells over which they are positioned (16). Chemokines and their GPCRs are promiscuous and can signal through multiple subtypes of G protein monomers, thus ensuring the regulated recruitment of the individual cells at a given period (7, 17). A wide range of proteins are involved in the synchronous administration of signaling pathways mediated by the chemokines and their receptors. Among them, chemokine activated intracellular “kinases” are the most crucial members (17, 18).
Kinases are the enzymes that catalyze the reaction in which the γ-phosphate from adenosine triphosphate (ATP) is transferred to the substrate having a hydroxyl group. The catalytic fraction of the kinase is highly conserved and comprises of ATP binding sites. Kinases play a significant role in signal transductions, and regulate wide range of cellular processes like differentiation, transcriptional activation, apoptosis, metabolism, etc.(19). As protein phosphorylation plays a significant role in intracellular communication, abnormal phosphorylation leads to the generation of numerous diseases including neurodegenerative diseases, rheumatoid arthritis, immunodeficiency, cardiovascular diseases, endocrine disorders and cancer (20). The deregulated activity of kinase is an indicator of inflammation and cancer; thus targeting the activity of kinase enzyme has become a promising therapy for numerous diseases. The therapies comprise of kinase inhibitors that are being profiled in screening of kinome (21, 22).
Given the ubiquitous function of GPCRs, the activation, expression and administration of the GPCR based signaling are tightly regulated by the action of different kinases which include phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK), G protein-coupled receptor kinase (GRKs), janus kinases (JAKs) etc. The commencement of the chemokine mediated intracellular signal flow is fundamentally through the activation of inactive heterotrimeric G proteins (23). Once chemokine binds to the receptor, the stimulation facilitates the GDP/GTP exchange, which in turn activates many signaling molecules, secondary messengers, as well as phosphorylation reactions catalyzed by the intracellular kinases to regulate the flow of chemokine-mediated signal. Signaling cascade induced by chemoattractants and their cognate receptors comprises of multiple downstream pathways. Upon binding of chemokines to GPCRs, associated G proteins get activated, and are dissociated into the GTP-linked-Gα and Gβγ subunits. Both these complexes are capable of activating different signaling cascades (Figure 2) (24). GTP-linked-Gα upon activation inhibits some isoforms of adenylyl cyclase, which in turn decrease the intracellular concentration of cAMP, and henceforth bring the reduction in the activity of cAMP-dependent protein kinase. Gβγ subunit is significantly required for chemotaxis and plays crucial role in the activation of different signaling effectors such as phosphatidylinositol specific phospholipase C (PLCβ2 and β3), and phosphatidylinositol-3-OH kinase (PI3Kγ). PI3Kγ catalyzes the rapid generation of phosphatidylinositol (3, 4, 5)-trisphosphate and also activates other kinases like protein kinase B (PKB/Akt) and MAPK (25, 26).
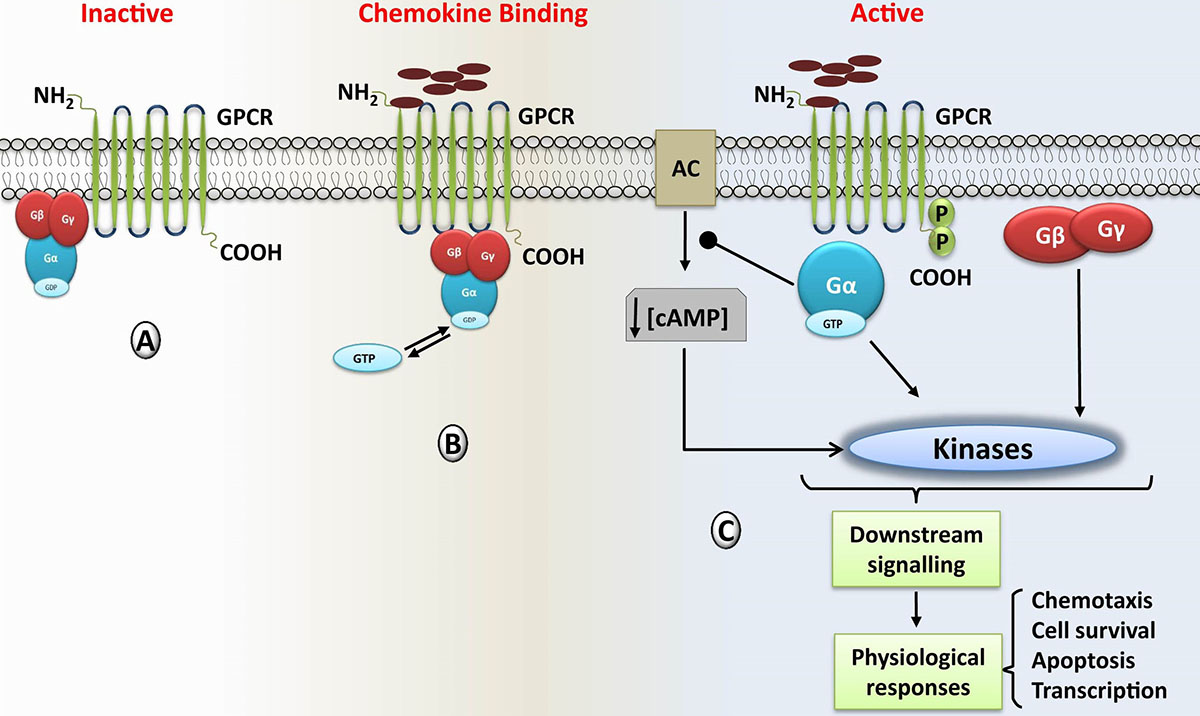 Figure 2
Figure 2Chemokine induced Signaling cascade mediated by the G protein-coupled receptors (GPCRs); A) inactive stage in which chemokine is not associated with its cognate receptor, and G-protein is intact and inactive state (bound with GDP). B) After binding of chemokine/agonist to its cognate GPCR, it triggers the bound G protein complex leading to GTP-GDP exchange and dissociation of the heterotrimeric G protein complex into Gα and Gβγ subunits. Upon dissociation, GTP remains with the Gα subunit. C) Both the Gα and Gβγ subunit complex activates downstream signaling cascades that ultimately regulate the physiological and pathological response of the cells.
The regulated flow of kinase mediated multiple signaling pathways are essential for the recruitment of immune cells to generate immune responses. However, indecorous signaling pathways are implicated in the development and progression of a broad range of pathophysiological conditions. There are several diseases that are incidental with the inadequate activation of the chemokine mediated signaling cascade; especially those characterized by an excessive cellular infiltrate, such as rheumatoid arthritis and systemic lupus, other inflammatory diseases, viral and bacterial infections, autoimmune diseases and cancer etc. (4, 27, 28). The present review throws light on the kinase-mediated signaling pathways of chemokines and their cognate GPCRs that are crucial in regulating various biological processes.
Phosphoinositide 3-kinases regulate multiple arrays of cellular responses such as mitogenesis, cell survival, glucose transport, membrane trafficking, superoxide production, membrane ruffling actin reorganization and chemotaxis (29, 30). Kinases of PI3K family phosphorylate the Phosphoinositide lipid (PI) at 3-OH position of the inositol head (31). Depending upon the substrate specificity and structural features, PI3K can be classified into three classes - class I, II and III. Class-I PI3Ks phosphorylate the phosphatidylinositol, phosphatidylinositol (4) phosphate, and phosphatidylinositol (4, 5) bisphosphate. There are four members in this class which can be further subdivided into IA and IB subclasses. The existence of these kinases in multiple isoforms provides an additional advantage of multiple signal pathway regulation. Subclass IA PI3Ks comprises of three kinases, PI3Kα, PI3Kβ, and PI3Kδ (32). The subclass IB contains the single-member PI3Kγ. Class-II PI3Ks utilize the phosphatidylinositol and phosphatidylinositol (4) phosphate as substrates. Class-III PI3Ks employ phosphatidylinositol as the substrate (33, 34). Enzyme phosphate and tensin homolog (PTEN) terminates the signaling of PI3K by dephosphorylating the PI (3, 4, 5) P3 (35). Studies using PI3K inhibitors, mouse models with gene knockout experiments, and overexpression of mutated forms of PI3K established that PI3Ks assimilate and transmit signals from diverse surface molecules such as chemokine receptors, B cell receptors, and adhesion molecules. Binding of ligands/signaling molecules (such as chemokines) to their cognate GPCRs activates the disintegration of receptor associated G protein into Gα and Gβγ subunit. Both these subunits can activate PI3Ks independently. This results in generation of many secondary messengers and effector molecules like phosphatidylinositol (3,4,5) trisphosphate (PIP3) that in turn can activate other protein kinases such as serine/threonine kinases (AKT/PKB and PDK1) to facilitate the flow of signal (31, 36) (Figure 3).
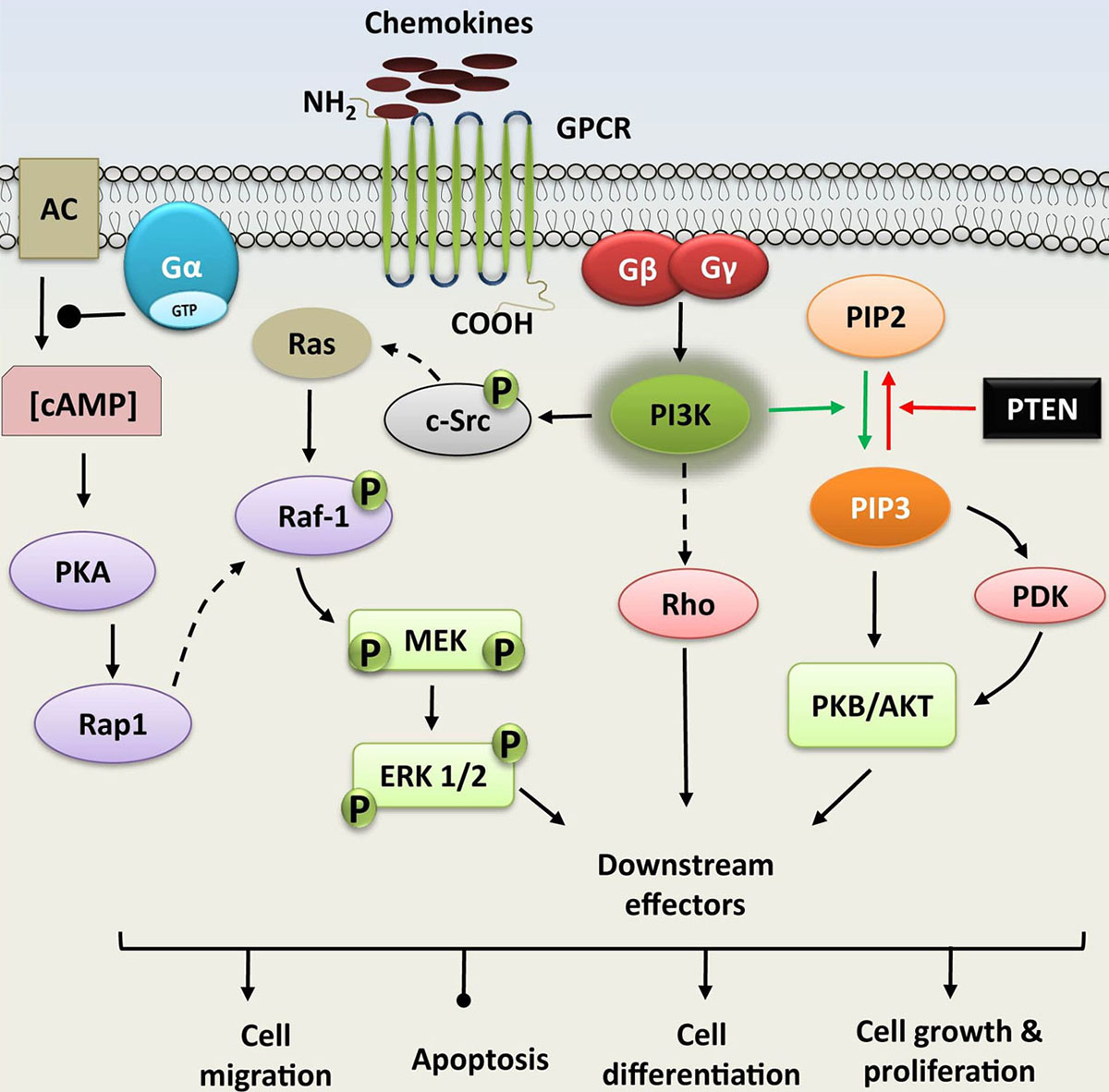 Figure 3
Figure 3Phosphoinositide 3-kinase (PI3K) and MAPK signaling; PI3K pathway is activated upon agonist binding to receptor G protein coupled receptors (GPCRs). GPCRs can activate PI3Ks via G proteins, such as Gβγ. PI3K phosphorylates the phosphatidylinositol (3, 4)-bisphosphate (PIP2), generating phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) which recruits other kinases like serine/threonine kinase (PDK/AKT). MAPK signaling cascade is also induced by the Gβγ subunit of G protein that activates the other successive component proteins (cytosolic Src, MEK, and ERK, etc.). Binding of chemokines to G-protein coupled receptors (GPCRs) also activates Ga subunit. This activation hampers the activity of adenylyl cyclase (AC), which lowers the concentration of cAMP, that subsequently regulates the proteins PKA, Rap1, MEK1/2, and ERK1/2. Both these signaling pathways modulate cellular functions, including proliferation, gene expression, cytoskeletal rearrangement, anti-apoptosis, and degranulation.
PI3Ks play a crucial role in regulating the signaling cascade induced by the chemokines (33). PI3K catalyzed signaling pathways induced by chemokines play a consequential role in signal activation of innate and adaptive immunity (35, 37). Activation of PI3K is a rugged signaling event of leukocyte migration and is applicable to the majority of homeostatic and inflammatory chemokine receptors present on the leukocytes (38, 39). The primary evidence for the involvement of PI3Ks in the chemokine mediated signaling cascade was reported using cellular migration experiments (40). It was demonstrated that the CCL5 induced chemotaxis and T cell polarization can be modulated by inhibiting the PI3K. In addition to this, various research groups have shown that chemokines CCL20, CCL3, CXCL1, and CXCL12, etc., stimulate cellular migration via PI3Ks, thus proving the fact that amplification and degradation of PI3K are significant steps in regulating the chemotactic pathways (41-43). Allergy and inflammatory diseases involve the recruitment of macrophages, dendritic cell, granulocytes, and mast cells. PI3Ks are crucial for the chemoattractant-mediated recruitment of these cells. In the murine model, it has been shown that mice devoid of the PI3Kγ have impeded migration towards chemokines at the site of inflammation (44). Impaired PI3K signaling causes severe defects in both innate and adaptive immunity that lead to different diseases like asthma, rheumatoid arthritis, and cancer (2, 37, 45). Many research groups have also demonstrated that, methods targeting PI3K and its different isoforms using inhibitors as a beneficial therapeutic strategy (Table 1).
| Inhibitor Compound | Target PI3K | Disease Target | Status | Reference |
|---|---|---|---|---|
| GSK2269557 | PI3Kδ | Asthma | Trial phase I | (46) |
| RV-1729 | PI3Kδ/PI3Kγ | Asthma/COPD | Trial phase I | (47) |
| IPI-549 | PI3Kγ | Advanced solid tumors | Trial phase I | (48) |
| Dezapelisib | PI3Kδ | B-cell malignancies | Trial phase I | (49) |
| Umbralisib | PI3Kδ | CLL | Trial phase I | (50) |
| PI-3065 | PI3Kδ | Advanced solid tumors | Trial phase I | (51) |
| SF1126 | pan PI3K | Neuroblastoma | Trial phase I | (52) |
| Pilaralisib | pan PI3K | Breast cancer | Trial phase I | (53) |
| AZD8186 | PI3Kβ | Breast cancer | Trial phase I | (54) |
| SAR260301 | PI3Kβ | Solid tumor | Trial phase I | (55) |
| Acalisib | PI3Kβ/ PI3Kγ | Lymphoid Malignancies | Trial phase I | (56) |
| CL27c | pan PI3K | Asthma/ pulmonary fibrosis | Trial phase I | (57) |
| AS-604850 | PI3Kγ | Multiple sclerosis | Trial phase I | (58) |
| IPI-145 | PI3Kδ/ PI3Kγ | Asthma | Trial phase II | (59) |
| TG100-115 | PI3Kδ/PI3Kγ | Asthma/COPD | Trial phase II | (60) |
| Pictilisib | pan-PI3K | Advanced solid tumors | Trial phase II | (61) |
| PX-866 | pan-PI3K | Prostate cancer | Trial phase II | (62) |
| GSK2636771 | PI3Kβ | Breast cancer | Trial phase II | (63) |
| Copanlisib | pan-PI3K | Breast cancer | Trial phase II | (64) |
| Alpelisib | PI3Kα | Advanced solid tumors | Trial phase III | (65) |
| Taselisib | PI3Kα/ PI3Kδ/ PI3Kγ | Advanced solid tumors | Trial phase III | (66) |
| Buparlisib | pan-PI3K | Advanced solid tumors | Trial phase III | (67) |
| Duvelisib | PI3Kδ/PI3Kγ | CLL | FDA approved | (68) |
| Idelalisib | PI3Kδ | CLL | FDA approved | (69) |
| BEZ235 | pan-PI3K | Solid tumors | Discontinued | (70) |
Asthma is a common clinical condition that has the characteristics of airway hyperresponsiveness (AHR), lungs infiltration with inflammatory cells, wheezing, shortness of breath and coughing. The pathological events in the asthma are mediated mainly by the airway smooth muscle (ASM) cells, and epithelial cells (71). PI3Ks play a pivotal role in the development and progression of almost all the above mentioned pathophysiological aspects of asthma. The activation of epithelial cells releases the chemokines and cytokines that attract the immune cells. PI3K activation is crucial for the regulation of ASM, as it upregulates the expression of chemokine CXCL8 that recruits different immune cells (72, 73). Further, it has been shown that T cells also contribute to the development of asthma by secreting the immune complexes that induce inflammation. Chemokine receptors recruit the p110γ and p110δ members of class-I PI3K family that contribute to activation, differentiation, and proliferation of the T cells (35, 74). PI3K mediated migration of neutrophils was also reported in some cases of asthma (75). The significance of PI3Ks in asthma makes it a potential therapeutic target for treatment of asthma (35). Indeed, various PI3K inhibitors have favorable pharmacokinetics, and are currently considered as promising drug candidates for the treatment of asthma (Table 1).
Mounting evidence reported by different research groups established the involvement of chemokine and chemokine receptors in various stages (development and progression) of tumorigenesis (76, 77). Chemokine receptors are majorly expressed by the tumor cells, along with generation of respective chemokines within the tumor microenvironment (45, 78, 79). The presence of large number of CCR10 chemokine receptors has been reported on the melanoma cells. Upon binding to its cognate ligand CCL27 , the PI3K pathway along with other intermediate pathways are initiated, and anti-apoptotic signals are drawn to protect the cells from dying (80). Moreover, the expression of the CCR7 receptor along with its ligands CCL19 and CCL21 has been observed to be involved in the growth and advancement of many types of cancer (58). In the case of non-Hodgkin’s lymphoma and pancreatic ductal adenocarcinoma, metastatic and lymphatic spreading is mediated through the PI3K/AKT signaling pathway induced by the high expression of CCR7 (81, 82). The PI3K pathway has been reported to be crucial in the growth and progression of prostate cancer. Binding of chemokine CXCL12 to its receptor CXCR4 induces PI3K pathway, which helps in promoting the proliferation of prostate cancer stem cells (CSCs). The CSCs help in re-emergence of tumor even after the primary treatment (79, 83). Furthermore, chronic lymphocytic leukemia (CLL) is a disease in which B cells are accumulated in the blood and tissue compartment. Many supporting cells like MSCs and NLCs secrete chemokines (CXCL12/ CXCL13) that play critical roles in the progression and retention of this disease (84, 85). According to a recent study the signaling pathway mediated by the PI3Kδ plays a significant role in the development and progression of chronic lymphocytic leukemia (CLL) (29). By using CAL-101, a potent inhibitor of PI3Kδ that inhibits the chemokine mediated signaling, the researchers observed a restriction in the B-cell receptor signaling and its reduced accumulation during CLL. Considering the different isoforms of PI3K, inhibitors have been developed for various isoforms that are currently in trial phase (Table 1).
G protein coupled receptor kinases are serine/threonine protein kinases that specifically identify and phosphorylate the GPCRs activated by the agonist. Receptor phosphorylation by GRK is one of the well-studied mechanisms of GPCR desensitization (86, 87). Binding of ligands to the GPCRs leads to the concurrent activation of G protein and β-arrestin-dependent signal transduction through a series of widely conserved biochemical steps (88). G protein dependent signaling is rapid and is dependent on secondary messengers, while arrestin signaling has slower onset and is signalosome dependent (89). Agonist activated GPCRs are phosphorylated by GRKs, which in turn regulates the binding of β-arrestin (90) (Figure 4). β-arrestin binding can initiate the desensitization of G protein signaling at various stages such as recruitment of enzyme that degrades secondary messengers (91, 92), steric hindrance of the receptor (93), and receptor internalization that prevents further binding of the cognate ligands to the receptor (94). β-arrestin molecule not only hinders the receptor associated G protein signaling, but also increases the desensitization and internalization of the GPCRs by shifting proteins such as PDEs and cytosolic Src (c-SRC ) to the receptors (89). These two proteins turn off the signaling pathway by degrading the cAMP once they reach the membrane. Protein kinase A (PKA), protein kinase B (PKB), protein kinase C (PKC) along with c-SRC also help in enhancing the activity of GRK by phosphorylating it (89, 95) (Figure 4). Due to the propensity of GRKs to interact with numerous proteins involved in the signal transduction process, along with their canonical role of GPCR internalization and desensitization, β-arrestin also initiates additional signaling processes regulated by other kinases including MAPK signaling (89, 95, 96), and receptor tyrosine kinase transactivation (97). GRKs are also involved in the regulation of cellular responses through phosphorylation independent pathways along with phosphorylation mediated processes (98).
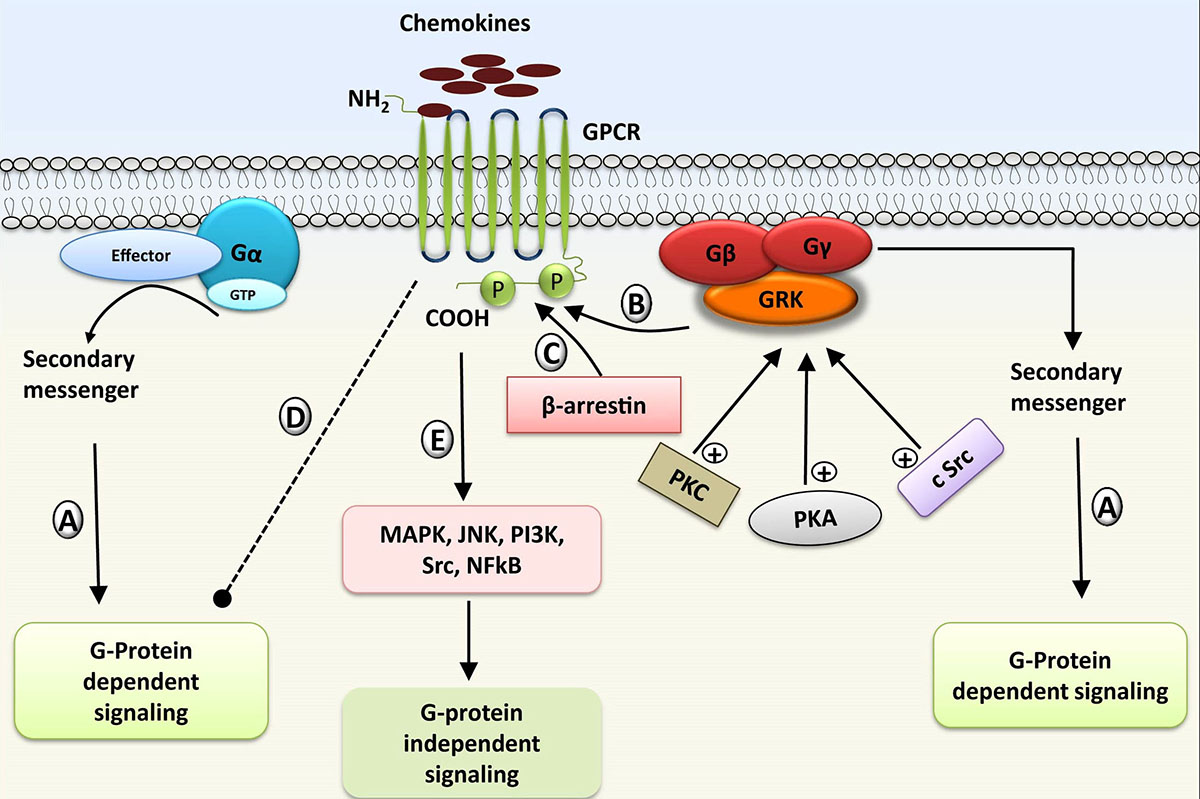 Figure 4
Figure 4Regulation and desensitization of GPCR by GRKs (G-protein receptor kinases): A) Activation of GPCR and G-protein dependent reactions. B) Recruitment of GRK for GPCR phosphorylation. C) Binding of β-arrestin to phosphorylated GPCR, and formation of signalosome complex leading to desensitization of receptor. (D-E) Inhibition of G protein dependent signaling and activation of G protein independent signaling cascade; Activity of GRK can be enhanced by other protein kinases including protein kinase A (PKA), protein kinase C (PKC), and c-Src.
There are seven isoforms of GRKs. All the GRKs are involved in the desensitization of GPCRs. The only little differences that have been observed are in the structure and function of GRKs. On the basis of the observed differences, they are categorized into three subfamilies (99). The first family, designated as GRK1 subfamily, comprises GRK1/7, second subfamily known as GRK2 subfamily constitutes GRK2/3, and third subfamily called GRK4 subfamily contains GRK4/5/6. GRK1 and GRK7 are called as visual GRK and are present in retina (100, 101), GRK4 is expressed in testis (102). In contrast, other isoforms like GRK2, GRK3, GRK5, and GRK6 are widely found in various cells across the body (103-105). GRKs are multidomain proteins, having a 25 residue N-terminal region that is unique to GRKs. It also comprises of a regulator of G-protein signaling homology domain (RH) followed by a catalytic (serine threonine-protein kinase) domain that is accountable for phosphorylating the substrates (106).
As GPCR signaling is essential for the proper functioning of the immune cells and also for maintaining homeostasis, dysregulation of the GRK can be deleterious to the cells like leukocytes and neutrophils (107). Such processes often lead to various pathological conditions including hypertension, chronic inflammatory disease, cardiac failure and rheumatoid arthritis etc. (108). Inflammatory mediators that regulate the expression of GRKs on immune cells via degradation and transcription might also alter the expression of GRKs in different disease conditions (109, 110). As novel interactions of GRKs with numerous proteins are being discovered, GRKs can be the potential target for many diseases (111, 112).
Chemokine mediated signaling is regulated by GRKs through chemokine receptor phosphorylation and desensitization (113, 114). GRK mediated desensitization is crucial in sensing the changes in the chemokine gradient, and in turn to control the cellular migration. Moreover, GRKs may also interfere in chemokine signaling by direct interaction with molecules such as MEK. It has been evidenced that CCR2 receptor mediated signaling of ERK1/2 was enhanced in splenocytes of GRK2 knockout mice (115). On a similar note, it was also reported in the GRK2 knockout mice model that CCR5 directly induced the signaling to protein kinase B (PKB), ERK 1/2 and calcium mobilization (116). Neutrophils play a major role in the different infectious and inflammatory diseases. Mobilization of neutrophils is highly controlled by the CXCR4 receptor mediated signaling, which is essentially regulated by GRK6 (116).
The concept of “biased agonism” is also well established in the case of chemokine mediated GRK signaling pathways. This is the phenomenon in which the effector pathways are selectively activated on the basis of the activity induced by a given ligand (117, 118). Some of the agonists may act as “G-Protein biased” i.e., specifically inducing G protein and not the arrestins, while the others may act as “β-arrestin biased” by selectively inducing the β-arrestins. Chemokine ligands CCL19 and CCL21 of the CCR7 receptor are observed to be biased; both the ligands have equal potency towards the receptor, but only CCL19 induced pathway selectively leads to receptor desensitization through β-arrestin activation (103, 119, 120). Based on this biased agonism phenomenon, many therapeutic agents were proposed for diseases such as Warts Hypogammaglobulinemia Infections Myelokathexis (WHIM) syndrome and HIV infection (121). Principally, the signaling pathway induced by CXCL12 that mediates chemotaxis and receptor internalization through chemokine receptor CXCR4 is regulated by β-arrestin2 and GRK3. In case of WHIM syndrome, a mutation is present at COOH tail of the receptor which leads to the defective CXCR4 signaling. In diseased condition, β-arrestin2 regulates the chemotactic migration of the defective/mutated leukocytes but does not induce receptor internalization (122, 123).
It is well established that the GRKs, specifically GRK2 is involved during various stages of tumorigenesis as it creates a suitable environment that supports tumor progression (124). As supported by cumulative shreds of evidence, GRK2 level has been reported to be upregulated and to alter the other pathways in case of breast cancer (124). This protein is considered to be the key hallmark of cancer as it acts as a crucial oncomodifier in different tissues (125). Deficiency of GRK6 promotes the CXCR2 moderated tumor metastasis and progression in lung carcinoma model (126).
GPCR mediates the most important signal flow for the functioning of cardiovascular system. The involvement of GRKs is also observed in the smooth and regulated flow of the signal in the andergenic system. Hyperactivation of the andergenic system is responsible for maintaining equanimity and progression of cardiovascular diseases like hypertension and heart failure (25, 114). GRK2, GRK3, and GRK5 are seen to be widely involved in most of the cardiovascular diseases (127, 128). A recent study suggested that development of promising compounds that inhibit the activity of GRK5 are handy in preventing and reversing the progression of heart failure (129). Atherosclerosis is a chronic, medically compromised condition that is responsible for the heart related transgression and stroke. It is well evidenced that chemokines of CC and CXC families are entangled in the atherosclerotic plaque in humans as well as in mice (130, 131). GRK5 is seen to impair atherosclerosis, as it inhibits the migration of monocytes by desensitizing the receptor CCR2 (132).
It is well established that the level of GRKs decreases during inflammatory diseases such as multiple sclerosis, sepsis and rheumatoid arthritis (133). In a sepsis patient, clearance of infectious bacteria is initiated by the migration of leukocytes (neutrophils) through CXCL8/IL8 family chemokine. However, due to the signaling by GRK5 and GRK2, the cognate receptor CXCR2 is down-tuned and the sepsis is pursued. Reports suggested that the hindrance in the expression of GRK2 in neutrophil activates the CXCR2 receptor signaling and in turn induces the migration of neutrophil to the site of infection in sepsis (134, 135).
Janus kinases are a small family of cytoplasmic kinases that are crucial in providing transmission signals from receptor to nucleus (136, 137). It consists of four members JAK1, JAK2, JAK3, and TYK2. Isoforms JAK1 and JAK2 of this family are crucial in administering a wide range of functions including growth, neural development and host defense (138, 139). JAK3 is mostly seen on the hematopoietic cells that are entangled in cellular signaling leading to immune responses. TYK2 is significant for the immune function and defense mechanism of the host (140). All the isoforms of JAKs possess a unique conserved region called JAKs homology domain (JH1-JH7). Along with the functional domain (JH1) at the C-terminal, they also possess a nonfunctional pseudokinase (JH2) (141).
Mechanistically, signaling through the JAK pathway is relatively simple, as limited molecular partners are involved (142). Binding of ligands with GPCRs induces the multimerization of receptor subunits. Due to this multimerization of receptor, two JAKs are brought into its close vicinity leading to the phosphorylation and activation of JAKs. Activated JAKs phosphorylate the C-terminal intracellular tail of both the receptors, thereby creating a docking site for signal transducer and activator of transcription (STATs). STATs are latent cytoplasmic transcription factors required by all the isoforms of JAK to mediate the signaling pathways (143). JAKs subsequently phosphorylate the tyrosine residue of STATs, which in turn initiates the dimerization of STATs. The activated STATs are then transported to the nucleus, where they bind to several places on the genome to modulate the gene expression (138, 144, 145). Currently, seven mammalian STATs have been recognized (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, STAT6). All these STATs are activated by a specific ligand (141, 144). A small protein family called suppressor of cytokine signaling (SOCS) is observed in the regulation of signaling through JAK/STAT pathway. It has been reported that the activation of the JAK-STAT pathway is regulated by SOCS protein, that interferes with the receptors required for relaying signal to the nucleus (138, 146, 147).
Dysregulation of the JAK/STAT-mediated signaling pathway was reported to be involved in different diseases including autoimmune diseases, inflammatory diseases, cancer, etc. (143, 148, 149). JAK/STAT pathways have also been identified as the major signaling pathway that activates the mechanism of wound healing (150, 151). Although JAK/STAT pathway is mostly implicated in cytokine signaling, its involvement in chemokine and its cognate receptor-mediated signaling has been well documented (152-154).
Among all the signaling pathways activated by chemokines, the involvement of phosphorylated JAK/STAT pathway is triggered soon after chemokines stimulate the GPCR (155). The very first evidence for the involvement of JAKs in chemokine mediated signaling was the manifestation that chemokine CCL2 (MCP-1) binding to CCR2 receptor induced the activation of JAK2/STAT3 leading to rapid phosphorylation of CCR2 receptor (156). Additionally, by using differentially tagged CCR2 recombinant receptors, it was also established that CCL2 induces the multimerization of CCR2. This finding suggests that the chemokine receptor has the propensity of multimerization upon ligand binding (157). Furthermore, the involvement of JAK/STAT pathways in numerous chemokine receptors signaling including CCR1 (158), CCR2 (156), CCR5 (159, 160), CCR7 (153), CCR9 (152), CXCR4 (155, 161, 162), CXCR3 (163) has been elucidated.
The association of JAK/STAT with malignancies was first identified in 1990 (168). Chemokines and their receptors participate in growth and progression of various cancers through JAK/STAT pathways (169). CXCR4/CXCL12 signaling mediated by JAKs is demonstrated to be crucial for the integrin affinity moderation and T lymphocyte homing. The alliance of this signaling is displayed in case of B-cell chronic lymphocytic leukemia (B-CLL). The obtained data corroborate with the theory that JAK2 is a significant regulator of B-cell adhesion by CXCL12 (154). As JAK/STAT pathways are involved in different types of cancers including colitis-associated cancer, solid-organ malignancies (143, 145), etc., cancer therapies comprising of pan-JAK inhibitors against various isoform of JAKs are under clinical trials (22, 145). Considering the importance of JAK mediated chemokine signaling, researchers have also been investigating them as potential therapeutic agents against a variety of other diseases (140). For example, Osteoarthritis is a chronic medical condition characterized by the degeneration of articular cartilages and synovial membrane inflammation (170). An elevated concentration of chemokines (CXCL8 and CXCL11) is observed in the synovial fluid collected from the Osteoarthritis victim. These chemokines help in disease progression through the JAK/STAT pathway and are also involved in the apoptosis of chondrocytes (171). Similarly, CXCL10 evidenced as the significant biomarker in the case of rheumatoid arthritis (RA), involves JAK/STAT signaling in the disease (172). Tofacitinib, a potent inhibitor of JAK mediated signaling pathway is demonstrated as a potential therapeutic agent against RA (144). The expression of CXCL10 was also observed in the case of primary Sjögren’s syndrome (pSS), a common chronic autoimmune disease. The CXCL9/CXCR3 and CXCL10/CXCR3 chemokine-receptor pairs induce JAK/STAT signaling to mediate the amassing of T cell infiltrate in the salivary gland of pSS victim (173). Cepharanthine controls pSS by inhibiting CXCL10 expression in human salivary gland ductal cells by obstructing the JAK2/STAT1 mediated signal flow (165). Several compounds have been synthesized and marketed to inhibit various diseases, where JAK/STAT is a regulatory signaling pathway (Table 2).
| Inhibitors | Disease | Chemokine/ chemokine receptor | JAK/STAT involved | References |
|---|---|---|---|---|
| Maraviroc | Acute Lymphoblastic Leukemia | CXCL12 |
JAK1/STAT3 |
(160) |
| Tofacitinib | Rheumatoid arthritis | CXCL2 |
JAK1/STAT1 |
(144, 164) |
| Cepharanthin | Primary Sjögren’s syndrome | CXCL10 | JAK2/STAT1 | (165) |
| ds‑echinoside A | Cancer | CXCR4 | JAK2/3‑STAT | (166) |
| Berberine | Cardiovascular Disease | CXCR4 | JAK2/STAT | (167) |
Mitogen-activated protein kinases are serine/threonine protein kinases that play crucial roles in the cellular processes including cell proliferation, differentiation, apoptosis and motility (174, 175). A number of studies have evidenced that the uncontrolled activation of MAPK pathways are involved in the initiation and advancement of different cancers, cardiovascular abnormalities and inflammatory diseases such as rheumatoid arthritis (176-178). MAPK pathways constitute a three tiered kinase cascade such as RAF, MEK and ERK family of kinases. Activation of this signal flow is initiated upon binding of ligands to the cell surface cognate receptor, which triggers the activation of RAS through the GTP-GDP exchange. Activated RAS-GTP engages and facilitates the activation of RAF that subsequently phosphorylates and activates the dual specific downstream loop of kinase MEK1 and MEK2. Phosphorylated MEK1/2, in turn, phosphorylate ERK1 and ERK2. Activated ERK1/2 act on its substrates in the cytosol and nucleus to regulate a broad range of cellular functions including cell growth, survival, differentiation, and cytoskeletal rearrangements, etc. (175, 179, 180) (Figure 3). Until now, three prime MAPK signaling pathways are identified in mammalian cells: They include extracellular signal-regulated protein kinases (ERK1/2), p38 MAPK, and c-Jun NH2-terminal protein kinases (JNKs) (181, 182). The extracellular response kinases (ERKs) are implicated in normal cellular development including proliferation, differentiation, cell death, etc. (183, 184). The activation of c-Jun N-terminal kinase (JNK) is majorly demonstrated to have involvement in response to stress signals including inflammatory cytokines (185, 186). The p38 MAPK pathway is firmly activated by environmental stressors such as lipopolysaccharide, which in turn increase the expression of cytokines/chemokines and their receptors, thus making it a crucial immune response regulator (187, 188). In addition, it is also well documented that signaling through MAPK is stimulated by arrestin in order to relay the signal through its three modules of signaling cascade (189, 190).
The chemokine mediated signaling pathway specificity determines the fortune of the cells. Chemokine induced signaling cascade through PI3K/Akt activation leads to the survival of the cells, while p38 MAPK pathway activation results in cell death (191). Apart from chemokines, there are other stimuli such as cytokines and growth factors that can also activate the MAPK/ERK signaling cascade for the migration of cells (192). The activity of the MAPK/ERK pathway is often upregulated in cancer as it plays a decisive role in oncogenesis (193, 194). The digressive activation of this pathway in cancer occurs through several mechanisms including a mutation in RAS/RAF and overexpression of receptor kinase (195). Chemokine CXCL8/IL8 induced MAPK pathway is demonstrated to have significant role in growth and proliferation of lung cancer, as the researchers reported that the proliferation and metastasis of lung cancer cells are fostered by integrin αvβ6, which enhance the expression of CXCL8/IL8 through the MEK pathway (196). There is accumulated evidence for the fact that chemokine CXCL12 and its receptor CXCR4 are significant for colon cancer (197). Comparably it has shown that silencing CXCL12 gene leads to the downregulation of MAPK signaling which in turn has inhibitory effect on the metastatic perspective of colon cancer (198).
Abnormal activity of P38 MAPK has been observed in most of the inflammatory diseases (199). Phosphorylation cascade by p38 MAPK is initiated by pathogens and inflammatory stimulus which ultimately leads to the production of anti-inflammatory cytokines and the phosphatase enzyme. This enzyme dephosphorylates and inactivates all three major MAP kinases such as p38MAPK, JNK, and ERK and triggers the initiation of multiple inflammatory diseases (200, 201). The JNK and p38 families of MAPKs have been identified to be involved in the pathogenesis of rheumatoid arthritis (202). The involvement of chemokine-induced MAPK pathway is also documented in RA patients. (203). Chemokine CXCL8 is present in large quantity in the synovial fluid of RA patients, whose expression is dependent on p38 MAPK. Moreover, reports established the involvement of MEK in chemokine induced signaling cascade (CCL4/CCR5/c-Jun and c-Fos/CCL2) in RA patients (204). Chemokines can potentially activate chondrocytes in order to release various inflammatory mediators. The high concentration of CXCL12 causes the death of chondrocytes via p38 MAPK activity (205). Further, signaling cascade induced by angiogenic chemokines and their respective receptors (CCL2/CCR2, CXCL8/CXCR2, and CXCL12/CXCR4) at the site of the wound is mediated by the MAPK/ERK pathway, which provides the base for formulations of various pharmacological inhibitors against MAPK (179, 181, 199, 206). Ulixertinib, an ERK1/2 inhibitor is having significant therapeutic potential against solid-tumor malignancies (194). Talmapimod (207) and pyrimido-pyridazinone (208) are p38 MAPK inhibitors formulated against RA are currently in clinical trials. Dilmapimod, a potent p38 MAPK inhibitor has displayed the inhibition of the MAPK pathway in asthma/COPD patients (209). Ralimetinib, another inhibitor of p38 MAPK has manifested its potential against advanced stages of cancer with acceptable pharmacokinetics (210).
Tec family kinases are non-receptor protein tyrosine kinases (PTKs), which are the critical components for signaling in lymphocytes (211, 212). Tec kinase family kinases are activated by a diverse range of surface receptors such as cytokines, chemokines, GPCRs, TLRs, integrins, as well as antigens (213). Kinases of this family are involved in various downstream signaling cascades related to proliferation, differentiation, actin reorganization, cell migration, Ca2+ influx, and apoptosis, etc. (214). Tec family member includes ITK (Interleukin-2 inducible T cell kinase), TEC (Tyrosine kinase expressed in hepatocellular carcinoma), RLK (Resting lymphocyte kinase), BTK (Burton’s tyrosine kinase), and BMX (Bone marrow tyrosine kinase on chromosome-X) (214, 215). Members of TFK family hold two differentiating characteristics that are unique to them. Firstly, they have Tec homology (TH) domain that is involved in regulatory interactions. Secondly, they possess a pleckstrin homology (PH) domain which initiates the molecular activation by binding with proteins and phospholipids including the subunits of heterotrimeric G proteins (216, 217).
TFKs are mostly seen on hematopoietic cells, where they regulate their response towards external stimuli. The flow of signal regulated by Tec kinases involves the recruitment of immune cells to the site of infection as they facilitate the activation of immune response (218). It is also evident that members of the Tec kinase family contribute to other signaling cascades such as PI3K and MAPK (219). ITK regulates the chemokine receptor signaling entangled to the migration processes through the stimulation of integrin and cell adhesion (220) It also regulates cytokine induced cell death in CD4+ T cells (221). Chemokine mediated migration of T cells in the case of allergic asthma is regulated by ITK. It was reported that quashing the activity of ITK leads to the reduced T cell migration, thus results in the prevention of allergic asthma (222).
Phosphorylation by Tec kinase at the tyrosine residue is associated with the stimulation of leucocyte (neutrophils) by chemokines (223, 224). The aberrant activation of this pathway may lead to various deleterious effects and pathophysiological conditions (225). Intestinal epithelial cells upon infection with bacteria or activation with other stimuli guide the production of chemokine CXCL8/IL8. Excessive neutrophils accumulated at the mucosal region facilitate the worsening of infection, and the chronic conditions may lead to life-threatening diseases like Crohn disease and ulcer, etc. (225, 226). Modification in the protein tyrosine kinases (PTKs) that regulate the CXCL8 production has been proposed as a novel therapeutic target (227).
BTK is identified as a significant factor in B-cell receptor signaling pathways and B-cell chemotaxis (228). It is evident that the BTK has a crucial role in the pathogenesis of B-cell chronic lymphocytic leukemia (B-CLL), as it plays a consequential role in the CXCL12 induced activation of integrin in both the normal as well as B-CLL condition. It has been demonstrated that BTK may act as the point of convergence between the B-cell receptor and chemokine induced signaling cascade (229)
Apart from the above mentioned kinase families, there are many other kinases that play pivotal roles in regulating the signaling pathways induced by the chemokines. These kinases are the members of serine threonine-protein kinase family, that belong to cAMP-dependent protein kinase which includes protein kinase A, protein kinase B/Akt, protein kinase C and, PDK (230, 231). Binding of chemokine to the receptors triggers the dissociation of G protein; Gα subunit regulates the production of cAMP from adenylyl cyclase (AC) (Figure 3). Protein kinase A (PKA) gets activated upon binding of cAMP to it; further it enters into the nucleus and subsequently phosphorylates the transcription factors (232, 233). PKB is one of the key mediators of PI3K pathway induced by the chemokines (234). Upon activation of PI3K pathway, a second messenger PIP3 is generated from PIP2, which facilitates the translocation of PKB to plasma membrane from cytoplasm, where it plays crucial role in cellular growth and proliferation (235). Chemokine mediated signaling initiates the generation of molecules such as diacylglycerol (DAG), which triggers calcium ion (Ca2+) mobilization, thus activating the Protein kinase C (PKC) for downstream signaling (236).
Src kinases are a small subgroup of the tyrosine kinase family and they act as the essential link between the effector molecules and the key transcription factors during cytoskeleton rearrangements, cell survival, cell growth, and proliferation (237). The Src family kinase regulation can be either activating or inhibitory. Kinases of this family play a crucial role in ITAM (immunoreceptor tyrosine based activation motif) dependent signaling cascade, utilized by immunoreceptors such as lymphocyte antigen receptors, Fc receptors and NK activating receptors. In addition, it also modulate ITIM (immunoreceptor tyrosine based inhibitory motif) dependent path, which negatively regulate both immunoreceptor function and host signaling activities, such as cytokines and integrin signal and regulate the activation of leukocytes (238).
The biological role of Src kinases is manifested in both leukocytes and hematopoietic cells as Gα subunit of G-protein associated with the chemokine receptor activates Src family kinases (237, 239). After getting stimulated by Gα subunit, Src subsequently phosphorylates multiple downstream substrates including Class IA PI 3-Kinase (PI3K), Interleukin-2 inducible T cell kinase (ITK), focal adhesion kinase (FAK), and proline-rich tyrosine kinase 2 (PYK2) (240-242). It is also evident that, once chemokine CXCR12 binds to its cognate receptor, MAPK signaling cascade activation is facilitated by both Lyn and Lck kinases which are the members of Src kinase family (243). PKA and PKB kinases along with Src are also reported to be involved in enhancing the activity of GRKs (Figure 4).
The current review has illustrated the significance of various kinases involved in the chemokine mediated signaling cascade and demonstrated their role in regulatory as well as inhibitory pathways on the basis of contemporary development in the research related to chemokines and kinases. After four to five decades of the persistent contribution of several research groups across the globe, it has been established that the kinase system induced by the chemokine/ chemokine receptor engrosses a central position in regulation of the immune system, thus playing a significant role in the initiation and progression of different pathophysiological conditions. Overwhelming pieces of evidence have established that kinases are associated with the complex networks of chemokine mediated signaling in which the consequential cross-talk among the components regulates multiple biological activities. However, it’s still a matter of debate regarding how to keep the other associated signaling pathway intact by inhibiting one kinase. A fundamental outline of how the chemokine and kinase systems work has been manifested by using mouse models, and there has been substantial progress in the application of this knowledge in clinical practice. Currently, these pathways are potential drug targets for many pathological conditions either involved in innate or adaptive immune components.
Many drug compounds which target kinases are in different phases of the clinical trials. Despite the fact that considerable success has been obtained by using chemokine and associated kinase systems as potential drug targets, satisfactory treatments for chronic immune mediated diseases remain a major unmet medical need. There is high biomedical demand for the development of novel potent kinase inhibitor with clinical specificity and lesser side effects. It is imperative to discern every meaningful interaction between the chemokine and the kinase system so that it can further aid in formulating alternative therapeutic compounds targeting chemokine/chemokine receptors and their associated kinases.
Apart from drugs targeting kinases, there exists compounds that show inhibitory effects on chemokine and GPCRs (244, 245). They include; engineered chemokines, synthetic GAGs and small molecule receptor targets, which can modulate and deactivate the kinase signaling pathway. Few noted examples include, chemically modified high affinity CCL5 ligands (246), synthetic monosaccharide, 2,4-O-di-sulfated iduronic acid (Di-S-IdoA) (247), selective inhibitors (LY294002 and MK-2206) of CXCR6 (248) etc. These studies establish that kinases, GPCRs, and chemokines are the crucial molecular partners, and are hot targets of pharmaceutical industry. Understanding the combinatorial mechanism of the action of inhibitor/drug molecules at molecular level will be useful in terms of system biology perspective, as they will elucidate the other key pathways involved, and aid us to formulate molecules with high specificity and minimal side-affects.
KMP acknowledges the receipt of Grant CRG/2018/001329 from SERB-DST, and DBT-IYBA fellowship – BT/07/IYBA/2013-19, DT acknowledge the receipt of MHRD fellowship from IIT-Roorkee.
GPCRs
G protein coupled receptors
Phosphoinositide 3-kinases
G protein coupled receptors kinase
Mitogen-activated protein kinases
Janus Kinases
signal transducer and activator of transcription
Extracellular signal-regulated kinases
Protein kinase A
Protein kinase B
Protein kinase C
Tec family kinases
Interleukin-2 inducible T cell kinase
Resting lymphocyte kinase
Burton’s tyrosine kinase
Tyrosine kinase expressed in hepatocellular carcinoma
Bone marrow tyrosine kinase on chromosome-X
Toll like receptor
Tec Homology
Pleckstrin Homology
Protein tyrosine kinase
B-cell chronic lymphocytic leukemia
Guanosine triphosphate
Guanosine diphosphate
Adenosine triphosphate
c-Jun NH2-terminal protein kinases
Epidermal derived growth factor
Rheumatoid arthritis
Primary Sjögren’s syndrome
Suppressor of cytokine signaling
JAKs homology
Warts Hypogammaglobulinemia Infections Myelokathexis
cytosolic Src
Chronic lymphocytic leukemia
Chronic obstructive pulmonary diseases
Adenylyl cyclase
Diacylglycerol
Phospholipase C
inositol (1,4,5) triphosphate
Food and drug associations
Multiple sclerosis
phosphatidylinositol-4-5-bisphosphate
Phosphatidylinositol-3-4-5-trisphosphate
phosphate and tensin homolog
Cyclic adenosine mono phosphate
Immunoreceptor tyrosine-based inhibitory motif
immunoreceptor tyrosine-based Activation motif