












Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Anatomy and Cell Science, Kansai Medical University, 2-5-1 Shinmachi, Hirakata-shi, Osaka-fu, 573-1010 Japan
2 Laboratory of Microbiology and Cell Biology, Department of Pharmacy, College of Pharmaceutical Sciences, Ritsumeikan University 1-1-1 Noji-Higashi, Kusatsu, Shiga 525-8577, Japan
Abstract
Recent evidence demonstrates that long non-coding RNAs (lncRNAs) regulate the expression of multiple genes in an epigenetic, transcriptional, or post-transcriptional manner. They are involved in various cellular phenomena, such as the recruitment of transcription factors, epigenetic chaperoning, control of alternative splicing, mRNA stability and translational activity, as well as acting as decoys against microRNAs. In this review, we summarize the pivotal roles of lncRNAs in regulation of the gene expression involved in neural cell differentiation, synaptogenesis and synaptic plasticity in the central nervous system (CNS). We also describe the aberrant expression of multiple lncRNAs involved in the pathogenesis of neurological diseases. The abnormal expression of lncRNAs leads to altered expression levels of target genes, which contributes to neurodegenerative diseases, such as in Alzheimer’s disease and Parkinson’s disease, and to the formation of tumors, such as glioma. Accordingly, we discuss recent findings for the modes of action of lncRNAs in normal CNS development and for aberrant lncRNA actions in the pathogenesis of neuronal diseases.
Keywords
- Long non-coding RNA
- Central nervous system development
- Neurological disease
- Alzheimer’s disease
- Parkinson’s disease
- Glioma
- Review
Messenger RNAs (mRNAs) play a pivotal role in converting genetic information into protein products. In addition, various forms of non-protein-coding RNA (ncRNA) regulate gene expression by epigenetic, transcriptional, and post-transcriptional processes (1). Recent studies based on new generation sequencing technologies, such as the Encyclopedia of DNA elements (ENCODE) project, show that ncRNAs are transcribed from approximately 80% of human and mouse genomes (2).
ncRNAs are classified into several types according to their functions. For example, tRNAs, rRNAs, and small nuclear RNAs (snRNAs) are housekeeping ncRNAs involved in normal physiological functions. In contrast, small ncRNAs, such as microRNAs (miRNAs), as well as long ncRNAs (lncRNAs) are regarded as regulatory ncRNAs (3). miRNAs are usually single-stranded RNAs of 20-23 nucleotides that regulate mRNA translation by directing target mRNAs to RNA-induced silencing complex (RISC)-dependent degradation or by temporarily stalling translation by isolating target mRNAs in cytoplasmic RNA-protein complexes, such as stress granules (4). lncRNAs are usually defined as non-protein-coding transcripts of at least 200 nucleotides. Recently, they have been recognized as key factors in a variety of processes, as listed in Table 1. In the nucleus, lncRNAs are predominantly involved with nuclear scaffolds, in gene dosage compensation, epigenetic chaperoning, and alternative splicing, while in the cytoplasm they are involved with mRNA stability, competing endogenous RNAs (ceRNAs), and translational activity (5-19).
| Roles | Roles of lncRNAs | Detailed actions | lncRNAs examples |
| Multiple roles in the nucleus | Nuclear scaffolds | Maintaining nuclear subcompartments | Malat1, NEAT1, Satellite III |
| Gene dosage compensation | Controlling the extensive transcriptional activity of the X chromosome | Xist, roX | |
| Epigenetic chaperons | Positively or negatively modulating gene transcription | HOTAIR, TUNA, Evf2 | |
| Alternative splicing | Modulating the splicing pattern | Gomafu, 51A | |
| Post-transcriptional roles in the cytoplasm | mRNA stability | Positively or negatively controlling mRNA degradation | BACE1-AS, 1/2-sbsRNAs, INF-alph1AS |
| ceRNAs | Sponging miRNAs and eventually increasing mRNA expression | linc-MD1, CRNDE, GAS5, MEG3, INF-alpha1AS | |
| Translational activity | Interacting with the translational machinery and affecting its activity | BC1, BC200, AS Uchl1 |
In this review, we primarily focus on how lncRNAs regulate the gene expression involved in central nervous system (CNS) development. The fundamental importance of lncRNAs in the CNS is supported by lncRNAs having high levels of expression in the brain compared with levels in other tissues (20) and by brain-specific lncRNAs being highly conserved among species (21). Indeed, emerging evidence shows that lncRNAs act as epigenetic chaperons (22-25), modulate alternative splicing (26), and regulate translational activity (27-29) during CNS development. Furthermore, we describe typical examples of aberrant gene expression caused by dysregulated lncRNA expression, which result in the onset of neurological diseases (30), including Alzheimer’s disease (AD), Parkinson’s disease (PD), and glioma. In AD, dysregulated lncRNA expression results in aberrant alternative splicing (11, 31), altered mRNA stability (12, 32), and abnormal translational activity (33). In PD, abnormally expressed lncRNAs act as epigenetic chaperons (34) and ceRNAs (35), and control mRNA stability (36). Moreover, a few lncRNAs have the potential to affect protein stability (37, 38), although interactions between lncRNAs and proteins have not been fully investigated. lncRNAs in gliomagenesis can act as ceRNAs that counteract miRNAs to modulate the expression levels of target mRNAs (39-48). Of note, it has been argued that the stoichiometric relationship between miRNAs and ceRNAs is crucial in the inhibition of miRNAs by ceRNAs (17, 49). Nevertheless, several lncRNAs certainly act as ceRNAs to modulate the expression of miRNA-targeted mRNAs, which alters cancer-related signaling pathways in glioma development.
In this review we present recent findings that shed light on the normal actions of lncRNAs in brain development and on how regulatory functions of lncRNAs are disrupted in neurological diseases.
CNS development is based on the differentiation of neural stem cells (NSCs) to form all the cell types of the mature CNS. This process is highly orchestrated and regulated by intrinsic/extrinsic cell factors such as transcription factors, secreted proteins and cell-cell interactions. Neural cell differentiation is divided into three stages; the self-renewal stage of NSCs, the commitment stage of NSCs to acquire a certain cell fate, and the maturation stage (Figure 1, A, B, C, respectively). At the commitment stage, the NSCs generate neuronal or glial progenitors. At the maturation stage, these neuronal or glial progenitors acquire more specific properties.
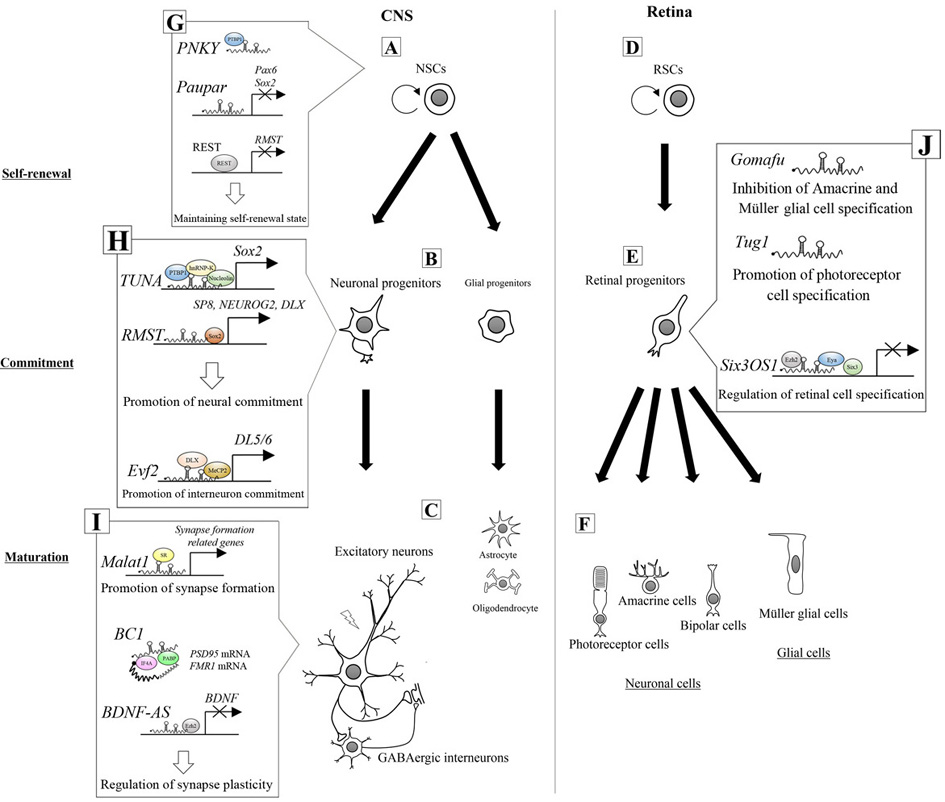 Figure 1.
Figure 1.Regulation of neuronal differentiation by lncRNAs in the CNS (A, B, C) and in the retina (D, E, F). (G) lncRNA PNKY maintains the self-renewal state of NSCs by interacting with PTBP1. lncRNA, Paupar, contributes to maintaining the self-renewal state by inhibiting Sox2/Pax6 gene expression. REST maintains the self-renewal state by inhibiting RMST transcription. (H) lncRNA, TUNA, forms a quadruplicate complex with PTBP1, hnRNP-K, and nucleolin, which enhances Sox2 transcription and promotes the commitment of neuronal progenitors. lncRNA, RMST, recruits SOX2 to the promoter regions of SP8, NEUROG2, and DLX genes, which promotes neural commitment. lncRNA, Evf2, recruits DLX and MeCP2 to the enhancer region of DL5/6, which promotes GABAergic interneuron commitment. (I) lncRNA, Malat1, recruits SR proteins to the transcription sites of synaptogenesis-related genes, which promote synapse formation. lncRNA, BC1, inhibits the translation of FMRP and PSD-95 mRNAs by interacting with eIF4A and PABP. lncRNA, BDNF-AS, recruits EZH2 to the promoter region of the BDNF gene, which represses BDNF transcription. (J) lncRNA, Gomafu, inhibits amacrine and Müller glial cell specification. lncRNA, Tug1, promotes photoreceptor cell specification. lncRNA, Six3OS1, represses the transcription of Six3 by interacting with EYA and EZH2, which regulates retinal cell specification.
As a relatively accessible part of the CNS, the retina has been employed to study neural cell differentiation (Figure 1, D, E, F). In the developing retina, retinal stem cells (RSCs) give rise to retinal progenitor cells (RPCs), which generate numerous post-mitotic retinal cell types, including both neuronal cells (photoreceptors, retinal ganglion cells, amacrine cells, horizontal cells and bipolar cells) and glial cells (Müller glial cells) (50).
Recent studies have shown that lncRNAs can regulate developmental processes in various tissues, such as skeletal muscle, blood vessels, pancreas and sperm, by modulating target gene expression to control cell differentiation (51-54). In the CNS, lncRNAs are involved not only in differentiation of neuronal and glial cells but also in synaptogenesis and synaptic plasticity. They are, therefore, of interest as emerging regulators of brain function (23, 55, 56).
An inhibitory mechanism for lncRNA transcription involving RE1-silencing transcription factor (REST) has been reported (57, 58). REST is a zinc finger protein that binds to a conserved 23 bp DNA motif, called RE1 (also known as neuron-restrictive silencer element or NRSE), that is found in promoter and enhancer regions of many neural genes (59). REST recruits chromatin remodeling components to RE1 and actively represses the transcription of a large number of neural genes and lncRNAs, such as DiGeorge Critical Region 5 (DGCR5) and rhabdomyosarcoma 2 associated transcript (RMST), via histone modification (60). In this section, we describe the various functions of lncRNAs by focusing on embryonic stem cell (ESC) or NSC differentiation and retinal cell specification. In addition, we focus on the modes of action of lncRNAs at each differentiation stage and on the lncRNA expression mechanisms regulated by REST.
Various in vitro approaches have been used for identification and functional analysis of lncRNAs. For example, a large number of genome-wide analyses have identified thousands of lncRNAs involved in pathological mechanisms, such as oncogenesis, metabolic abnormalities, and infectious diseases (61). Candidate lncRNAs are generally analyzed using Gene Ontology (GO) and pathway analysis and further validated by in situ hybridization or quantitative RT-PCR (RT-qPCR). These in vitro approaches have identified neural differentiation-related lncRNAs, such as Paupar, RMST, and sine oculis-related homeobox 3 opposite strand 1 (Six3OS1) (58, 62, 63).
The neural-specific lncRNA, PNKY, is expressed in NSCs in the subventricular zone (SVZ), which is located in the lateral walls of the ventricles and acts as germinal region. Here, PNKY acts as a repressor of neural commitment (64) (Figure 1 G). This function was verified by an in vivo knockdown (KD) experiment employing in utero electroporation of mouse embryonic brain with a shRNA expression vector. The PNKY KD led to an expansion of neurogenic progenitors in the SVZ, indicating that PNKY can inhibit the commitment stage of the NSCs (64). Associated investigation of PNKY binding proteins by RNA immunoprecipitation (RIP) experiments using control SVZ lysates revealed that PNKY interacts with the splicing regulator, polypyrimidine tract-binding protein 1 (PTBP1). Immunohistochemical staining of PTBP1 showed that it is expressed in the NSCs at the SVZ. PTBP1-KD in NSCs produced a similar phenotype to that observed in PNKY-KD cells, indicating that both PNKY and PTBP1 can regulate a common set of genes related to cell-cell adhesion, synaptogenesis and neurogenesis, which are necessary to maintain the self-renewal state. Furthermore, results from double KD of PNKY and PTBP1 indicated the possibility that they act in the same pathway because neuron- specific gene expression levels did not show any synergistic changes compared with those in the individual KDs. These results indicate that PNKY can negatively regulate the neural commitment of NSCs by coordinately interacting with PTBP1 (64).
Many studies employ ESCs as an alternative to NSCs, but as an in vitro model for neural differentiation. As shown in Figure 1, G, H, these studies show that a transcription factor, SRY-box 2 (Sox2), is essential for ESCs to maintain the self-renewal stage (65) and for transition to the commitment stage (66, 67). Intriguingly, several lncRNAs are involved in the regulation of Sox2 gene expression and in the action of the SOX2 protein during the transition phase (58, 62, 63). Paupar is one of such lncRNA that represses Sox2/Paired box 6 (Pax6) gene expression, thereby maintaining ESCs in the self-renewal state (Figure 1 G); the genome-wide binding profiles of Paupar, identified using Capture Hybridization Analysis of RNA Targets-Sequencing, revealed that the lncRNA bound to promoter loci of Sox2 in Neuro2a neuroblastoma cells. GO analysis of genes harboring Paupar binding sites indicated that Paupar can regulate the transcription of genes involved in stem cell development, such as Sox2 and Pax6. Furthermore, knockdown of Paupar expression in Neuro2a cells caused the up-regulation of Pax6 mRNA levels and increased Sox2-reporter expression, causing neurite outgrowth and up-regulation of a neuronal differentiation marker, class III beta-tubulin (Tubb3) (62).
In contrast to Paupar, the lncRNA TUNA enhances Sox2 transcription in ESCs (Figure 1 H). TUNA was originally identified as a novel lncRNA controlling the pluripotency of mouse ESCs by a study using a genome-scale short hairpin RNA (shRNA) library that targets lncRNAs (22). TUNA forms a quadruplicate complex (TUNA-RBP complex) with the RNA-binding proteins PTBP1, hnRNP-K, and nucleolin. Chromatin isolation by RNA purification revealed that TUNA binds to the Sox2 promoter and activates transcription by recruiting PTBP1, hnRNP-K, and nucleolin. Knockdown of TUNA resulted in decreased Sox2 expression and repression of ESC neural differentiation. These results indicate that TUNA can promote ESCs to transition from the self-renewal state to the commitment stage by activating Sox2 gene expression (22).
In the commitment stage, RMST recruits SOX2 to promote neuronal differentiation (Figure. 1 H). RMST was identified as a neuronal lncRNA by studies comparing gene expression profiles of human ESCs and differentiated human neurons, using microarray screening in association with RT-qPCR (58) Ng and colleagues showed by RIP that in NSCs, RMST associated with SOX2 and recruited it to the promoter regions of genes encoding trans-acting transcription factor 8 (SP8), neurogenin 2 (NEUROG2), and distal-less homeobox (DLX) (23). Knockdown of RMST in NSCs reduced the number of TUBB3-positive neurons, whereas RMST overexpression resulted in increased levels of TUBB3 and increased the number of TUBB3-positive neurons. These results implicate RMST as a key player in NSCs acquiring a neural cell fate in the commitment stage (23, 58). These reports thus showed that TUNA and RMST can collaborate to promote neuronal differentiation by controlling expression of both Sox2 and Sox2-downstream genes.
REST can regulate the transcription of RMST during the differentiation of human ESCs into neurons (23, 58). The underlying mechanisms of REST action were examined by a chromatin immunoprecipitation (ChIP) assays in combination with siRNA-mediated knockdown. REST was shown to bind to the RMST locus and to repress transcription in human ESCs (58). These results suggest that REST acts as a negative regulator controlling RMST-dependent differentiation of NSCs into neurons. This was supported by the observation that the levels of REST decreased as differentiation proceeded from NSCs to neuronal cells, while, conversely, RMST expression levels increased (23). Although the mechanism by which the expression levels of REST decrease during neuronal differentiation is not fully elucidated, phosphorylation- or ubiquitination-dependent REST degradation in response to extracellular stimuli, such as growth factors or retinoic acids, is associated with neuronal differentiation (68, 69). These findings indicate that as the amount of REST, which is normally abundant, decreases the expression level of RMST rises, resulting in promotion of neuronal commitment.
In the developing brain, accurate differentiation of GABAergic interneurons, which have inhibitory functions on postsynaptic neurons, is essential for neural circuit formation (Figure. 1 C), and dysfunction of interneurons is associated with neurological disorders (24). During interneuron specification, the lncRNA, Evf2, is key for promoting commitment to GABAergic progenitors (24) (Figure. 1 H). Evf2 has been characterized using Evf2 loss-of-function mice that possess a triple polyadenylation site that leads to the premature termination of Evf2 transcription. Quantitative chromatin IP-PCR showed that loss of functional Evf2 expression prevents DLX and methyl-CpG binding protein 2 (MeCP2) transcription factors from binding to intergenic enhancer loci in DLX5 and DLX6 genes. While DLX5 induces the expression of glutamic acid decarboxylase (GAD), which synthesizes gamma-aminobutyric acid (GABA), Evf2 loss-of-function mice show reduced levels of GAD67 protein and a reduced number of GAD67-positive GABAergic interneurons in the postnatal brain (24). These results suggest that Evf2 plays an important role in the development of GABAergic progenitors from NSCs in the developing mouse brain.
The regulatory roles of lncRNAs in CNS development have also been investigated by employing the developing retina as a model, where RPCs can generate all retinal cell types, including both neuronal and glial cells (70). The lncRNA, Gomafu, is expressed in a specific set of retinal neurons (Figure 1 J). shRNA-mediated knockdown of Gomafu in the postnatal retina increased the numbers of amacrine cells and Müller glial cells, suggesting that Gomafu can negatively regulate the commitment of multiple retinal cell types (25, 71). On the other hand, in utero knockdown of the lncRNA taurine up-regulated 1 (Tug1) in the rat postnatal retina resulted in the loss or malformation of the outer segment of photoreceptor cells, whereas other cell types, including bipolar cells, amacrine cells, and Müller glial cells, were not affected (Figure 1 J). These observations suggest that Tug1 can positively regulate the commitment of RPCs to retinal photoreceptors (72). The molecular basis of the action of each of these lncRNAs remains to be elucidated; however, these results indicate that Gomafu and Tug1 are required for the commitment stage in the developing retina.
In contrast, the mechanism of action of lncRNA, Six3OS1, on retinal cell type specification has become clearer. Six3OS1 is transcribed from the reverse strand of Six3, which encodes a transcription factor required for mammalian eye development (25) (Figure 1 J). Both knockdown and overexpression of Six3OS1 show that it does not affect Six3 expression levels. RIP analysis then revealed that Six3OS1 associates with EYA family proteins, which are protein tyrosine phosphatases that function as transcriptional co-regulators. The Six3OS1-EYA ribonucleoprotein complex then interacts with SIX3 family transcription factors as well as enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a component of the polycomb-repressive complex 2 (PRC2), resulting in silencing of SIX3 target genes through methylation of histone 3 regions (25, 73). Furthermore, in utero knockdown of Six3OS1 in the mouse postnatal retina reduced the number of bipolar cells and increased the number of Müller glial cells, whereas overexpression of SIX3 protein rescued the Six3OS1 knockdown phenotype (25). These results suggest that the Six3OS1-EYA ribonucleoprotein complex can modulate SIX3 activity through PRC2-dependent histone modification of its target genes, resulting in the regulation of RPC specification to the particular retinal cell type.
Overall, various investigations have revealed that lncRNAs are involved in the regulation of neural cell differentiation observed both in vivo, such as in the SVZ and retina, and in vitro, such as in ESCs and neural cell lines. These lncRNAs are key players that regulate the interactions of chromatin remodeling enzyme subunits, such as PRC2 and EZH2, with target genes or that recruit transcriptional machinery, such as SOX2 and MeCP2, to specific loci. Considering that distinct lncRNAs act to maintain each differentiation stage, lncRNAs have emerged as important components in the processes of neural differentiation.
During development, neuronal cells extend processes, such as dendrites and axons, and organize synapses that function as platforms for neurotransmitter-dependent signal transmission between neurons. This process is termed synaptogenesis. It has been known for a long time that regulated synaptogenesis is a fundamental process in the organization of neural circuits during development and in the processes of learning and memory in the mature brain (74). There is, however, a limited number of studies that report on the functional correlation between lncRNAs and synaptogenesis. From among these studies, we first discuss the possible action of the lncRNAs, Malat1, and fragile X mental retardation 4 (FMR4) on synaptogenesis.
Malat1 is highly abundant in neurons and localizes to nuclear speckles in a transcription-dependent manner (75) (Figure 1 I). Knockdown experiments with the active transcription sites-visualization technique revealed that Malat1 recruited SR family splicing factors to transcription sites. DNA microarray and GO analyses using Malat1-depleted Neuro2a cells showed that Malat1 regulates the expression levels of genes involved in synapse and dendrite development. Interestingly, knockdown of Malat1 caused a significant reduction of synapse formation between dendrites and axons, whereas Malat1 overexpression increased the synaptic density in cultured hippocampal neurons (75). These results strongly suggest that Malat1 regulates synaptogenesis by recruiting splicing factors to synapse formation-related gene loci. However, an in vivo study using Malat1 knockout mice produced conflicting results because the mice did not show any defects in pre- or post-natal, or mature stages (26). Moreover, loss of Malat1 expression did not cause any significant changes in the expression levels or phosphorylation status of SR proteins. RNA-sequencing (RNA-seq) experiments using brain cortex of Malat1 knockout mice showed that loss of Malat1 did not positively or negatively affect global pre-mRNA splicing in the adult mouse brain (26). Although these results indicate that Malat1 can be involved in synaptogenesis through pre-mRNA splicing, more detailed in vivo analyses are required to fully elucidate the role and relevance of Malat1 in synaptogenesis.
The lncRNA, FMR4, is a natural antisense RNA transcribed from the reverse strand of the fragile-X retardation 1 (FMR1) gene. This lncRNA is classified as a member of a trans-acting lncRNA family that includes Malat1 and HOX transcript antisense RNA (HOTAIR), which function at distant genomic loci from the region of origin. It has been reported that FMR4 somehow prevents apoptosis and has no effect on expression of FMR1 mRNA (76). Microarray analysis using HEK293T cells with knockdown or overexpression of FMR4 identified 238 mRNAs, whose expression levels were affected. Subsequent pathway analysis revealed that FMR4 transacts with genes related to synaptogenesis, and regulation of cell cycle and apoptosis (77). These results indicate the possibility of FMR4 involvement in synaptogenesis during neuronal development by modulating the expression levels of synaptogenesis-related genes in a trans-acting manner.
As synaptogenesis proceeds, neurons generate action potentials in response to synaptic excitation. Neurons modulate the strength and duration of action potentials in response to external stimuli. This is termed synaptic plasticity and plays a key role in neuronal development. Long-term potentiation (LTP) and its opposite phenomenon, long-term depression (LTD) are fundamental properties of synaptic plasticity and are defined as a persistent increase or decrease of synaptic strength, respectively, induced by high frequency stimulation (78). These processes are essential for neural circuits and, therefore, for brain functions such as learning and memory (79). There are several examples of lncRNAs that modulate this synaptic plasticity.
Brain cytoplasmic RNA 1 (BC1) was one of first lncRNAs to be identified and is abundantly expressed in the rodent brain (Figure. 1 I). BC1 is actively transported to neuronal dendrites and can act as a translational repressor, interacting directly with eIF4A and poly-A binding protein (PABP) (27, 28). BC1 usually represses the translation of post-synaptic density protein 95 (PSD-95) mRNA and fragile-X retardation protein (FMRP) mRNA in neuronal dendrites. When the group I metabolic glutamate receptor (mGluR) was stimulated in neurons by glutamate, the translational repression was canceled and both PSD-95 and FMR1 mRNAs were translated. BC1 knockout mice display constitutive de-repression of translation of these mRNAs. Subsequent mGluR activation was suggested to enhance excessive translation of PSD-95 and FMR1 mRNAs, and to lead to neuronal hyperexcitability in the BC1 knockout mice (29). Behavioral tests subsequently revealed that BC1 knockout mice show decreased levels of exploration and higher levels of anxiety compared with controls but normal spatial memory functions (80). This is an elegant experiment that correlated lncRNA function with synaptic plasticity at the levels of molecular mechanism and higher in vivo brain function.
Brain-derived neurotrophic factor (BDNF) is essential for differentiation, long-term survival, and signal transmission in neuronal cells (81). BDNF is also required for synaptic plasticity (79). BDNF-AS is an lncRNA transcribed from the reverse strand of the BDNF gene (82) (Figure 1 I). BDNF-AS represses BDNF transcription in an epigenetic manner. BDNF-AS-specific knockdown in combination with ChIP assays indicated that BDNF-AS enhances H3K27 methylation levels within the BDNF promoter region by recruiting EZH2 without affecting BDNF mRNA stability (82). Intriguingly, repeated depolarization by potassium chloride, which induces LTP, enhanced the levels of BDNF transcription in the human SH-SY5Y neuroblastoma cell line, whereas the levels of BDNF-AS expression were repressed (83). These results suggest that BDNF-AS is involved in synaptic plasticity through the epigenetic regulation of BDNF expression in response to neuronal excitation.
Conversely, synaptic plasticity can modulate lncRNA expression in response to external stimulation. Global transcriptome analysis was recently performed using adult rat hippocampal dentate gyrus (DG) to identify novel lncRNAs whose expression levels were altered in response to synaptic stimulation and LTP formation (84). RNA-seq and subsequent bioinformatic analyses revealed that the expression levels of 71 novel lncRNAs were significantly changed in the adult rat DG upon in vivo LTP formation, of which the majority were upregulated (84). These results indicate that lncRNAs and synaptic plasticity can be mutually regulated in the CNS. However, the target genes of these LTP-dependent lncRNAs are unidentified (84) and how these lncRNAs act on their targets to regulate neuronal development remains to be elucidated.
Considering that both synaptogenesis and synaptic plasticity play important roles in the formation of functional neural circuits and in the acquisition of CNS functions, Maag et al suggest that lncRNAs can directly contribute to nervous system development by controlling synaptogenesis (85). However, this contribution receives negative feed-back regulation from synaptic plasticity-dependent lncRNA expression.
Neurological diseases are important causes of human illness and mortality. They are usually classified by their pathology into several groups; including cerebrovascular diseases, dementia, demyelinating diseases, and neurodegenerative diseases (Table 2). Most such neurological diseases develop after completion of CNS development, that is, after the maturation stage in Figure 1. However, neurological diseases that result from developmental abnormalities that arise in the self-renewal or commitment stages can be caused by structural abnormalities in the CNS formed in utero.
| Disease condition |
| • Cerebrovascular diseases |
| • Dementia |
| • Demyelinating diseases |
| • Neurodegenerative diseases |
| • Infectious diseases |
| • Neuromuscular diseases |
| • Epilepsy, Seizure and Headache |
| • Phakomatosis |
| • Brain tumor |
| • Hydrocephalus |
| • Peripheral nervous system disorders |
Dysregulation of lncRNAs has been observed in various diseases, including cognitive disorders, cardiovascular diseases, and cancer (86-88). As described in section 3, lncRNAs are involved in the formation of the CNS; therefore, it is plausible that dysregulation of lncRNAs can cause neurological diseases. In this section, we focus on the mode of action of lncRNAs involved in neurological diseases. We discuss major neurological diseases, in which the correlation between dysregulated lncRNA expression and the onset and pathogenesis has been described (Table 3). Such neurological diseases include glioma and neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease. The pathogenic involvement of lncRNAs in secondary neurological diseases has been reviewed elsewhere (89, 90).
| Neurological disease | Related lncRNAs |
| Neurodegenerative disease | |
| Alzheimer’s disease | BACE1-AS, 51A, BC200, 17A |
| Parkinson’s disease | Malat1, MAPT-AS1, HOTAIR, NEAT1 |
| Brain tumor | |
| Glioma | CRNDE, HOTAIR, MEG3, GAS5 |
| Diseases causing abnormal synaptic plasticity | |
| Fragile X syndrome | FMR4 |
AD is one of the most common neurological diseases, leading to memory and cognitive dysfunction. The majority of cases are sporadic, with approximately 10% of cases exhibiting familial heritability. Many AD-related genes have been identified, whose mutation or aberrant expression can lead to the onset of AD. These include, amyloid beta precursor protein (APP), beta-secretase 1 (BACE1), and sortilin related receptor 1 (SORL1). The protein products from the latter two genes are implicated in aberrant processing of APP and the subsequent formation of extracellular amyloid beta (Abeta) plaques, which are also known as the senile plaques. Enhanced aggregation and deposition of Abeta lead to neuronal loss and brain contraction, which can cause dementia (91).
BACE1 encodes beta-secretase, a single-pass transmembrane protein involved in the processing of APP. BACE1 cleaves off the extracellular domain of APP to produce the proteolytic products Abeta 1-40 or Abeta 1-42, which are the major components of Abeta plaques. Aberrant BACE1-dependent APP processing is likely to be a critical step in AD pathogenesis (92).
SORL1 encodes a type-1 membrane protein, SORL1, which is involved in the endocytosis of cell surface APP and in its subsequent transport to the Golgi apparatus. SORL1 then traps APP in the Golgi apparatus and reduces the amount of APP that is transported back to the cell surface to form Abeta plaques (93). SORL1 can interact with APP and affect both APP-trafficking and BACE1-dependent proteolytic processing. In addition, overexpression of SORL1 reduces the amount of cell surface APP and suppresses Abeta plaque formation, whereas reduced expression of SORL1 causes aberrant APP trafficking and increased APP processing and Abeta plaque formation (93). These results indicate that dysregulation of intracellular APP trafficking can enhance AD symptoms.
Several lncRNAs have been implicated in the regulation of BACE1 and SORL1 expression in a post-transcriptional manner (12, 94). The lncRNA, BACE1-AS, is transcribed from the reverse strand of the BACE1 gene and forms a duplex with BACE1 mRNA. Formation of this duplex enhances BACE1 mRNA stability, resulting in increased levels of BACE1 protein, and abnormally increased levels of Abeta 1-42 (Figure 2). Indeed, the levels of both BACE1 mRNA and BACE1-AS were up-regulated in both AD patients and APP transgenic mice (12).
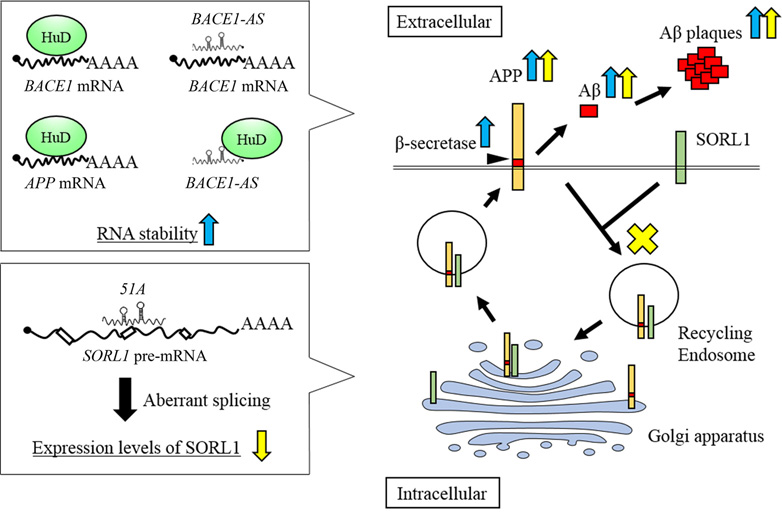 Figure 2.
Figure 2.AD-related lncRNAs in amyloid beta (Abeta) plaque formation. The lncRNA, BACE1-AS, and HuD interact with BACE1 mRNA and increase mRNA stability to promote beta-secretase activity, resulting in enhancement of Abeta plaque formation (indicated by blue arrows). lncRNA, 51A, reduces the levels of SORL1 protein by altering the pre-mRNA splicing pattern, which leads to inhibition of SORL1-dependent APP endocytosis and enhance the Abeta plaque formation (indicated by yellow arrows).
In addition, RNA stability of BACE1-AS is increased by neural RNA-binding protein Hu antigen D (HuD), which promotes APP synthesis (32). HuD also binds to BACE1 and APP mRNAs by recognizing U- or AU-rich elements, which prevents these mRNAs from being degraded (32) (Figure 2). Indeed, in HuD transgenic mice, HuD raised the expression levels of BACE1-AS, and BACE1 and APP mRNAs, resulting in increased levels of Abeta 1-40 (32). In AD brains, both HuD and BACE1 protein levels are also significantly increased (32). These findings indicate that BACE1-AS in combination with HuD can stabilize BACE1 mRNA in a post-transcriptional manner, leading to aberrant APP processing, which might contribute to AD symptoms (12).
The lncRNA, 51A, is transcribed from the reverse strand of SORL1 intron 1. Overexpression of 51A in human neuroblastoma SKNBE2 cells leads to greatly reduced levels of SORL1 splice variant A mRNA and its protein product, SORL1 variant A (94). Enhanced 51A expression also significantly increases the secretion of both Abeta 1-40 and Abeta 1-42. Indeed, the level of 51A was up-regulated in the cerebral cortices of AD patients compared with control individuals (94). These results suggest that 51A can change the SORL1 pre-mRNA splicing pattern and negatively affect the expression levels of SORL1 protein by forming an RNA duplex with the SORL1 pre-mRNA at canonical splice sites. The reduction of SORL1 protein levels then causes aberrant SORL1-dependent intracellular APP processing (Figure 2).
In recent years, transcriptome analyses have been performed to comprehensively identify all lncRNAs involved in AD pathogenesis. These analyses were largely based on microarray screening and RNA sequencing using brain samples of AD patients or AD model mice. These approaches enabled the expression levels of individual lncRNAs to be determined and the identification of target genes regulated in cis or in trans by the lncRNAs (95, 96).
Faghihi and colleagues (95) employed the RNA-seq approach and identified 143 protein-coding genes, 31 natural antisense transcripts (NATs), and 89 lincRNAs specifically expressed in the brains of AD patients. Intriguingly, subsequent pathway analysis of the 143 protein-coding genes revealed dysregulation of Abeta clearance. Indeed, the associated RT-qPCR analysis revealed that the mRNA levels of serpin family E member 1 (SERPINE1), encoding the plasminogen activator inhibitor type-1 (PAI-1), were significantly up-regulated in AD brains. PAI-1 binds to plasminogen activators and reduces their serine protease activity, which inhibits the conversion of plasminogen to plasmin. Plasmin digests Abeta peptides both in vitro and in vivo and is involved in Abeta clearance in AD mice (97). These results indicate that increased SERPINE1 mRNA levels can cause impairment of Abeta clearance in AD brains. Faghihi and colleagues also specified two novel NATs and lincRNAs that might contribute to AD pathogenesis (95). The NATs, HAO2-AS and EBF3-AS, were transcribed from the reverse strand of the hydroxyacid oxidase 2 (HAO2) gene and the early B cell factor 3 (EBF3) gene, respectively. The two lincRNAs were AD-linc1 and AD-linc2 (XLOC_753726 and XLOC_612449, respectively). RT-qPCR analysis revealed that the expression levels of these four lncRNAs were up-regulated in AD brains. Furthermore fractionation of brain tissues showed that these lncRNAs were abundantly localized in the nucleoplasm and chromatin fractions (95) (Figure 3).
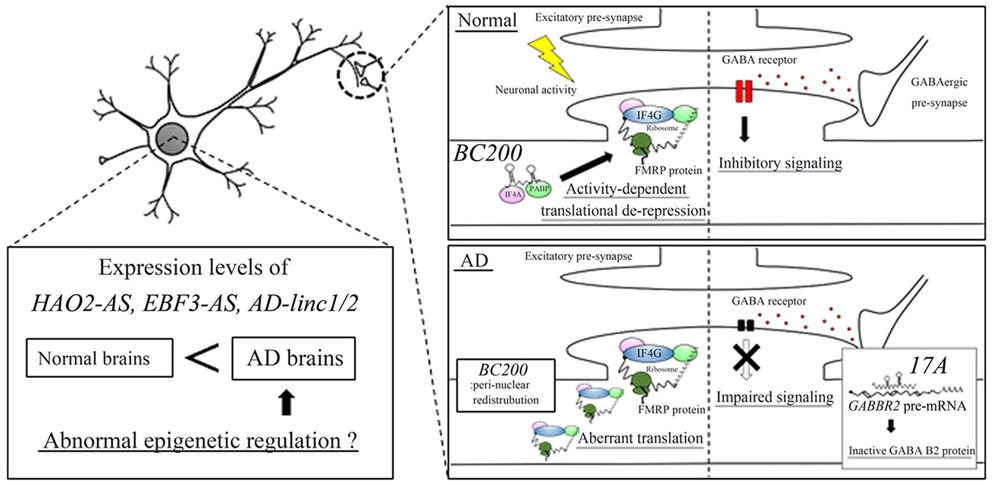 Figure 3.
Figure 3.lncRNAs that are possibly related to AD pathogenesis. The expression levels of HAO2-AS, EBF3-AS, and AD-linc1/2 are abnormally up-regulated in the AD brain, which may epigenetically alter the expression levels of protein-coding genes that contribute to AD pathogenesis. lncRNA, BC200, is localized in the post-synaptic region and represses translation of target mRNAs, including FMR1 in normal brain. In the AD brain, BC200 shows abnormal peri-nuclear localization, which aberrantly enhances FMRP translation in dendrites and causes the impaired plasticity. lncRNA, 17A, produces an inactive GABA B2 protein by altering the splicing pattern of GABBR2 pre-mRNA, resulting in impaired GABA receptor-mediated signaling.
As described in the section 3, lncRNAs are involved in multiple epigenetic processes, such as DNA methylation and histone modification, by regulating the interactions of chromatin remodeling enzyme subunits with target genes. The nuclear localization of these aberrantly expressed NATs and lincRNAs might epigenetically alter the expression levels of protein-coding genes, such as SERPINE1, which may contribute to AD pathogenesis by dysregulating Abeta clearance.
Lee et al (96) reported the presence of unique lincRNAs specifically expressed in an AD transgenic mouse (3xTg-AD) that has mutations in presenilin 1 (PSEN1), microtubule associated protein tau (MAPT) and APP. They employed microarray analysis to compare the expression levels of lincRNAs between 9-month-old 3xTg-AD mice and control mice, and found that 205 lincRNAs were specifically expressed in the AD model mice. Among these lincRNAs, 27 had target genes that showed significantly dysregulated expression patterns in a concordant manner. Subsequent GO analysis showed that the most enriched biological process category associated with these candidate target genes was DNA-dependent regulation of transcription. These results suggest that the 27 lincRNAs might regulate the expression levels of their target protein-coding genes, which are involved in the transcriptional regulation of downstream genes (96).
To more fully understand the modes of action of novel lncRNAs involved in AD pathogenesis, further stepwise investigations are required. Primarily the target genes of novel lncRNAs should be identified by searching preexisting transcriptome databases or by direct analyses using RNA-seq and/or microarray approaches. Secondly, comparative studies of target gene expression levels between AD patients and control individuals may provide valuable information to evaluate whether or not an lncRNA is actively involved in AD pathogenesis. Thirdly, target gene candidates may be analyzed by GO or pathway analysis to integrate the genetic information of the genes. This would allow researchers to investigate how the target genes cause AD pathogenesis. Loss or gain of function analyses of the target genes may then verify the results. Finally, it is crucial to examine the relationship between protein levels of the target genes and the levels of Abeta peptide synthesis or Abeta plaque formation to confirm involvement in AD pathogenesis. These investigations would contribute to the identification of genes modulated by lncRNAs and to understanding their modes of action in Abeta peptide synthesis or Abeta plaque formation.
Other lncRNAs, such as 17A, and BC200, have been implicated in the AD pathogenesis; however, their functional roles are poorly understood (31, 33). 17A is transcribed from the reverse strand of the gamma-aminobutyric acid type B receptor subunit 2 (GABBR2) gene that encodes the GABA B2 receptor (31). Overexpression of 17A in SH-SY5Y cells induced alternative splicing of GABBR2 pre-mRNA, which led to the synthesis of inactive GABA B2 protein and impairment of GABA-mediated inhibitory signaling and enhancement of Abeta peptide secretion by an unknown mechanism. Furthermore, in the AD brain, expression levels of 17A were up-regulated compared with control brains (31). These results suggest that enhanced expression of 17A can inhibit GABA-mediated signaling by changing the splicing pattern of GABBR2 pre-mRNA, leading to enhanced Abeta peptide synthesis in the AD brain (Figure 3).
BC200 is a 200 nucleotide-long lncRNA predominantly expressed in the human brain (98). BC200 may be involved in synaptic plasticity and can act as a translational repressor by interacting directly with eIF4A and PABP, which is similar to rodent BC1 RNA (29). In healthy neurons, BC200 RNA is localized in the post-synaptic region and represses translation of target mRNAs, including FMR1 mRNA (99). With human aging, the expression levels of BC200 RNA decrease, which can lead to de-repression of FMR1 mRNA translation, resulting in decreased synaptic plasticity and subsequent impaired learning and memory (33). In the neurons of AD brains, the expression levels of BC200 RNA are aberrantly increased. FMR1 mRNAs are predominantly expressed in the post-synaptic region of neurons; therefore, redistribution of BC200 RNAs from this region can cause abnormal depression of FMR1 mRNA translation, resulting in the impaired plasticity (33) (Figure 3). These results suggest that BC200 plays a role in memory impairment in AD patients.
Several lines of evidence demonstrate the modes of action of lncRNAs involved in APP synthesis and subsequent Abeta deposition, revealing that lncRNAs can contribute to AD pathogenesis. These lncRNAs can modulate the expression levels of target genes at various stages in the gene expression process (Table 3). Further studies are required to clarify how aberrant regulation of the processes mentioned above can lead to APP synthesis and subsequent Abeta deposition, and investigations of AD-related lncRNAs will help to understand AD pathogenesis.
PD is a progressive neurodegenerative disease exhibiting motor symptoms, including bradykinesia, rigidity, resting tremor, and posture instability. PD also presents non-motor symptoms, such as autonomic neuropathy, depression and dementia. PD is characterized by the appearance of Lewy bodies, cytoplasmic inclusions composed of alpha-synuclein-ubiquitin complexes, and the degeneration of nigrostriatal dopamine neurons (100).
The clinical diagnosis of PD requires the presence of the motor symptoms, called parkinsonism (101), whereas when dementia occurs before the onset of parkinsonism, Lewy body dementia is diagnosed. Lewy body dementia presents visual hallucination, dementia, and parkinsonism and Lewy bodies develop throughout the brain (102).
To elucidate PD pathogenesis, experimental in vivo and in vitro PD models have been employed (103, 104). An in vivo PD mouse model was established by the administration of the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-hydrogen chloride (MPTP-HCl), by intraperitoneal injection, while in vitro PD models have been prepared by the treatment of cell lines, such as SH-SY5Y neuroblastoma cells, with 1-methyl-4-phenyl- pyridinium ion (MPP+), which is the metabolite of MPTP. In PD model animals, MPP+ generated from MPTP is transported into dopaminergic neurons by the dopamine transporter (DAT). This impairs mitochondrial energy production by inhibiting complex I activity, resulting in neuronal degeneration. The MPTP-treated animals are then evaluated using behavioral tests to determine whether their motor phenotype represents the clinical symptoms observed in PD patients (105).
Several PD-related genes have been identified by genetic association studies of autosomal dominant and recessive PD families. These genes include synuclein alpha (SNCA), leucine rich repeat kinase 2 (LRRK2), parkin RBR E3 ubiquitin protein ligase (PRKN), phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1), and microtubule associated protein tau (MAPT) (106). Mutations in these genes can lead to multiple intracellular abnormalities, including formation of cytotoxic aggregates, impaired actin remodeling, dysfunctional autophagy, and enhanced pro-apoptotic signaling (107), all of which can lead to neuronal degeneration and PD pathogenesis. In addition, emerging evidence indicates that lncRNAs are involved in PD pathogenesis. Altered expression of lncRNAs was first identified in leukocytes of PD patients by Soreq et al. using whole transcriptome RNA sequencing, although the detailed functions of the identified lncRNAs were not fully elucidated (108).
In the following paragraphs, we describe the canonical functions of PD-related proteins and the aberrant molecular events caused by PD-related mutations, which eventually lead to neuronal degeneration. We highlight the relevant lncRNAs identified so far and their roles in regulating the expression of PD-related genes and their products.
SNCA encodes alpha-synuclein, the first PD-related gene to be identified and the major component of Lewy bodies. Alpha-synuclein inhibits the endocytosis of muscarinic receptors by inhibiting phospholipase D2 activity, and can reduce the cytoplasmic levels of both 3,4-dihydrophenylalanine (L-DOPA) and dopamine (DA) by suppressing the phosphorylation of tyrosine hydroxylase (109, 110). These findings suggest that alpha-synuclein might be involved in the regulation of neurotransmitter release.
Missense mutations in SNCA, such as Ala53Thr, create alpha-synuclein mutants that are likely to form a beta-sheet structure, which can lead to aberrant aggregation of alpha-synuclein (Figure 4) (111). Furthermore, SNCA locus triplication or duplication can increase the levels of alpha-synuclein protein, leading to aberrant aggregation and neuronal degeneration (112, 113). It is not fully understood how alpha-synuclein aggregation causes cytotoxicity; however, recent reports suggest that an abnormal alpha-synuclein abundance can cause aberrant microtubule assembly in pre-synaptic regions, resulting in impaired vesicle recycling, impaired endocytosis, reduced vesicle pool size, and diminished exocytosis of neurotransmitters (114-116). These studies suggest that aberrant alpha-synuclein aggregation can lead to neuronal degeneration by dysregulating neurotransmitter release.
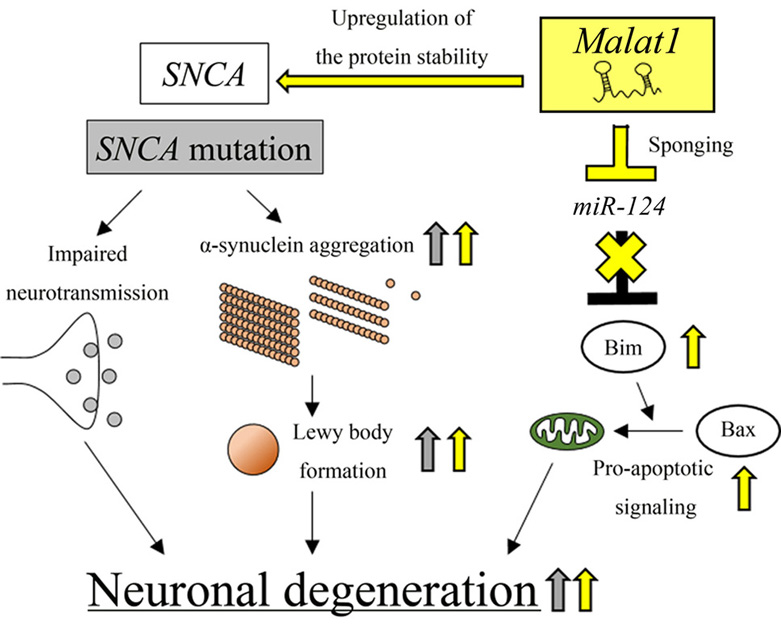 Figure 4.
Figure 4.lncRNA Malat1-mediated neuronal degeneration in PD pathogenesis. Malat1 enhances alpha-synuclein protein stability, which leads to aggregation and Lewy body formation, resulting in neuronal degradation. Malat1 acts as a decoy to repress miR-124, leading to enhanced apoptotic signaling. This effect causes neuronal degeneration. Malat1-dependent effects are indicated by yellow arrows. SNCA mutation-dependent effects are indicated by gray arrows.
The lncRNA, Malat1, is involved in PD pathogenesis by enhancing alpha-synuclein protein stability and inhibiting miR-124, which acts as a neuroprotective miRNA (Figure 4) (35, 37). In the MPTP+-treated SH-SY5Y cells, the expression levels of Malat1 are increased. Furthermore, Malat1 associates with alpha-synuclein protein and prevents the protein from undergoing proteasome-dependent degradation, resulting in impaired cell viability (37). In addition, Malat1 enhances neuronal apoptosis by acting as an miR-124 sponge in both in vitro and in vivo PD models (35). miR-124 was reported to exert a neuroprotective effect by reducing the expression levels of Bim, a BCL2 homology region 3 (BH3)-only protein. This accelerated apoptosis through the mitochondrial translocation of BCL2 associated X protein (BAX) (117). In MPTP+-treated SH-SY5Y cells, the expression levels of Malat1 are significantly elevated, whereas those of miR-124 are reduced. Knockdown of Malat1 increases miR-124 expression, which causes a reduction in the number of apoptotic dopaminergic neurons, which was verified by measuring activated caspase-3 activity (35). These results suggest that, in addition to the positive regulation of alpha-synuclein protein stability, Malat1 can act as a ceRNA against miR-124 (see also Section 2 above) and promote neuronal degeneration by inducing apoptosis, which eventually leads to PD pathogenesis.
LRRK2 encodes a protein kinase that can regulate neuronal cell morphology, growth and motility through the phosphorylation of ezrin/radixin/moesin (ERM) proteins, which play roles in actin remodeling by anchoring actin filaments to the plasma membrane (118) (Figure 5). In familial PD, several genetic variations of LRRK2 are frequently observed (119). These mutations cause enhanced kinase activity of LRRK2 (120), leading to increased phosphorylation of ERM proteins and subsequent shortening of neuronal processes (118). LRRK2 mutations cause other cellular dysfunctions, including enhanced autophagic activity through extracellular signal regulated protein kinase/mitogen activated protein kinase (ERK/MAPK) signaling (121) and increased apoptosis by activating caspase-3 activity (122). Given that the overexpression of wild-type (WT) LRRK2 in transgenic mice results in shortened neuronal processes (122), the increased phosphorylation of LRRK2-target proteins may lead to neuronal degeneration.
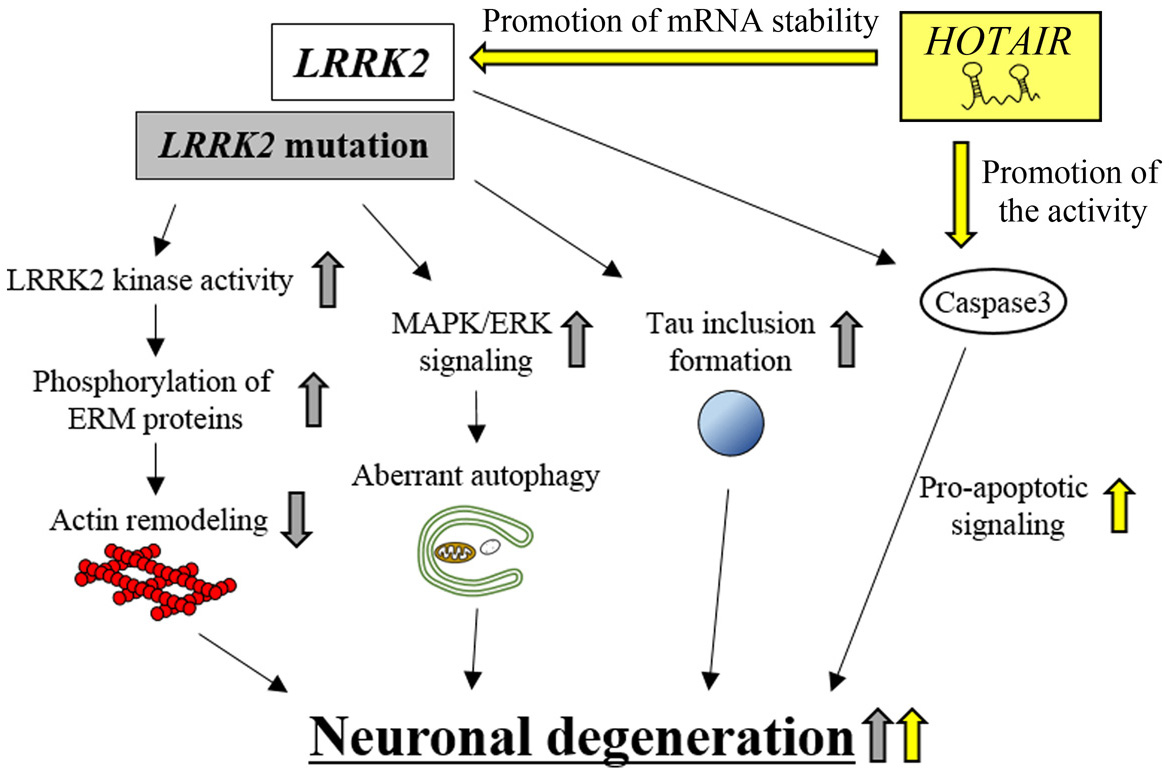 Figure 5.
Figure 5.HOTAIR-mediated neuronal degeneration in PD pathogenesis. HOTAIR promotes LRRK2 mRNA stability and, hence, up-regulates LRRK2 kinase activity. This effect eventually increases neuronal degeneration. HOTAIR enhances caspase-3 activity and, thereby, increases apoptotic signaling. This effect also contributes to eventual neuronal degeneration. HOTAIR-dependent effects are indicated by yellow arrows. LRRK2 mutation-dependent effects are indicated by gray arrows.
The lncRNA, HOTAIR (HOX transcript antisense RNA), is transcribed from the reverse strand of the intergenic region between HOXC11 and HOXC12 genes. HOTAIR was originally reported to regulate the expression levels of target genes, such as HOXD and cadherin 1 (CDH1) in an epigenetic manner (123-126). Several studies using in vivo and in vitro PD models revealed that the expression levels of HOTAIR are up-regulated both in the midbrain of MTPT-treated mice and in MPP+-treated SH-SY5Y cells (36). Up-regulated HOTAIR enhances the protein levels of LRRK2 by inhibiting the degradation of LRRK2 mRNA (36), which leads to neuronal degeneration through various subcellular processes, including increased apoptosis (122). In contrast, knockdown of HOTAIR in MPP+-treated SH-SY5Y cells reduces LRRK2 mRNA and protein levels, and inhibits neuronal degeneration (36). Moreover, this neuroprotective effect of HOTAIR knockdown was abolished by the additional overexpression of WT LRRK2 in MPP+-treated SH-SY5Y cells (127). It is interesting to note that HOTAIR knockdown produces another neuroprotection phenotype by inhibiting caspase-3 activity, which causes a reduction of apoptosis (36). These results suggest that HOTAIR can promote neuronal degeneration by controlling the expression levels of LRRK2 mRNA and caspase-3 enzymatic activity; however, the molecular mechanism of HOTAIR action remains to be elucidated.
MAPT encodes microtubule-associated protein tau, which stabilizes microtubule networks and regulates the transport of vesicles or organelles along the microtubules (128). In transgenic (TG) mice, WT MAPT overexpression causes hyperphosphorylation of tau present in neuronal cell bodies, resulting in the formation of cytotoxic neurofibrillary tangles and enhanced neuronal degeneration compared with control WT mice (129) (Figure 6). In Norwegian PD patients, a particular set of sequential single nucleotide polymorphisms (SNPs) in MAPT, which is termed the H1 haplotype, was observed significantly more frequently than in control individuals (130). The SNPs caused enhanced expression of MAPT and was confirmed as a PD risk factor (131).
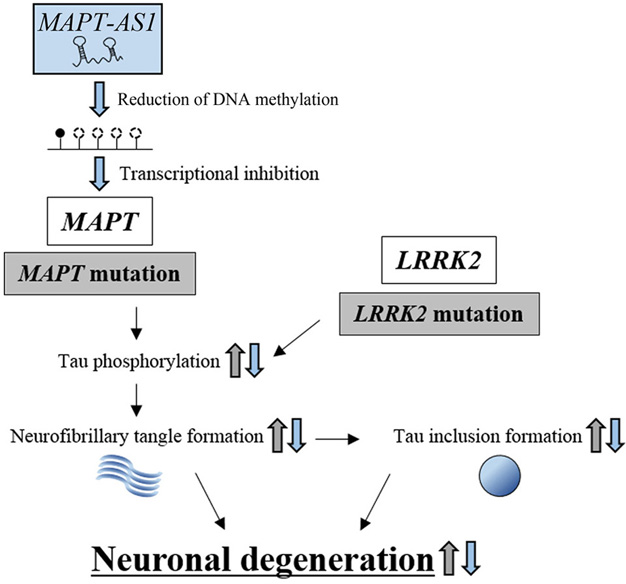 Figure 6.
Figure 6.The protective effect of lncRNA, MAPT-AS1, against neuronal degeneration. Mutations in MAPT and LRRK2 increase the levels of phosphorylated tau, which enhances neurofibrillary tangle formation and tau inclusion formation, resulting in neuronal degeneration (indicated by gray arrows). MAPT-AS1 inhibits MAPT transcription by reducing the DNA methylation levels within the promoter region, resulting in repression of neuronal degeneration. MAPT-AS1-dependent effects are indicated by blue arrows.
These studies suggest that increased MAPT expression and subsequent hyperphosphorylation of tau can cause neuronal degeneration by generating cytotoxic neurofibrillary tangles, which are a hallmark of tauopathy. Although PD is recognized as an alpha-synucleinopathy that exhibits aberrant aggregation of alpha-synuclein (111), the above studies, the H1 haplotype in particular, provide another possibility; PD can be induced by tauopathy with cytotoxic neurofibrillary tangle formation.
The lncRNA, MAPT-AS1, is transcribed from the reverse strand of the MAPT promoter region (34), and is involved in the epigenetic regulation of the MAPT promoter (Figure 6). In vitro methylation analysis in HEK293 cells showed that the overexpression of MAPT-AS1 reduced the DNA methylation levels within the MAPT promoter region, while knockdown of MAPT-AS1 increased methylation of the MAPT promoter region (34). Intriguingly, luciferase analysis indicated that overexpression of MAPT-AS1 reduced MAPT promoter activity, whereas knockdown of MAPT-AS1 increased promoter activity. It follows that the expression levels of MAPT are reduced or increased by the overexpression or knockdown of MAPT-AS1, respectively. Indeed, increased methylation in MAPT promoter regions is associated with reduced MAPT-AS1 expression in PD patients (132).
In general, DNA methylation promotes the association of methyl-CpG-binding domain (MBD) proteins with a promoter region, which prevents transcription factors from recognizing the promoter region, resulting in reduced transcription and conformational change of chromatin into silent chromatin (133). However, the above MAPT-AS1 regulation of CpG methylation at the MAPT promoter region and the resulting effects on MAPT mRNA expression appears to violate this rule. It has been previously shown that methyl-CpG-binding protein 2 (MeCP2), a member of the MBD family, can bind to methylated DNA and activate or repress transcription, which depends on the type of target gene (134, 135). On a methylated promoter region, MeCP2 modulates the expression levels of downstream genes by selecting transcriptional activators or repressors. For example, MeCP2 interacts with cAMP responsive element binding protein 1 (CREB1) to activate brain-derived neurotrophic factor (Bdnf) gene transcription. However MeCP2 can also interact with SIN3 transcription regulator family member A (Sin3A) to repress Hairy2a gene transcription (136-139). To clarify the mechanism by which MAPT-AS1 reduces MAPT expression through the suppression of DNA methylation, the transcriptional regulators that interact with the methylated MAPT promoter region need to be identified, e.g. by employing ChIP assays with antibodies against MeCP2. Promoter-associated lncRNAs are usually scaffolds for nuclear processes or guides for ribonucleoprotein complexes; therefore, the above results suggest that MAPT-AS1 may be involved in the epigenetic regulation of the MAPT promoter, resulting in the down-regulation of MAPT expression. MAPT-AS1 might therefore play a pivotal protective role against neuronal degeneration in PD pathogenesis.
Under physiological conditions, mitochondrial energy production simultaneously generates reactive oxygen species (ROS) that can impair mitochondrial functions. Generally, antioxidant enzymes, such as glutathione peroxidase-1, superoxide dismutase-2, and peroxiredoxin-1, remove the ROS to protect mitochondria from oxidative stress (140). However, a small portion of ROS escape the antioxidant enzymes and may damage the mitochondria (141). The impaired mitochondria, which can continue to produce ROS to inflict intracellular oxidative stress, are usually removed by the ubiquitin-proteasome system (142). The system requires PINK1 and Parkin, encoded by PINK1 (PTEN-induced kinase 1) and PRKN, respectively (143). Although under physiological conditions, PINK1 is constitutively synthesized and localized to the mitochondrial outer membrane, the kinase is usually degraded by membrane-voltage sensitive proteolysis (144). However, in impaired mitochondria, turn-over of PINK1 is inhibited by reduced membrane voltage, resulting in increased accumulation of PINK1 (144). The accumulated PINK1 then phosphorylates Parkin which moves from the cytosol to the mitochondria. Phosphorylated Parkin can ubiquitinate mitochondrial outer membrane proteins via its E3-ubiquitin ligase activity (143). The ubiquitinated proteins then enhance the degradation of impaired mitochondria by the ubiquitin-proteasome system (143). These results indicate that PINK1 and Parkin regulate mitochondrial homeostasis, which protects neurons from the oxidative stress produced by impaired mitochondria, and subsequent neuronal degeneration. Indeed, the PINK1 or PRKN mutations found in autosomal recessive juvenile PD (145, 146), reduce the degradation of impaired mitochondria (142). The mutations reduce the efficiency of PINK1 for phosphorylating Parkin. Non-phosphorylated Parkin then works in an inefficient way to ubiquitinate the impaired mitochondria (142), resulting in neuronal degeneration (147).
Meanwhile, enhanced nuclear paraspeckle assembly transcript 1 (NEAT1) expression can induce aberrant autophagy of both healthy mitochondria and impaired mitochondria by modulating PINK1 protein stability, resulting in neuronal degeneration (Figure 7) (148, 149). NEAT1 is transcribed from the NEAT1 locus and was originally reported to be involved in paraspeckle formation in the nucleus (150). In PD model cells (MPP+-treated SH-SY5Y cells), the expression levels of NEAT1 are increased. Overexpressed NEAT1 further increases the expression levels of PINK1 by inhibiting the degradation of the kinase, resulting in enhanced accumulation of PINK1 not only in the impaired mitochondria but also in intact mitochondria. Accumulated PINK1 directly interacts with LC3-II, the phosphatidylethanolamine-conjugated form of LC3 that is essential for the initiation of autophagy, and increases the accumulation of LC3-II in mitochondria, resulting in the aberrant mitochondrial autophagy (148). Intriguingly, the NEAT1/PINK1/LC3-II axis acted not only in the degradation of impaired mitochondria in PD model cells but also in the aberrant elimination of surviving intact mitochondria (149). The NEAT1-mediated overexpression of PINK1 may encourage the degradation of intact mitochondria, resulting in reduced ATP production, which can lead to neuronal degeneration. How NEAT1 inhibits PINK1 protein degradation is still uncharacterized. The above results, however, suggest that the lncRNA can act by stabilizing PINK1, an upstream regulator of LC3-II mobilization, to induce aberrant autophagy. NEAT1 therefore plays a role in PD pathogenesis.
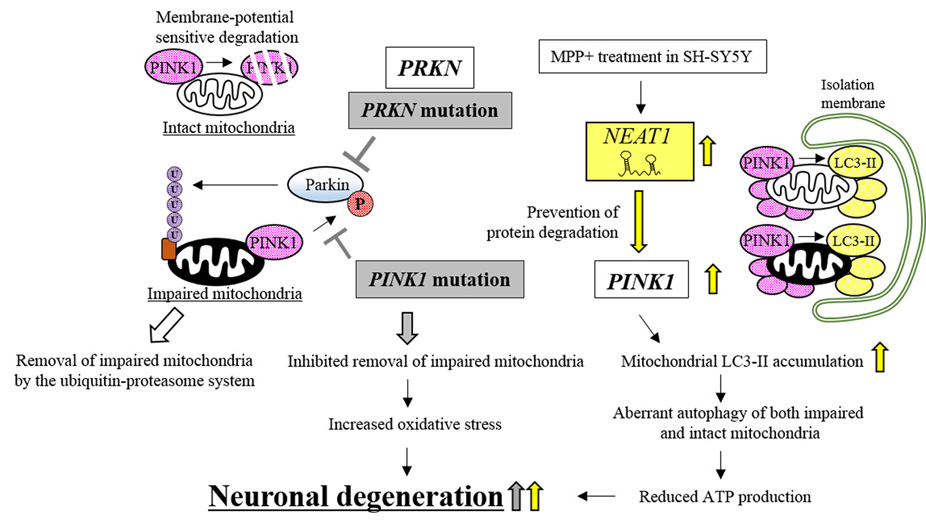 Figure 7.
Figure 7.Neuronal degeneration caused by lncRNA, NEAT1, contributes to PD pathogenesis. Mutations in PRKN and PINK1 reduce the levels of phosphorylated Parkin, which inhibits the removal of impaired mitochondria and causes neuronal degeneration (indicated by gray boxes and arrows). NEAT1 prevents PINK1 degradation, which causes neuronal degeneration by inducing the aberrant autophagy of mitochondria. NEAT1-dependent effects are indicated by yellow arrows.
It is widely accepted that PD is caused by mutations in various genes that cause intracellular dysfunction and eventual neuronal degeneration. Recent studies have clarified the modes of action of several lncRNAs that can positively or negatively modulate the expression of causative PD genes and the subsequent intracellular dysfunction (Figure 8). Although further studies are required to clarify the issues listed in Table 4 that are related to PD pathogenesis, it is increasingly evident that the lncRNAs involved in neuronal degeneration play a pivotal role in the onset of PD.
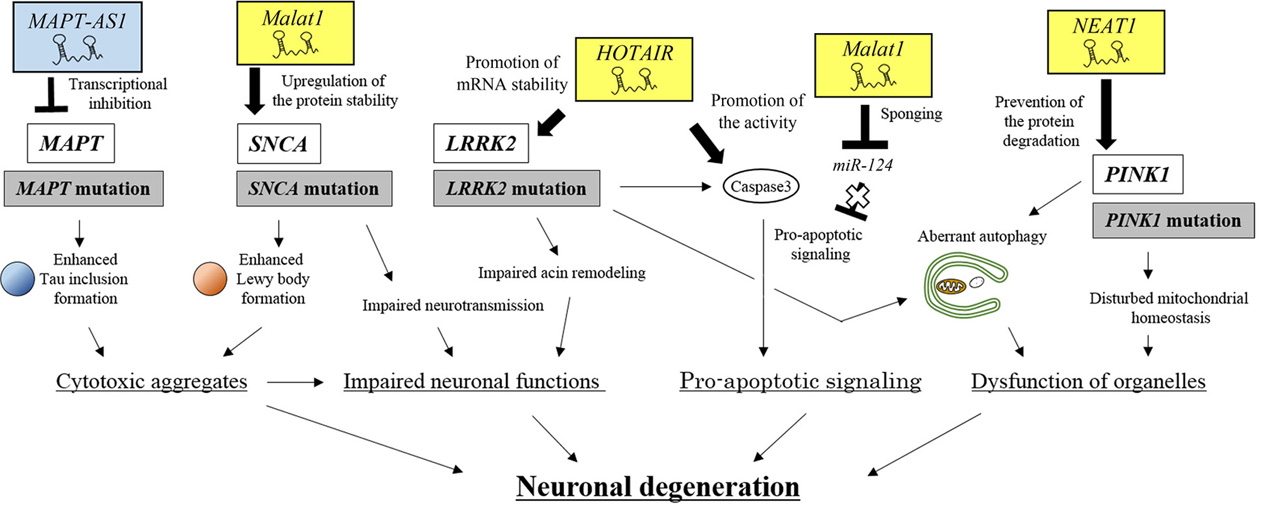 Figure 8.
Figure 8.An integrated view of neuronal degeneration in PD pathogenesis. PD-related lncRNAs positively or negatively regulate signaling pathways, which leads to neuronal degeneration by inducing intracellular dysfunction, such as the formation of cytoplasmic aggregates, impaired neuronal functions, pro-apoptotic signaling and organelle dysfunction.
| lncRNAs | Involved process | Mechanism of action |
| HAO-2AS, EBF3-AS, AD-linc1/2 | Transcription | Epigenetic regulation? |
| 51A, 17A | Co-transcription | Control of alternative splicing |
| BACE1-AS | Post-transcription | Regulation of RNA stability |
| BC200 | Translation | Interaction with translational machinery |
Glioma is a type of primary brain cancer, originating from glial cells, such as astrocytes and oligodendrocytes, and accounts for eighty percent of malignant cancers in the CNS (151). The five-year survival rate of glioblastoma multiforme, the most malignant form of glioma, is less than 5% (152). To improve this poor clinical outcome, the molecular mechanisms of glioma development have been extensively investigated.
Hanahan and Weingerg proposed a series of cancer hallmarks, which has been widely accepted to encompass the molecular mechanisms of cancer development (153). The cancer hallmarks rationalized the individual processes in cancer development; (1) sustaining proliferative signaling; (2) evading growth suppressors; (3) enabling replicative immortality; (4) activating invasion and metastasis; (5) inducing angiogenesis; (6) resisting cell death; (7) deregulating cellular energetics; (8) avoiding immune destruction; (9) genome instability and mutation and (10) tumor-promoting inflammation. During cancer development, cancer initiation includes hallmarks (1), (2), and (10), while cancer progression includes hallmarks (3), (4), and (5). Hallmark (6) is closely related to both initiation and progression of cancer. Hallmarks (7), (8), and (9) are related to other processes, such as regulation of the cancer microenvironment (153).
Intriguingly, recent evidence shows that lncRNAs are involved in cancer development by modulating the cancer hallmarks. Gutschner and Diederichs highlighted the functions of lncRNAs that are involved in the epigenetic regulation of genes whose protein products positively or negatively affect the cancer hallmarks (154). Furthermore, Yoshida and Kimura clarified the modes of action of pathogen-associated non-coding RNAs, which act for or against the individual hallmarks in infectious oncogenesis caused by viruses or bacteria (61).
In the following paragraphs, we first describe the major signaling pathways that are modulated by miRNAs and lncRNAs during glioma initiation and progression. We then discuss the detailed roles of these pathways with respect to the cancer hallmarks. We then refer to miRNAs and lncRNAs that modulate the hallmarks. We put emphasis on the particular sets of lncRNAs that counteract miRNAs as ceRNAs. These lncRNAs include colorectal neoplasia differentially expressed (CRNDE), HOTAIR, maternally expressed 3 (MEG3), and growth arrest specific 5 (GAS5). CRNDE and HOTAIR play pivotal roles in glioma oncogenesis, while MEG3 and GAS5 act as tumor suppressors. We then discuss the modes of action for the lncRNAs that positively or negatively contribute to glioma oncogenesis.
In glioma cells, aberrant expression of signaling ligands and their cell surface receptors causes the abnormal activation of downstream signaling pathways that generally depend on the phosphorylation of their components. Recent evidence shows enhanced expression levels of fibroblast growth factor 1 (FGF1), interleukin 6 (IL6), and EPH receptor A2 (EphA2) in glioma cells (41, 155, 156). Increased levels of FGF1 activate both ERK/MAPK and PI3K/Akt/mTOR signaling pathways (41), while increased IL6 activates the JAK/STAT signaling pathway (155). The ligand-dependent activation of these signaling pathways causes enhanced proliferation, which leads to cancer initiation. This activation also enhances migration and invasion and inhibits apoptosis, which contributes to cancer progression.
In contrast, overexpression of EphA2, a receptor tyrosine kinase, is frequently observed in human cancers, including glioma (156). Upregulated EphA2 can interact with other cell-surface receptors, such as EGFR and HER2/ErbB2, which amplifies MAPK, Akt and Rho family GTPase activities (157, 158). These effects have been linked to cancer progression and poor overall survival (157). In addition, convincing evidence shows that the efficiencies of these signaling pathways are positively or negatively modulated by miRNAs and lncRNAs (39-48). These studies suggest that aberrant activation of multiple pathways can induce the cancer hallmarks, particularly hallmarks (1), (4), and (6), and can contribute to caner initiation and progression of glioma.
The molecular mechanisms by which aberrantly activated signaling pathways lead to cancer hallmarks have been elucidated (Figure 9). We put particular emphasis on hallmarks (1), (4), and (6), which are induced by signaling pathways that are modulated by miRNAs and lncRNAs in glioma oncogenesis. Other cancer hallmarks, such as (2), (5), and (10), also contribute to glioma development; however, the relationships between the signaling pathways and these hallmarks have not been fully investigated with respect to whether miRNAs and lncRNAs regulate signaling efficiency.
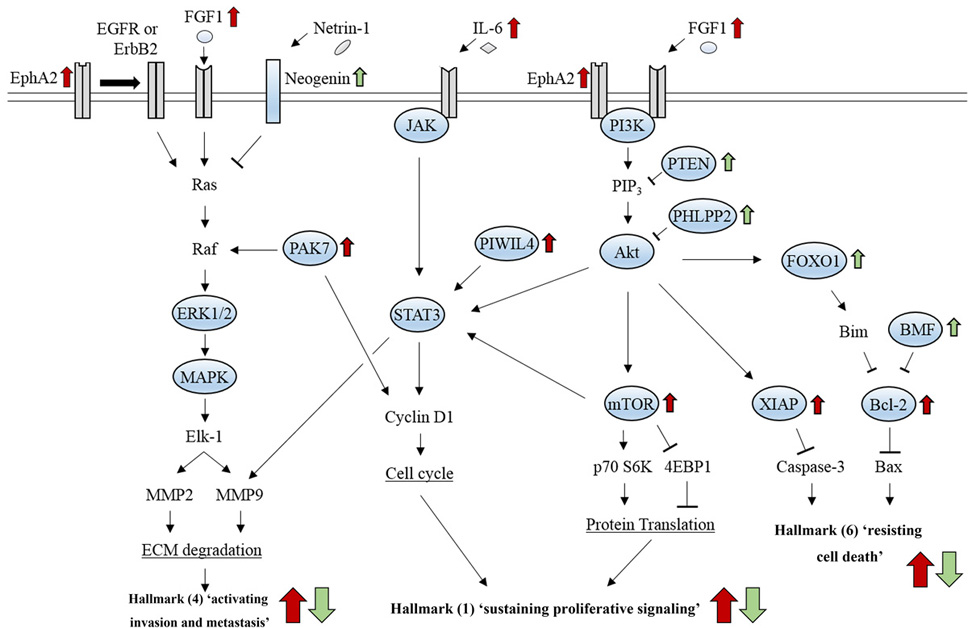 Figure 9.
Figure 9.Signaling pathways related to cancer hallmarks that contribute to glioma development. Hallmark (1) is caused by enhanced cell cycle progression through JAK/STAT signaling and by increased protein synthesis through PI3K/Akt/mTOR signaling. Hallmark (4) is induced by ECM degradation through ERK/MAPK signaling and JAK/STAT signaling, which can cross-talk with the PI3K/Akt/mTOR pathway. Hallmark (6) is induced by inhibition of apoptosis, which results from reduced signaling activity of either caspase-3 or Bax through the PI3K/Akt pathway.
Hallmark 1 ‘sustained proliferative signaling’ can be caused by aberrant activation of several pathways: accelerated cell cycle progression induced by Cyclin D1 activation through JAK/STAT signaling pathway (159); increased protein synthesis by either ribosomal protein S6 kinase (p70S6K) activation or suppression of eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) through the PI3K/Akt/mTOR signaling pathway (160) (Figure 9). Hallmark 4 ‘activated invasion and metastasis’ is mainly induced by extracellular matrix (ECM) degradation which results from increased expression of matrix metallopeptidase 2 (MMP2) and MMP9 through ERK/MAPK signaling and JAK/STAT signaling that can cross-talk with the PI3K/Akt/mTOR pathway (161) (Figure 9). Hallmark 6 ‘resistance to cell death’, can be caused by inhibition of apoptosis, which results from reduced signaling activity of either caspase-3 or Bax through the PI3K/Akt pathway (162) (Figure 9). These findings suggest that activation of these signaling pathways promotes processes, such as ECM degradation, cell cycle progression, and inhibition of protein translation and apoptosis, which are directly linked to hallmarks (1), (4), and (6) in glioma oncogenesis. Moreover, recent studies show signaling pathways that are aberrantly activated in glioma development can be positively or negatively modulated by a particular set of lncRNAs that counteract miRNAs (Table 5).
| lncRNA | Target molecule | Involved process | Mechanism of action | Issues to be elucidated |
| MAPT-AS1 | MAPT | Transcription | Regulation of DNA methylation | Role in DNA methylation / identification of transcription factors |
| HOTAIR | LRRK2 | Post-transcription | Regulation of RNA stability | The mechanism of LRRK2 mRNA stability |
| Caspase-3 | Post-translation | Regulation of enzyme activity | The mechanism of caspase-3 activation | |
| Malat1 | SNCA | Post-transcription | miRNA sponging | The mechanism of α-synuclein protein stability |
| miR-124 | Post-translation | Regulation of protein stability | Expression levels in the case of miR-124 down-regulation | |
| NEAT1 | PINK1 | Post-translation | Regulation of protein stability | The mechanism of PINK1 protein turnover |
As mentioned above, sustained proliferative signaling can be caused by aberrant activation of JAK/STAT and PI3K/Akt/mTOR signaling pathways. The lncRNAs that are related to these signaling pathways can act as ceRNAs to modulate the proliferation of glioma cells by counteracting their target miRNAs (Figure 10).
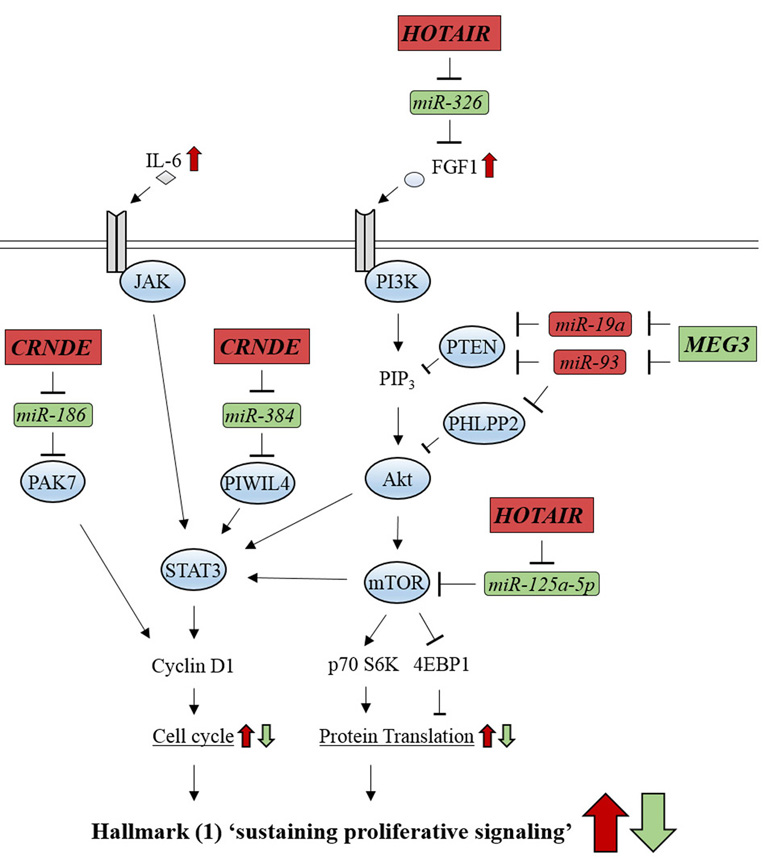 Figure 10.
Figure 10.Glioma-related ncRNAs regulate the signaling pathways involved in cancer hallmark (1) ‘sustaining proliferative signaling’. lncRNA, CRNDE, activates JAK/STAT signaling by counteracting miR-186 and miR-384. lncRNA, HOTAIR, activates PI3K/Akt/mTOR signaling by inhibiting miR-326 and miR-125a-5p. lncRNA, MEG3, represses PI3K/Akt/mTOR signaling by inhibiting miR-19a and miR-93. Oncogenic-ncRNAs and their effects are indicated by red boxes and arrows, respectively. Tumor suppressive ncRNAs and their effects are indicated by green boxes and arrows, respectively.
The lncRNA, CRNDE, can modulate JAK/STAT signaling by acting as an upstream regulator of miR-384/PIWIL4 and miR-186/PAK7 axes (39, 40). In glioma cells, miR-384 interacts with the 3′-UTR of Piwi-like 4 (PIWIL4) mRNA and reduces its expression through RNA destabilization (40). PIWIL4 is an Argonaut family member, originally identified to be involved in post-transcriptional silencing during spermatogenesis, but PIWIL4 is highly expressed in various tumors (163). miR-384-mediated reduction of PIWIL4 mRNA levels suppresses STAT3 phosphorylation, although the mechanism of phosphorylation by PIWIL4 remains unclear (40). Reduced levels of phosphorylated STAT3 attenuate the proliferation of glioma cells through reduced transcription of Cyclin D1, which normally accelerates cell cycle progression and thus promotes cell proliferation (164).
miR-186 interacts with the 3′-UTR of p21-activated kinase (PAK7) mRNA. PAK7 is involved in various cellular phenomena, including proliferation and differentiation (165). miR-186 reduces the levels of PAK7, leading to suppressed phosphorylation and activity of Cyclin D1 and suppressed proliferation of glioma cells (39). CRNDE is located on human chromosome 16 and was first identified in colorectal cancer to display oncogenic properties (166). In primary glioma tissues, CRNDE is overexpressed compared with the normal brain tissues (39). CRNDE prevents miR-384 and miR-186 from interacting with PIWIL4 and PAK7 mRNAs, respectively, and abolishes the tumor suppressive effects of these miRNAs by acting as an onco-ceRNA (39, 40). CRNDE can strongly activate JAK/STAT signaling and enhances sustained proliferative signaling though accelerated cell cycle progression.
In PI3K/Akt/mTOR signaling, HOTAIR can exert oncogenic effects by acting as an upstream regulator of both miR-326/FGF1 and miR-125a-5p/mTOR axes (41, 42). In contrast, the lncRNA MEG3 functions as tumor suppressor by inhibiting onco-miR-19a and onco-miR-93 (43, 44). HOTAIR can act as an onco-lncRNA that accelerates sustained proliferative signaling, while MEG3 acts as a tumor suppressor (Figure 10). miR-326 recognizes the 3′-UTR of fibroblast growth factor 1 (FGF1) mRNA, resulting in destabilized mRNA and reduced FGF1 levels. The miRNA thereby suppresses FGF-dependent PI3K/Akt/mTOR signaling, which suppresses proliferation of glioma cells (41). HOTAIR prevents miR-326 from interacting with FGF1 mRNA and abolishes the tumor suppressive effects of miRNA. HOTAIR thus activates PI3K/Akt/mTOR signaling and enhances sustained proliferative signaling (41) (Figure 10).
The expression levels of miR-125a-5p are reduced in glioma cells (42). The administration of Schisandrin B (Sch B), a chemical compound derived from the traditional Chinese medicinal herb, Schisandra chinesis Baill, increases the expression of miR-125a-5p in a dose-dependent manner. Although its mechanism of action on phosphorylation is unclear, overexpression of miR-125a-5p inhibits the phosphorylation of mechanistic target of rapamycin (mTOR), a serine/threonine kinase that enhances overall protein translation by activating p70S6K or inhibiting 4EBP1. This inhibited the proliferation of glioma cells (42). Overexpression of HOTAIR canceled out the tumor suppressive effects of Sch B by inhibiting miR-125a-5p expression in glioma cells, while knockdown of HOTAIR produced tumor-suppressive effects (42). These results suggest that HOTAIR can prevent miR-125a-5p from suppressing the phosphorylation of mTOR and can abolish the tumor suppressive effects of the miRNA. HOTAIR might thereby increase the efficiency of PI3K/Akt/mTOR signaling and cause sustained proliferative signaling.
Intriguingly, both miR-19a and miR-93 interact with the 3′-UTR of PTEN mRNA and reduce its levels through RNA destabilization in glioma cells (43, 44). PTEN dephosphorylates phosphatidylinositol-3-phosphate (PIP3), which suppresses PI3K/Akt/mTOR signaling. PTEN is a well-characterized tumor suppressor that is frequently mutated in various cancers (167). miR-19a and miR-93 reduce the levels of PTEN, which activates PI3K/Akt/mTOR signaling by inhibiting the dephosphorylation of PIP3 (43, 44). In addition, miR-93 reduces the levels of the PH domain and leucine rich repeat protein phosphatase 2 (PHLPP2), which can directly dephosphorylate phosphorylated Akt and inhibit PI3K/Akt/mTOR signaling (168).
MEG3 is located on human chromosome 14 and increases the levels of p53, a well-known tumor suppressor, although the precise mechanism of its action remain unclear (169). The levels of MEG3 are reduced in glioma cells (43). MEG3 normally acts as a ceRNA and prevents both miR-19a and miR-93 from interacting with their target mRNAs and abolishes their oncogenic effects. MEG3 thus increases levels of PTEN and PHLPP2 and suppresses PI3K/Akt/mTOR signaling, which suppresses proliferative signaling in glioma cells (43, 44). These studies suggest that HOTAIR and CRNDE can initiate proliferative signaling by counteracting multiple tumor suppressive miRNAs and that MEG3 can counteract sustained proliferative signaling by inhibiting oncogenic miRNAs.
Invasion and metastasis is mainly activated by aberrant activation of ERK/MAPK signaling. The aberrant activation of JAK/STAT and PI3K/Akt/mTOR signaling pathways also contributes to activation of this hallmark through the STAT3/MMP9 axis (Figure 9). lncRNAs that are related to these signaling pathways can modulate the invasion and metastasis of glioma cells by counteracting target miRNAs (Figure 11).
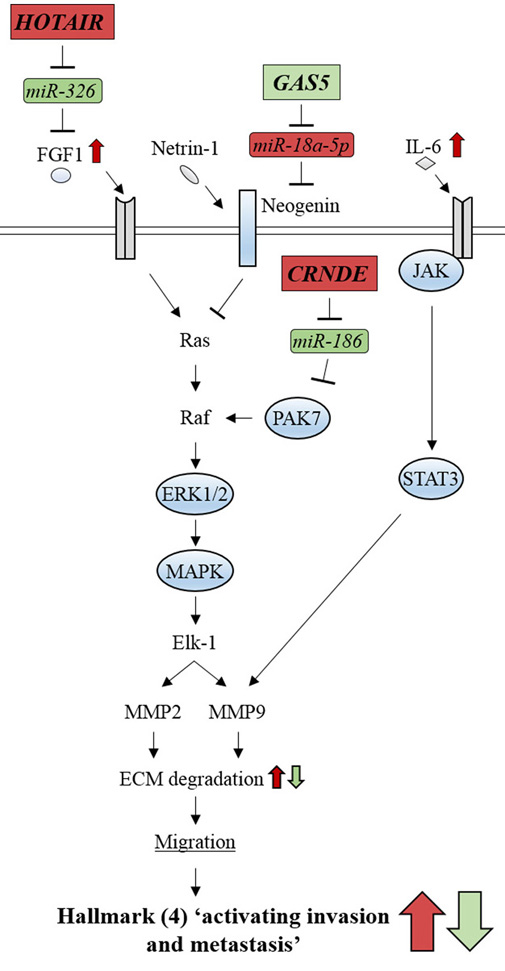 Figure 11.
Figure 11.Glioma-related ncRNAs modulate the signaling pathways involved in cancer hallmark (4) ‘activating invasion and metastasis’. lncRNAs, CRNDE and HOTAIR, activate ERK/MAPK signaling by inhibiting miR-186 and miR-326, respectively. lncRNA, GAS5, represses ERK/MAPK signaling by inhibiting miR-18a-5p. Oncogenic-ncRNAs and their effects are indicated by red boxes and arrows, respectively. Tumor suppressive ncRNAs and their effects are indicated by green boxes and arrows, respectively.
In the ERK/MAPK signaling pathway, the lncRNAs, GAS5, HOTAIR and CRNDE modulate the signaling efficiency by targeting miR-18a-5p, miR-326, and miR-186, respectively, which contribute to the activation of invasion and metastasis of glioma cells (39, 41, 45) (Figure 11). miR-18a-5p functions as an oncogenic miRNA by reducing the levels of Neogenin mRNA by destabilization (170). Neogenin is a Netrin-1 receptor that represses ERK/MAPK signaling, which leads to reduced migration and invasion of glioma cells (170). Indeed, miR-18a-5p de-repressed the Neogenin-dependent reduction of migration and invasion of glioma cells (45). The lncRNA, GAS5, was identified as a spliced form of an lncRNA in which introns encode small nucleolar RNAs (snoRNAs) (171). The expression levels of GAS5 are reduced in glioma tissues compared with normal brain tissues (172). GAS5 prevents miR-18a-5p from interacting with Neogenin mRNA and abolishes the oncogenic effects of the miRNA. GAS5 attenuates the miR-18a-5p-dependent over-activation of ERK/MAPK signaling, resulting in suppression of the hallmark, activation of invasion and metastasis (45). In contrast, miR-326 and miR-186 act as tumor suppressors in ERK/MAPK signaling similar to miR-186 and miR-384 in the JAK/STAT pathway and miR-326 and miR-125a-5p in the PI3K/Akt/mTOR pathway, as described above. HOTAIR up-regulates ERK/MAPK signaling by suppressing the miR-326/FGF1 axis (41). Furthermore, CRNDE regulates the miR-186/PAK7 axis and indirectly enhances PAK7-dependent phosphorylation of Raf, a signal transducer in the ERK/MAPK pathway (39). HOTAIR and CRNDE can thus activate the invasion and metastasis of glioma cells (Figure 11).
Meanwhile, PI3K/Akt/mTOR and JAK/STAT signaling pathways communicate with the ERK/MAPK pathway by Akt/mTOR and STAT3-dependent modulation of MMP9 expression (161) (Figure 9). These observations indicate that MEG3/miR-19a/miR-93, HOTAIR/miR-125a-5p (PI3K/Akt/mTOR pathway) and the CRNDE/miR-384 (JAK/STAT pathway) axes can affect not only the ‘sustaining proliferative signaling’ hallmark but also the ‘activating invasion and metastasis’ hallmark. Activated STAT3 can increase MMP9 expression and can thus enhance ECM degradation of glioma cells, which results in enhanced activation of invasion and metastasis (Figure 9). These studies suggest that the activation of invasion and metastasis is predominantly accelerated by increased ECM degradation, which is modulated by HOTAIR, CRNDE, and GAS5 through ERK/MAPK signaling (Figure 9). In addition, HOTAIR and CRNDE can strongly activate invasion and metastasis by STAT3-dependent modulation of MMP9 expression through JAK/STAT and PI3K/Akt/mTOR signaling pathways (Figure 9).
Hallmark (6) ‘resisting cell death’ is mainly induced by aberrant activation of PI3K/Akt signaling in glioma cells. The lncRNAs that are related to this signaling pathway can modulate apoptotic signaling by acting as ceRNAs to inhibit their target miRNAs (Figure 12). Downstream of Akt, GAS5 and CRNDE can modulate the efficiency of signaling by inhibiting their target miRNAs, such as miR-196a-5p, miR-222, miR-186, and miR-136-5p, which contributes to hallmark (6) ‘resisting cell death’ (39, 46-48). In contrast, upstream of Akt, common sets of miRNAs/lncRNAs, such as MEG3/miR-19a/miR-93 act for hallmarks (1) and (6) (see Figures 10 and 12).
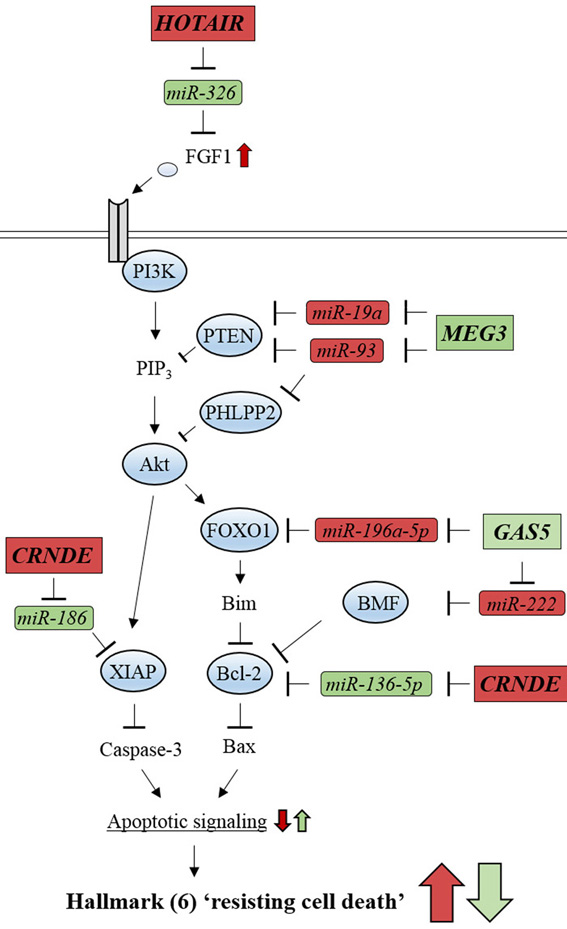 Figure 12.
Figure 12.Glioma-related ncRNAs regulate the PI3K/Akt signaling pathway involved in cancer hallmark (6) ‘resisting cell death’. lncRNA, HOTAIR, activates PI3K/Akt signaling by inhibiting miR-326. lncRNA, MEG3, represses PI3K/Akt signaling by inhibiting miR-19a and miR-93. lncRNA, CRNDE, inhibits apoptotic signaling by repressing miR-186. lncRNA, GAS5, enhances apoptotic signaling by inhibiting miR-196a-5p and miR-222. Oncogenic-ncRNAs and their effects are indicated by red boxes and arrows, respectively. Tumor suppressive ncRNAs and their effects are indicated by green boxes and arrows, respectively.
Both miR-196a-5p and miR-222 play essential roles in the oncogenesis of glioma cells by inhibiting apoptotic signaling (46, 47) (Figure 12). miR-196a-5p interacts with the 3′-UTR of forkhead box O1 (FOXO1) mRNA. FOXO1 is a transcription factor that is phosphorylated and activated by Akt. Reduced levels of FOXO1 mRNA and, therefore, FOXO1 protein production, result in suppressed expression of Bim, which binds to and prevents Bcl-2 from antagonizing Bax (47). Bcl-2 is an apoptosis suppressor that inhibits Bax activity to promote the release of cytochrome c from mitochondria (173). Indeed, miR-196a-5p-mediated reduction of FOXO1 prevents glioma cells from undergoing apoptosis (47). miR-222 interacts with the 3′-UTR of Bcl2 modifying factor (Bmf) mRNA and reduces its expression in glioma cells (46). BMF can induce apoptosis by repressing Bcl-2 (174). Indeed, miR-222-mediated reduction of BMF also prevents glioma cells from undergoing apoptosis (46).
Intriguingly, GAS5 inhibits both miR-196a-5p and miR-222 from interacting with FOXO1 and Bmf mRNAs, respectively, which suppresses cancer hallmark (6) ‘resisting cell death’ (46, 47). These studies thus indicate that GAS5 can function as a potent inducer of apoptosis by inhibiting the onco-miRNAs involved in anti-apoptotic signaling in glioma. This results in hallmark (6) ‘resisting cell death’. In contrast, miR-136-5p and miR-186 enhance apoptosis and thus function as tumor suppressors (39, 48). miR-136-5p interacts with the 3′-UTR of Bcl-2 mRNA and reduces expression through mRNA destabilization in glioma cells (48). miR-186 recognizes the 3′-UTR of X-linked inhibitor of apoptosis (XIAP) mRNA and reduces its expression through mRNA destabilization (39). XIAP is a member of the IAP family and inhibits apoptotic signaling by ubiquitination and subsequent degradation of caspase-3 protein (162). The expression levels of XIAP were increased in glioma cells, which leads to a reduction of apoptosis (175). Interestingly, CRNDE prevents both miR-136-5p and miR-186 from acting on Bcl-2 and XIAP mRNAs, respectively, resulting in resistance to cell death in glioma cells (39, 48). In this sense, CRNDE is an extensive suppressor of apoptosis. These studies indicate that hallmark (6) ‘resisting cell death’ can be mediated by HOTAIR, CRNDE, MEG5 and GAS5, which inhibit apoptosis through the PI3K/Akt/ signaling pathway (Figure 11).
lncRNAs function as upstream regulators of multiple cancer hallmarks; therefore, it is important to clarify the molecular mechanisms of aberrant lncRNA expression in glioma cells. Although these mechanisms have not been fully elucidated, a few recent studies indicate that epigenetic modifications of promoter regions are involved (176, 177). As described above, MEG3 functions as a tumor suppressor lncRNA, whose expression is reduced in glioma tissues (43, 178). Recently, Lee et al demonstrated that DNA methyltransferase 1 (DNMT1) causes hypermethylation of the MEG3 promoter region in glioma cells. Knockdown of DNMT1 reduced MEG3 promoter methylation levels and increased expression, resulting in reduced proliferation and increased apoptosis of glioma cells (176). Meanwhile, Pastori et al showed that Bromodomain Containing 4 (BRD4) can increase the expression levels of HOTAIR in glioblastoma (177). BRD4 is a member of the bromodomain and extraterminal (BET) family that recognizes acetylated histones, which is a key process in RNA polymerase-II-dependent transcription (179). The expression levels of BRD4 are elevated in glioblastoma cells (180) and ChIP assays show that BRD4 localizes to the promoter region of HOTAIR. Furthermore, inhibition of BRD4 by I-BET151, a specific inhibitor of BET family proteins, reduced the expression levels of HOTAIR, causing reduced proliferation and enhanced apoptosis of glioblastoma cells (177). These studies suggest that epigenetic modifications can control the expression of several lncRNAs and can play a role in glioma development. It will also be interesting to reveal the mechanisms controlling lncRNA expression levels in a post-transcriptional manner and their roles in glioma development. In addition, considering that lncRNAs, such as HOTAIR and CRNDE, can exert extensive effects on glioma oncogenesis by modulating multiple cancer hallmarks, these oncogenic lncRNAs might be possible therapeutic targets in the treatment of glioma. To establish clinically feasible strategies that negatively affect the actions of oncogenic lncRNAs, oligonucleotide therapeutics, such as antisense oligonucleotides and RNA interference need to be explored. In addition, administration of specific inhibitors, such as I-BET151, has potential and warrants further study. .
Overall, it is plausible that glioma development proceeds through excessive activation of multiple signaling pathways followed by induction of the cancer hallmarks (1) ‘sustaining proliferative signaling’, (4) ‘activating invasion and metastasis’, and (6) ‘resisting cell death’. Emerging evidence suggests that lncRNAs can positively or negatively modulate these cancer hallmarks by acting as upstream regulators (Table 5). It is possible that novel findings regarding lncRNAs and the modulation of cancer hallmarks (2) ‘evading growth suppressors’, (5) ‘inducing angiogenesis’ and (10) ‘tumor-promoting inflammation’, which are not discussed in this section, may be reported in future studies; however, it is readily apparent that lncRNAs/miRNAs are related to cancer hallmarks (see Table 5) and play essential roles in glioma development.
| Cancer development process | lncRNAs | Target gene | Downstream signaling | Cancer phenotype | |
| Onco-lncRNAs | Suppressors | Onco-lncRNAs | Suppressors | ||
| Cancer initiation (hallmarks 1, 2, 6 and 10) | CRNDE | miR-136-5p | Bcl-2 | Proliferation↑, Migration↑, Invasion↑, Apoptosis↓ | |
| CRNDE | miR-384 | PIWIL4 | Proliferation↑, Migration↑, Invasion↑, Apoptosis↓ | ||
| CRNDE | miR-186 | XIAP and PAK7 | Proliferation↑, Migration↑, Invasion↑, Apoptosis↓ | ||
| HOTAIR | miR-326 | FGF | Proliferation↑, Migration↑, Invasion↑, Apoptosis↓ | ||
| HOTAIR | miR-125a-5p | mTOR | Proliferation↑, Migration↑, Apoptosis↓ | ||
| Cancer progression (hallmarks 3, 4, 5 and 6) | MEG3 | miR-93 | PTEN and PHLPP2 | Proliferation↓, Tumor growth in vivo↓, Apoptosis↑ | |
| MEG3 | miR-19a | PTEN | Proliferation↓, Migration↓, Invasion↓, Apoptosis↑ | ||
| GAS5 | miR-18a-5p | Neogenin | Proliferation↓, Migration↓, Invasion↓ | ||
| GAS5 | miR-222 | BMF | Proliferation↓, Migration↓, Invasion↓, Apoptosis↑ | ||
| GAS5 | miR-196a-5p | FOXO1 | Proliferation↓, Migration↓, Invasion↓, Apoptosis↑ | ||
lncRNAs are now recognized as crucial players in CNS development and brain function. Pivotal roles of lncRNAs in neural cell differentiation and circuit formation have become apparent, as described in this review. lncRNAs regulate gene expression involved in these processes in temporal or spatial manners via epigenetic, transcriptional, post-transcriptional and translational modes of action. Furthermore, aberrant expression of multiple lncRNAs, leading to altered expression levels of target genes, is involved in the pathogenesis and pathophysiology of many neurological diseases.
Elucidating the modes of action of lncRNAs is useful to not only better understand of roles of lncRNAs in CNS neurogenesis, but also to enable the regulatory mechanisms of lncRNAs to be exploited for clinical applications, e.g. for the regeneration of damaged nervous tissue. Indeed, knockdown of the lncRNA BDNF-AS, which is involved in neuronal differentiation and synaptic plasticity through the epigenetic regulation of BDNF expression, attenuated hypoxia-induced neuronal cell death in a model mouse of cerebral infarction (181). Furthermore, expression profiles of lncRNAs in human induced pluripotent stem (iPS) cell-derived neurons have been analyzed (182), with the aim of identifying candidate lncRNAs responsible for regeneration of the human CNS. Moreover, disease-related lncRNAs may be useful as therapeutic targets or as biomarkers for diagnostic aims. For example, the lncRNAs responsible for aberrant Abeta synthesis, e.g. BACE1-AS and 51A, are up-regulated in AD brain neurons and it may, therefore, be possible to reverse the clinical stages of AD by suppressing expression of such lncRNAs. As a method to substantiate this aspiration, oligonucleotide-mediated natural antisense transcript-targeted regulation (NATRE) technology is of great interest. NATRE uses sense oligodeoxynucleotides designed from the lncRNA target sequence in an mRNA and, thereby, inhibits the target mRNA-lncRNA interaction in a specific manner (183). Although targeted drug delivery systems that can specifically deliver NATRE oligodeoxynucleotides to neurons in the AD brain are still to be developed, it is worth evaluating AD-related lncRNAs as therapeutic targets. In contrast, in diseases caused by multiple pathways, such as PD or glioma, a number of lncRNAs is intricately involved in each pathway. In such diseases, lncRNAs may be relevant as diagnostic marker(s) rather than as therapeutic targets.
Recent developments in the field of RNA biology have revealed that lncRNAs play pivotal roles in regulating gene expression and may, therefore, contribute to the pathogenesis of CNS diseases. Further detailed studies of the mechanisms of action of these critical lncRNAs will clarify their roles in the molecular pathogenesis of neurological disease and contribute to the development of efficient diagnostic markers.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. 16K08480 to H. Y. and No. 18K15009 to S. O. We thank Jeremy Allen, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Abbreviations: ncRNA: non-coding RNA, lncRNA: long non-coding RNA, mRNA: messenger RNA, miRNA: microRNA, CNS: central nervous system, ENCODE: Encyclopedia of DNA elements, tRNA: transfer RNA, rRNA: ribosomal RNA, snRNA: small nuclear, RISC: RNA-induced silencing complex, ceRNA: competing endogenous RNA, AD: Alzheimer’s disease, PD: Parkinson’s disease, NSC: neural stem cell, RSC: retinal stem cells, RPC: retinal progenitor cells, REST: RE1-silencing transcription factor, NRSE: neuron-restrictive silencer element, DGCR5: DiGeorge Critical Region 5, RMST: rhabdomyosarcoma 2 associated transcript, ESC: embryonic stem cell, GO: Gene Ontology, RT-PCR: reverse transcription-polymerase chain reaction, RT-qPCR: quantitative RT-PCR, Six3OS1: sine oculis-related homeobox 3 opposite strand 1, SVZ: subventricular zone, KD: knockdown, RIP: RNA immunoprecipitation, PTBP1: polypyrimidine tract-binding protein 1, Sox2: SRY-box 2, Pax6: paired box 6, TUBB3: class III beta-tubulin, TUNA: Tcl1 upstream neuron-associated lincRNA, shRNA: short hairpin RNA, siRNA: small interfering RNA, hnRNP: heterogeneous nuclear ribonucleoprotein, SP8: trans-acting transcription factor 8, NEUROG2: neurogenin 2, DLX: distal-less homeobox, ChIP: chromatin immunoprecipitation, GABA: gamma-aminobutyric acid, MeCP2: methyl-CpG binding protein 2, GAD: glutamic acid decarboxylases, Tug1: taurine up-regulated 1, EZH2: enhancer of zeste 2 polycomb repressive complex 2 subunit, PRC2: polycomb-repressive complex 2, FMR4: fragile X mental retardation 4, RNA-seq: RNA sequencing, FMR1: fragile X mental retardation 1, HOTAIR: HOX transcript antisense RNA, HEK: human embryonic kidney, LTP: long-term potentiation, LTD: long-term depression, BC1: brain cytoplasmic RNA 1, eIF: eukaryotic initiation factor, PABP: poly-A binding protein, PSD-95: post-synaptic density protein 95, FMRP: fragile-X retardation protein, mGluR: group I metabolic glutamate receptor, BDNF: brain-derived neurotrophic factor, BDNF-AS: BDNF antisense RNA, DG: dentate gyrus, APP: amyloid beta precursor protein, BACE1: beta-secretase 1, SORL1: sortilin related receptor 1, Abeta: amyloid beta, BACE1-AS: BACE1 antisense RNA, HuD: Hu antigen D, NAT: natural antisense transcript, lincRNA: large intergenic non-coding RNA, SERPINE1: serpin family E member 1, PAI-1: plasminogen activator inhibitor type-1, HAO2: hydroxyacid oxidase 2, EBF3: early B cell factor 3, PSEN1: presenilin 1, MAPT: microtubule associated protein tau, GABBR2: gamma-aminobutyric acid type B receptor subunit 2, MPTP-HCl: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-hydrogen chloride, MPP+: 1-methyl-4-phenyl- pyridinium ion, DAT: dopamine transporter, SNCA: synuclein alpha, LRRK2: leucine rich repeat kinase 2, PRKN: parkin RBR E3 ubiquitin protein ligase, PTEN: phosphatase and tensin homologue, PINK1: PTEN-induced putative kinase 1, DOPA: dihydrophenylalanine, DA: dopamine, BH3: BCL2 homology region 3, Bax: BCL2 associated X, ERM: ezrin/radixin/moesin, ERK: extracellular signal regulated protein kinase, MAPK: mitogen activated protein kinase, WT: wild type, CDH1: cadherin 1, TG: transgenic, SNP: single nucleotide polymorphism, MBD: methyl-CpG-binding domain, CREB1: cAMP responsive element binding protein 1, Sin3A: SIN3 transcription regulator family member A, NEAT1: nuclear paraspeckle assembly transcript 1, ROS: reactive oxygen species, CRNDE: colorectal neoplasia differentially expressed, MEG3: maternally expressed 3, GAS5: growth arrest specific 5, FGF1: fibroblast growth factor 1, IL6: interleukin 6, EphA2: EPH receptor A2, PI3K: phosphoinositide-3-kinase, mTOR: mechanistic target of rapamycin kinase, JAK: Jak family tyrosine kinases, STAT: signal transducer and activator of transcription, EGFR: epidermal growth factor receptor, 4EBP1: 4E binding protein 1, ECM: extracellular matrix, MMP: matrix metallopeptidase, UTR: untranslated region, PIWIL4: Piwi-like 4, PAK7: p21-activated kinase, Sch B: Schisandrin B, PIP3: phosphatidylinositol-3-phosphate, PHLPP2: PH domain and leucine rich repeat protein phosphatase 2, FOXO1: forkhead box O1, BMF: Bcl2 modifying factor, XIAP: X-linked inhibitor of apoptosis, DNMT1: DNA methyltransferase 1, BRD4: Bromodomain Containing 4, BET: bromodomain and extraterminal, iPS: induced pluripotent stem, NATRE: natural antisense transcript-targeted regulation