
Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.
 , Nikolaos Papanas 2, Andreas Melidonis 1
, Nikolaos Papanas 2, Andreas Melidonis 11 Diabetes Centre, First Department of Internal Medicine, Tzaneio General Hospital of Piraeus, Piraeus, Greece
2 Diabetes Centre, Second Department of Internal Medicine, Democritus University of Thrace, University Hospital of Alexandroupolis, Greece
Abstract
Numerous micro-organisms naturally reside in the human body assuming a symbiotic, or, at times, even a dysbiotic relationship with the host. These microbial populations are referred to as the human microbiota. Host microbial populations are an important mediator of gastro-intestinal mucosal permeability, bile acid metabolism, short-chain fatty acids synthesis, fermentation of dietary polysaccharides and FXR/TGR5 signaling. Variations in the composition and function of gut microbiota have been observed in type 2 diabetes mellitus, insulin resistance and obesity, as well as in inflammatory bowel diseases. The microbial imbalance induced by such pathological processes is described as dysbiosis. In this review, we describe the pathophysiological links between type 2 diabetes mellitus and gut microbiota, explore the effect of anti-diabetic drugs on gut microbiota and suggest possible therapeutic targets.
Keywords
- Anti-diabetic drugs
- Dysbiosis
- Gut microbiota
- Type 2 diabetes mellitus
- Review
The human gut microbiota (GM) consists of ~105 billion micro-organisms, further classified into 500 different bacterial species per individual of which most are anaerobic and form a symbiotic relationship with the host (1). Each individual presents with extensive microbial diversity for the first 3-5 years of his life, whereas the adult GM is relatively stable as far as its composition is concerned (1). Normal GM is mainly constituted by four major phyla, Bacteroidetes, Firmicutes, Actinobacteria and Proteobacteria (1). As the gastrointestinal tract extends from the esophagus to the rectum, the composition of the microbial population varies (1). The esophagus, duodenum and jejunum are mostly populated by members of the Streptococcus genus while Helicobacter genera are predominant in the stomach. The greatest diversity is noted in the large intestine where various genera co-exist. This ecosystem forms a symbiotic relationship with the host and has numerous functional aspects (2). GM is of uttermost importance for the fermentation of carbohydrates and indigestible oligosaccharides. This process can provide the human organism with essential metabolic precursors, specifically short chain fatty acids (SCFA) (3). Additionally, both lipid and protein metabolism are enhanced by the presence of GM (1-3).
The microbial populations of the gut protect the host through their many different properties (1,4). GM can affect drug pharmacokinetics in numerous ways by activating or neutralizing therapeutics, or even modifying half-life and drug-mucosal interactions (4) Shifts in microbiota population stability incrementally increase the risk of disease, a phenomenon best interpreted as a form of homeostatic dysregulation (5). Alterations in GM can be commonly induced by bacterial infections, use of antibiotics or proton pump inhibitors, lifestyle and long-term diet changes, surgical procedures or polypharmacy regimens (5-7). In turn, dysbiosis has been linked with the pathogenesis of obesity, insulin resistance and diabetes mellitus, chronic heart diseases, inflammatory bowel diseases, cancer, autism, liver diseases and human immunodeficiency virus infections (8).
The prevalence of type 2 diabetes mellitus (T2DM) has quadrupled in the last 3 decades, with the International Diabetes Federation (IDF) reporting that approximately 415 million adults were diagnosed with diabetes mellitus worldwide in 2015: of these, the majority have T2DM. The epidemic escalation seems to be multi-factorial and stemming from an entanglement of population aging, socio-economic development, poor dietary habits and unhealthy lifestyle (9).
Numerous studies have explored the link between human GM and T2DM (10-15). While findings seem to vary among the different studies and populations, the co-existence of a dysbiotic GM and T2DM has been established and several mechanisms have now been proposed in order to explain this relationship (16).
In this context, the aim of the present review is to describe the alteration that the human GM undergoes when metabolic dysregulation and T2DM occur, as well as the effects of anti-diabetic medication on GM.
One of many pathophysiological processes described as a substrate of dysbiosis in T2DM is termed “metabolic endotoxemia”(17). The “metabolic inflammation” hypothesis states that lipopolysaccharide (LPS), a highly antigenic, gram-negative bacterial cell wall component, can amplify the inflammatory response of the host, a reaction often observed in metabolic dysregulation. In this context, LPS should be discussed in combination with CD14, lymphocyte antigen 96 (MD-2) and toll-like receptor 4 (TLR4), as those four molecules form a pathway of great importance in metabolic processes (18). LPS binds to TLR4 using CD14 and MD-2 as accessory proteins to activate myeloid differentiation primary response 88 (MyD88) independent or dependent signaling (17, 18). Ultimately, all pathways converge downstream on NF-κB-mediated upregulation of pro-inflammatory cytokines such as tumor necrosis factor α, interleukin-1 (IL-1), IL-6, IL-8, platelet activating factor, tissue factor and more, giving rise to inflammation. Researchers have described LPS as a possible causative factor for insulin resistance, noting that high-fat diet regimes triggered a rise in LPS concentration that was followed by fasting glycemia, insulinemia and weight-gain, while similar effects have been observed in subjects undergoing continuous LPS infusion (19).
Furthermore, mucosal permeability appears to be affected in a research model whereby high-fat diet is used, mainly due to epithelial tight junction protein decrease (20). As such, gut mucosal integrity is prone to change when GM composition changes, allowing for endotoxemia to occur (20). Another pathway that is tightly associated with the aforementioned is the endocannabinoid (eCB) system. The eCB and LPS regulatory loops affect gut both mucosal integrity and the host’s adipose tissue, the latter being through lipogenesis dysregulation (16).
A molecule facilitating lipid gut absorption, intestinal alkaline phosphatase (IAP), normally prevents inflammation by countering LPS and is affected by the dietary habits and GM population of the host. High-fat diet causes a decrease in IAP and triggers, via one of many pathways, homeostatic dysregulation and subsequent inflammation (21).
GM can also modulate the energy produced by host metabolism, allowing for increased energy harvesting from food intake in obese subjects when compared with lean subjects. Three pathways can allow for such a phenomenon to take place, polysaccharides otherwise unable to be digested are fermented by altered microbial populations, more potent gut uptake of monosaccharides is performed or lipid metabolism is dysregulated by the GM of obese subjects (16, 22).
Of note, all aforementioned mechanisms and pathways intertwine in numerous ways. Hence, T2DM is closely connected with the GM and its alterations by manifold mechanisms (16, 22). This connection may enable novel therapeutic approaches, which will diverge from the well-established path of glycemic control and will rather target major underlying pathologies, notably obesity and insulin resistance.
Metformin is the most widely used first-line therapy for T2DM and a member of the biguanides. Metformin’s mechanisms of action have not been fully elucidated, but they are mainly exerted in the liver, skeletal muscle and gut (23). Reduced hepatic gluconeogenesis and glycolysis, increased peripheral glucose uptake, as well as increased glycogen synthesis are the main actions of metformin: these are mediated via adenosine monophosphate-activated protein kinase (AMPK)-dependent and -independent pathways (24). Numerous actions are performed by metformin in the gastrointestinal tract, namely GM modification, increased glucose uptake by enterocytes, increased lactate production and GLP-1 levels, as well as enrichment of bile-acid pool (25).
Both in rodent and in human studies, metformin alters the GM composition in correlation with diet, leading to a decrease in diversity of the microbial population (26–28). Studies have consistently shown an increase in Akkermansia muciniphila, a bacterium inversely associated with obesity, T2DM, cardiovascular disease and inflammation (28, 29).
Moreover, Forslund et al. analyzed data from 784 human gut metagenomes, attempting to decode the treatment signature of metformin on GM in T2DM patients (30). SCFA-producing bacteria were found to facilitate the pharmacological actions of metformin by amplifying butyrate and propionate production. Interestingly, decreased metformin tolerance due to intestinal side effects could be related to an increase of Escherichia species (30).
In a double-blind study in treatment-naive subjects with T2DM, Wu et al. randomized participants to placebo or metformin for 4 months (31). Whole-genome shotgun sequencing of fecal samples was carried out. Both groups underwent diet modification, resulting in uniformly decreased BMI among all study participants (31). In the placebo group, only a single bacterial strain was altered, despite the reduction of BMI. In the metformin group, 81 strains differed following 2 months of treatment and 86 after the complete 4-month period. Most modified strains were characterized as either γ-proteobacteria or Firmicutes (31). Importantly, subgroup of patients from the placebo arm of the study were switched to metformin and they showed similar results after treatment. Again in this study, there was an increase of A. muciniphila in the metformin group, as suggested by other researchers (26, 28, 29).
In summary, metformin regulates numerous metabolic pathways via interaction with the GM and confounds, in part, the dysbiosis induced by T2DM. Shifts in microbial population proliferated by metformin administration are tightly connected to both efficacy and tolerance of this agent. Nevertheless, more data is needed to clarify the direct and indirect actions of metformin and to further exploit how these could turn into therapeutic advantages.
Alpha-glucosidase inhibitors (α-GIs) are a class of anti-diabetic drugs acting in the epithelium of the small intestine, delaying the digestion of carbohydrates by reversible and competitive inhibition of intestinal alpha-glucosidases, thereby diminishing the rate of glucose absorption and attenuating postprandial hyperglycemia (32). They are bacterial products, mostly employed for competitive survival in a microbial population, due to their ability to affect an important source of nutrients, such as carbohydrates (33). This class comprises acarbose, miglitol and voglibose.
Since α-GIs are metabolized exclusively in the intestinal tract, it would be expected that they interact with the host GM. A double-blind, controlled crossover trial by Zhang et al. included patients with prediabetes and showed that acarbose treatment altered the microbial population in comparison with placebo, suggesting that acarbose-induced GM alterations may be related to this drug’s efficacy (34). Specifically, acarbose alters the GM in a manner dependent of the baseline host GM composition. Acarbose treatment results in increased concentration of Lactobacilli and Bifidobacteria, combined with a decrease of Bacteroides (35–37).
In a study on rodents, miglitol showed signs of improving insulin resistance and preventing development of non-alcoholic steatohepatitis (NASH) induced by a high-fat, high-sucrose diet (HFHSD), along with shortening intestinal transit time and reducing inflammation (38). Such results could be achieved by modulating the metabolic endotoxemia and LPS-induced low grade inflammation attributed to dysbiosis caused by the HFHSD. However, this explanation is speculative and more research is required.
Similarly to miglitol, voglibose was studied in mice (39). Do et al. sought to determine its effect on metabolic dysregulation (39). Voglibose treatment succeeded in preventing obesity, dyslipidaemia and impaired glucose metabolism. Potential underlying mechanisms included altered GM population, altered bile acid and hepatic lipid metabolism and increased GLP-1 levels, which reduced appetite (39). GM analysis revealed a reduced Firmicutes/Bacteroidetes ratio in the voglibose group. This finding that underlines the beneficial effects of voglibose in a dysbiotic microbial population, given that metabolic dysregulation and high-fat diet increase this ratio(40, 41).
Glucagon-like peptide-1 (GLP-1) is an incretin hormone that delays gastric motility, suppresses appetite, stimulates glucose-dependent insulin secretion and decreases glucagon secretion (42). It has a very short half-life, because it is rapidly cleaved and deactivated by the enzyme dipeptidyl peptidase-4 (DPP-4). There are now DPP-4 inhibitors (DPP-4is: alogliptin, linagliptin, saxagliptin, sitagliptin and vildagliptin), which inhibit both GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) degradation, thereby increasing their plasma concentrations. There also pharmacological GLP-1 receptor agonists (GLP-1 RAs: exenatide, liraglutide, lixisenatide, exenatide long-acting release, dulaglutide, albiglutide and semaglutide). DPP-4is and GLP-1 RAs represent the latest additions to the therapeutic armamentarium acting via the incretin pathway (42).
An investigation into the effect of sitagliptin on gut microbiota of high fat/high carbohydrate fed rodents (43) showed that the phyla Firmicutes and Tenericutes increased and Bacteroidetes decreased, despite the diet-induced glucose intolerance. Sitagliptin partially reversed the experimentally induced dysbiosis and altered the population of SCFA-producing bacteria (43).
In a similar experiment, vildagliptin treatment was associated with decreased Firmicutes/Bacteroidetes ratio, along with increased Bacteroidetes and decreased Firmicutes (44). SCFA-producing bacteria were also more abundant in the vildagliptin group (44). In a recent study on DPP-4 inhibitors, vildagliptin was linked with decreased Oscillibacter spp. along with increased Lactobacilli spp. and propionate production (45). These findings were attributed to a direct effect of DPP-4 inhibition in the gastrointestinal tract, a suggestion further supported by the findings of Olivares et al. proposing the existence of significant intrinsic DPP-4-like activity in the gut microbial populations (46).
In contrast to the other DPP-4is, saxagliptin did not appear to alter GM diversity in comparison with liraglutide or placebo in an 8-week trial in mice (47). Given that saxaglitpin did not exhibit significant results and that data on linagliptin and alogliptin are limited, there is a clear need for additional research with DPP-4is in this field.
GLP-1 RAs are divided into short- (exenatide and lixisenatide) and long-acting (liraglutide, exenatide long-acting release, dulaglutide, albiglutide and semaglutide) compounds (48). T2DM-induced dysbiosis has been suggested to attenuate autonomic intestinal neuropathy along with GLP-1 resistance, suggesting a potential reciprocal relationship between GLP-1 RAs and changes in GM expression (49).
Short-acting compounds are expected to affect gut microflora composition by delaying gastric emptying. However, there are no studies to date to confirm or refute this proposition.
Similarly, there may be a favorable effect of long-acting GLP-1 RAs, but again there are no studies. The role of liraglutide in intestinal microflora modulation is currently under investigation.
Wang et al. randomized mice to saxagliptin or liraglutide (47). The latter appeared to alter the relative concentrations of bacterial phylotypes affecting weight regulation. Indeed, phylotypes of the families Erysipelotrichaceae, Lachnospiraceae, Lactobacillaceae and Porphyromonadaceae were increased while others of Clostridiales and Bacteroidales were diminished (47). This is not surprising, given the effect of liraglutide on weight reduction (47).
Another head-to-head study compared liraglutide with metformin (50). T2DM subjects on metformin were randomized to 2 groups, the first being switched to liraglutide and the second continuing their initial treatment (50). Microbial analysis was performed via stool sample 16S rRNA sequencing. GM profiles differed between the 2 groups. In the liraglutide group, bacteria of the Akkermansia genus were increased and those of the Sutterela genus were reduced in comparison with to the metformin group (50). Importantly, this GM pattern seen in the liraglutide group is the exact opposite of that which is characteristic of the metabolic syndrome (51). This finding underlines the beneficial effects of liraglutide on GM and metabolism (50).
Liraglutide-related intestinal microflora remodeling has been confirmed in an experimental study by Zhang et al. (52). After liraglutide treatment, diabetic mice exhibited an enrichment of SCFA-producing bacteria, a reduction of Firmicutes/Bacteroidetes ratio and an increase of Tenericutes, indicating successful reversal of dysbiosis (52). These results are promising and call for additional experience.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors (dapagliflozin, empagliflozin, canagliflozin, tofogliflozin, sotagliflozin, luseogliflozin, ertugliflozin, ipragliflozin and others being developed) are a novel class of anti-diabetic substances (53, 54). They inhibit glucose reabsorption by inhibiting the sodium-glucose cotransporter 2 in the renal proximal tubule. Thus, they induce glycosuria, thereby reducing serum glucose (53, 54).
A study in diabetic mice demonstrated that the addition of 60mg/kg dapagliflozin on a regular diet improved vascular function and GM at 8 weeks in comparison to regular diet only (55). GM composition of subjects was assessed by use of 16s rRNA sequencing. Diabetic mice receiving dapagliflozin exhibited a favorable reduction of the Firmicutes/Bacteroidetes ratio, as compared to the diet-only group (55).
Du et al. examined the effect of dual inhibition (inhibition of intestinal SGLT-1, leading to reduced intestinal absorption, in addition to SGLT-2 inhibition) in diabetic mice (56). Dual inhibition was more efficacious in promoting glycosuria and in improving glycemic control. However, it had no effect on GM diversity. The latter could perhaps be attributed, at least partly, to the short treatment period (6 days), which may have been insufficient for GM remodeling (56).
In rodents with chronic kidney disease, a condition often exacerbated by the uremic toxins produced by the GM, Mishima et al. examined the effect of 2-week canagliflozin regimen (10mg/kg) (57). Treatment succeeded in promoting intestinal carbohydrate fermentation, reducing uremic toxins (p-cresyl sulfate and indoxyl sulfate) and improving GM synthesis.
A summary of the interraction between gut microbiome and different antidiabetic substances is provided in Table 1.
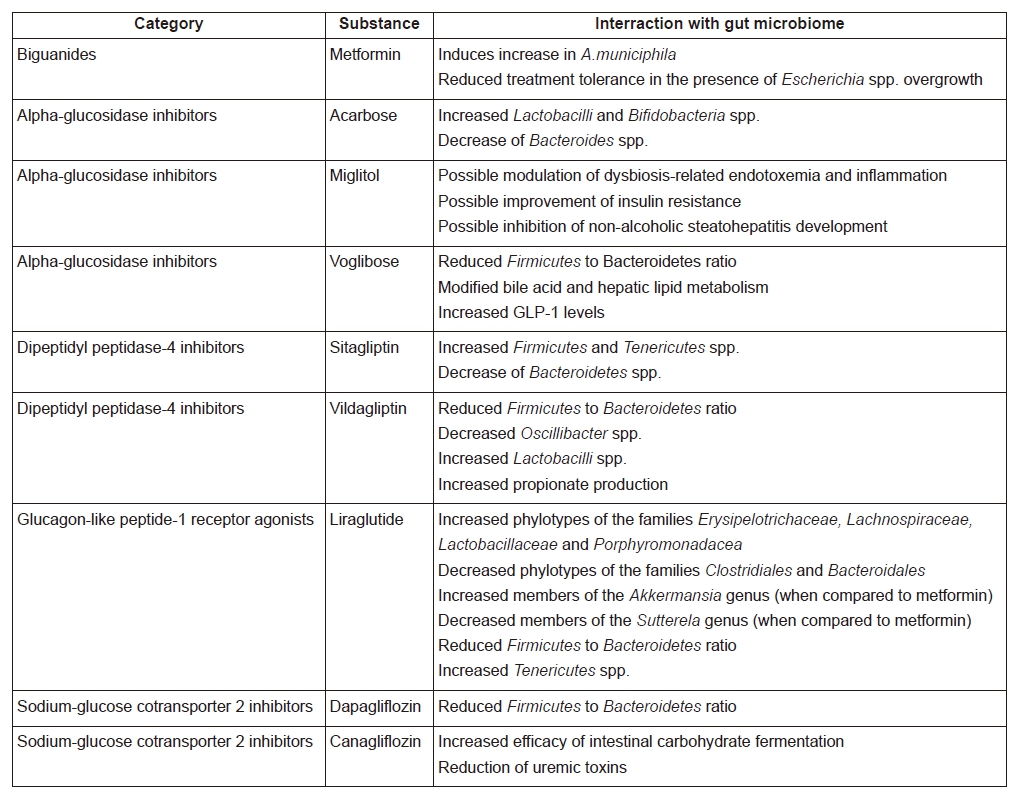
There is now ample evidence on the complex relationship between intestinal microbial destabilization, T2DM and reversal of dysbiosis via anti-diabetic treatment. The main effect produced by the majority of anti-diabetic agents was a decrease in the Firmicutes/Bacteroidetes ratio and an increase in efficient carbohydrate fermentation (28, 29, 34, 44). These treatment-induced changes indicate a reversal of dysbiosis. The extent of the clinical impact of the aforementioned treatment-induced shifts in GM populations has not been clarified, yet we can expect ameliorated metabolic functionality in those subjects that have underwent marked GM remodeling. Further study is required in order to compare carbohydrate and oligosaccharide fermentation efficacy, insulin sensitivity, immune capacity and other functions mediated directly or indirectly by the GM in subjects receiving treatment when compared to controls, in order to accurately discern the differences related to the various treatment regimens. Moreover, the information on the effects of new agents (SGLT-2is, DPP-4is, GLP-1RAs) is very limited, and relevant studies are eagerly awaited.
We also need additional data on the precise mechanisms by which T2DM interacts with GM. Such new research will offer new insights into pathogenesis of aberrant GM in T2DM and offer new therapeutic vistas. Moreover, new knowledge may enable the use of intestinal bacteria patterns as biomarkers of response to treatment. Indeed, the field of GM in relation to disease is still relatively unexplored, and understanding the full potential of the intricate and complex interactions remains a major challenge.
There is no funding or conflict of interest. All authors equally contributed to this paper with conception and design of the study, literature review and analysis, drafting and critical revision and editing, and final approval of the final version.