




Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Signal Transduction in Cancer and Stem Cells Laboratory, Division of Cancer Biology and Inflammatory Disorder, Council of Scientific and Industrial Research-Indian Institute of Chemical Biology (CSIR-IICB), 4 Raja S.C. Mullick Road Kolkata- 700032 and CN-06, Sector-V, Salt Lake, Kolkata-700091, India
Abstract
In cerebral tissues, due to continuous and high metabolic demand, energy is produced exclusively by mitochondrial oxidative phosphorylation (OXPHOS). Obstruction of blood flow leads to cerebral ischemia, hypoxia and decreased cellular ATP production. The reactive oxygen species (ROS) generated as by-product of OXPHOS alter many intracellular signaling pathways and result in damaged cellular components. Under such hypoxic conditions, a key factor known as hypoxia inducible factor 1 (HIF1) is stabilized and activated and such activation induces expression of a defined set of target genes which are required for cell survival and angiogenesis. Reperfusion that follows such ischemia alters signaling pathways which are involved in cellular fate. Here, we will review the role of ROS, HIF-1 alpha and other signaling network in mitochondrial dysfunction and cell fate determination in ischemia-reperfusion models in the brain. We will also address both current and future therapeutic strategies for clinical significance that are being developed for treatment of cerebral ischemia.
Keywords
- Cerebral ischemia
- Stroke
- ROS
- Hypoxia
- Mitochondria
- Electron transport chain
- Signaling
- Therapy
- Reperfusion
- Review
Brain is the most important organ in human body and also the most susceptible organ for ischemic injury. In cerebral ischemia blood flow to the brain is obstructed which causes damage or death of cerebral tissues. The overall damage caused by ischemic preconditioning can be recovered by reperfusion therapy within a time not beyond the critical level. Several molecular regulatory pathways take regulatory roles in fate determination of brain tissues after blood flow obstruction in ischemic preconditioning for survival of the cells in the ischemic penumbra.
Mitochondrial apoptotic and necrotic pathways are the main causes for brain tissue damage in cerebral ischemia (1-2). Ischemic stroke results in Na+/K+ pump dysfunction as well as cellular calcium homeostasis breakdown by inducing oxidative stress and mitochondrial swelling (3, 4). Immediately after stroke oxidative stress in cerebral tissue stimulates mitochondrial ROS generation and cytosolic NADPH production that causes damage to cellular macromolecules such as lipids, proteins and DNA (5). These excessive intracellular ROS levels show their patho-physiological effects in case of several neurodegenerative diseases as well (6-8). Nitrosative stress in ischemic stroke also triggers protein misfolding and aggregation of mitochondrial components that leads to several neurodegenerative diseases, aging and even cancer.
The hypoxia inducible factors (HIFs) are oxygen regulated transcription factors that play important roles in hypoxia sensing and adaptation (9). They act as critical effectors in response to depleted oxygen levels and a plethora of genes are under their control. HIF-1α expression along with mitochondrial ROS generation are boosted in response to ischemic oxidative stress (10). In hypoxia, HIF-1α is stabilized by accumulated high ROS levels generated from complex III in mitochondria. The mechanism behind this is the oxidative inactivation of non-heme iron in the catalytic site of Prolyl hydroxylase (PHD) enzyme (11). HIF-1α accumulation in neuronal cells is protective whereas endothelial HIF-1α induction is implicated in blood brain barrier (BBB) disruption (12). It is important to keep in mind that hypoxia-driven ROS activates NF-kB (13) and other transcription factors such as NRF2 (14) which play a vital role in regulating transcription of proteins involved in antioxidant defense. ROS can also activate the MAPK pathway leading to the increased expression of VEGF (15). Increased expressions of VEGF and its receptors VEGF-R1 and R2 are also due to the activation of HIF-1α by ROS and have fundamental roles in increased cell survival (16-17). ROS also activates other intracellular signaling pathways that include MAPK, NF-kB and upstream of MMPs. Herein, the authors are majorly discussed the pathophysiological features of cerebral ischemia and reviews some of the major human trials under the current treatment scenario.
Cerebral ischemia occurs when there is insufficient blood flow to the brain (18) so that cerebral hypoxia develops through limited oxygen supply which leads to death of the cerebral tissues. This sudden destruction of brain tissue is called as cerebral infarction or ischemic stroke in the brain (19). Transient cerebral ischemic attack in young adults is commonly considered as a benign event. However, severity increases significantly with aging and is the second leading cause of death (20). According to its origin, there are two major types of cerebral ischemic stroke: focal and global (fig1) (21). Cerebral stroke can be further divided into thrombotic, embolic and hypo-perfusion. Thrombotic and embolic are generally focal in nature whereas hypo-perfusion affects the brain globally.
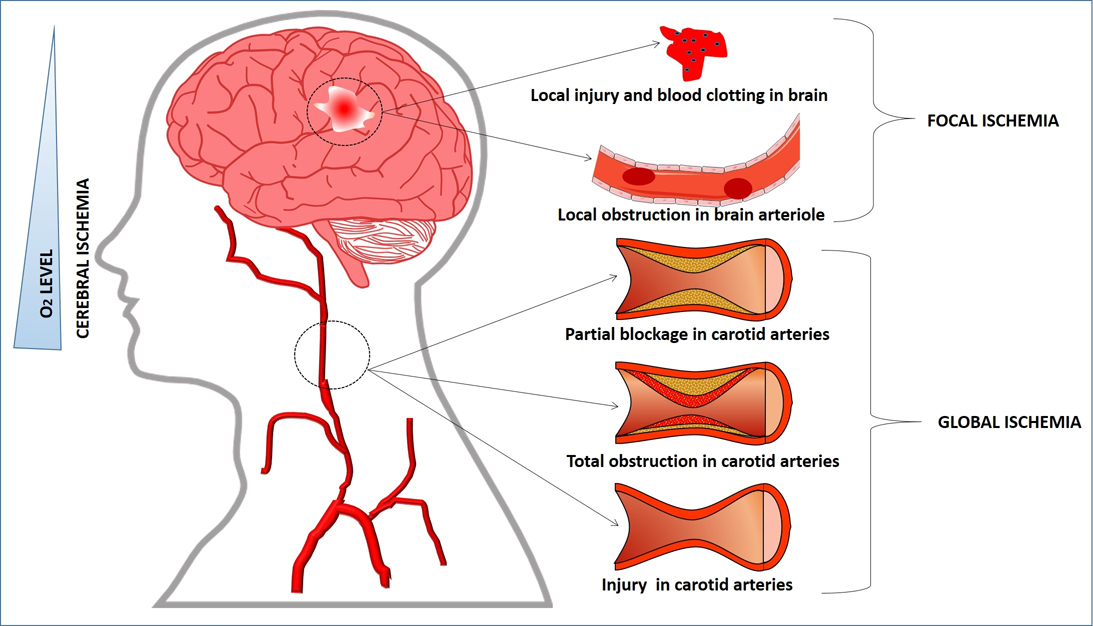 Figure 1
Figure 1Pictorial representation of the type of cerebral ischemia.
In focal type, blood flow is restricted to the specific region of the brain and the cell death limited to that particular area. In this type, there is an “ischemic core” and “ischemic penumbra” with severe cerebral blood flow (CBF) reduction in the vascular bed. When a blood clot has formed in a particular region of the brain, it decreases blood flow as well as increases the risk of cell death in that specific area. In focal cerebral ischemic stroke, blood flow stopped in the central core but some blood flows in that area via collateral circulation i.e., a gradient of blood flow occurs from inner core to the ischemic region. Reversible injury to irreversible cellular infarction after focal ischemia largely depends on ischemic severity and duration (22). Focal ischemia reduces CBF in a specific vascular territory and is clinically known as “ischemic stroke”.
In global cerebral ischemic stroke blood flow is obstructed due to blockage of carotid arteries in the head and neck region. Severity of brain damage depends on the time required for restoration of blood flow after blockage. If circulation is restored within a short period of time damage is not severe otherwise brain damage can be permanent and fatal. In complete global ischemia, blood flow has stopped completely whereas in case of incomplete type, blood flow is reduced so unable to maintain metabolism and other functions of the brain. Global stroke leads to a cascade of physiological processes culminating with the development of seizures. After cerebral ischemia, excitatory neurotransmitters are also released and play an important role in neuronal ischemic injury. Extracellular glutamate level is elevated after cerebral stroke that leads to ischemic brain damage. Excitatory neurotransmitters are released which play an important role in neuronal ischemic injury (23). Excitatory N-methyl-D-aspartate (NMDA), a sub type of glutamate receptor antagonist and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), the ligand for a distinct non NMDA glutamate receptor subtype are over stimulated by glutamate activation due to extracellular glutamate accumulation in the vulnerable regions of brain especially hippocampus and neo-cortex by promoting Na+ influx and K+ efflux through glutamate receptor activated membrane channels. So, neuronal death can be delayed by blocking NMDA receptor after global cerebral stroke (24).
In cerebral stroke, mitochondrial ROS are generated via electron transfer to oxygen through change in mitochondrial membrane potential by which the potential energy of proton motive force (PMF) drives protons into the mitochondrial matrix for ATP synthesis by oxidative phosphorylation (25).
In cerebral stroke, mitochondrial redox potential decreases after oxidative stress that leads to generation of ROS from mitochondrial ETC at cytochrome III level (26-28). Cellular ROS, the by-products of respiratory chain reaction are hence accumulated. Under normal conditions, low level of ROS are generated by leaking electrons from the respiratory chain enzyme complexes I and III and reaction with oxygen molecules results in the formation of superoxide radicals (O2.-). These radicals are converted to form H2O2 spontaneously by the enzyme known as superoxide dismutase (SOD) at low pH (29-30). Moreover ETC proteins which are rich in iron-sulfur clusters and heme proteins form highly reactive hydroxyl radicals (OH?) from hydrogen peroxide by providing necessary cofactors (31). In the mitochondrial respiratory chain most of the oxygen is reduced to water by the cytochrome C oxidase through four steps of electron reduction. Only a small amount of oxygen is reduced partially in the midway of ETC to generate O2.- (32). Superoxide is mostly produced at the Q cycle in complex III. A small flux of ROS is generated by ETC under normal condition. But after cerebral ischemic stroke, mitochondrial ETC impairs due to oxidative stress and huge amount of ROS is produced which results in lipid peroxidation, protein oxidation and ultimately DNA damage (33-34).
Under oxidative stress after ischemic stroke, mitochondria produces several ROS as metabolic by-products including superoxide radicals (O2.-), hydrogen peroxide (H2O2) and hydroxyl radicals (OH?) (fig2). Superoxide dismutase detoxifies O2.- to H2O2 and ultimately it is converted to H2O by catalase or glutathione peroxidase. Highly reactive hydroxyl radicals (OH?) are also produced from H2O2 and O2.- radicals. Pro-oxidant enzymes such as xanthine oxidase (XO) and NADPH oxidase (NOX) are also responsible for generation of O2.- radicals (35). Furthermore, nitric oxide (NO) reacts with O2.- radicals to form highly reactive peroxynitrate (ONOO?) anion which also damages cellular DNA, proteins and lipids. Additionally, NO reacts with specific protein thiol groups to induce protein misfolding and mitochondrial fission-fusion process that leads to neurotoxicity in cerebral stroke (36) as NO irreversibly modifies several proteins and lipids (37-38).
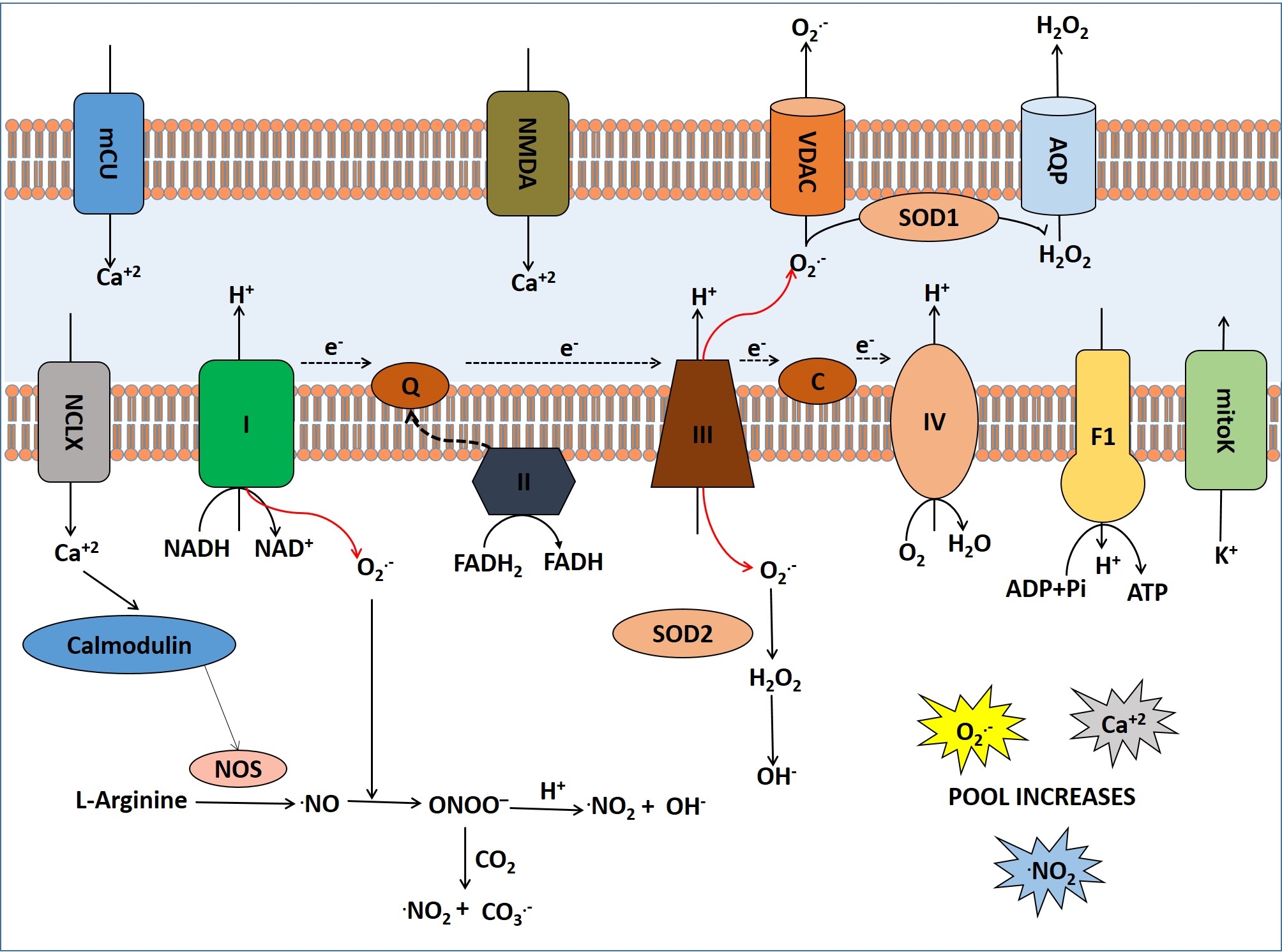 Figure 2
Figure 2Generation, transport and effects of ROS and NOS inside the mitochondria in cerebral ischemia.
Many biochemical and physiological changes occur as result of interruption of blood flow to the brain including loss of ATP generation, production of lactate anaerobically, compression of brain capillaries and reduced reflow of blood due to the swelling of astrocytes. Electrolyte balance of brain tissue cells gets disturbed with the fall in ATP level which results in potassium leakage from the intracellular compartment and influx of sodium and calcium along with the remarkable increase in cellular water content due to Na+/K+ ATPase and voltage gated calcium channel dysfunctions (39). The reduced cellular ATP in cells by glycolytic mechanism uses glucose and glycogen from the reservoir of the brain cells and accumulates proton and lactate leading to intracellular acidification (40-41).
In cerebral ischemia, mitochondrial ultrastructure is changed due to calcium overloading and free radical generation as well as decrease in adenine nucleotide translocase. Due to the loss of membrane integrity, the efficiency of calcium pump is reduced (41). The increased intracellular calcium activates membrane phospholipases and protein kinases that leads to accumulation of free fatty acids (FFA) and arachidonic acids (AA). FFAs, acyl Co-A and long chain acyl-carnitines are accumulated among which acyl Co-A is deleterious to mitochondrial enzyme systems. AA and FFAs can cause tissue irritation as well as platelet aggregation, vasospasm, clotting and oedema (42). Lactic acidosis injures neuronal cells by increasing lactate levels and decreasing the cellular pH. Lactic acid also degrades NADH as well as it inhibits the synthesis of ATP during post-ischemic events (43). All these conditions can cause irreversible tissue death which ultimately leads to permanent damage of brain tissue in the affected area.
In ischemia excitatory, amino acid particularly glutamate is released excessively to the extracellular compartment and neurotransmitter reuptake is decreased (44-45). As a result, Na+ influxes more to the neurons via monovalent ion channels which also decreases ionic gradient by consuming ATP. Blood flow reduction for a longer time releases more glutamate resulting in acute ischemic stroke of the patients that leads to excitotoxicity induced brain damage and the excessive glutamate concentration destroys central neurons. In the synapses, activation of ionotropic glutamate receptors is known as “excitotoxicity” (46). This excitotoxicity can cause neuronal death and disturbances in mitochondrial function because of increased production of free radicals. This also diminishes antioxidant defense and induces apoptotic cascade. Neuronal insult is also defined as cell death resulting from the toxic actions of excitatory amino acids. Intense exposure of glutamate in the cell leads to injury and death of neurons as well as occurrence of ionic imbalance. Mitochondrial membrane may become more permeable and cytochrome C is released resulting in activation of apoptotic cascade by increased cytosolic calcium concentration (47).
Ischemic hypoxic situation causes depolarization of membrane due to cellular K+ efflux, Na+ influx and activation of the voltage gated calcium channel. Increased intracellular calcium concentration leads to chronic response to hypoxic situation (48).
At lower concentrations, although ROS is beneficial for various normal cellular function, such as regulations of vascular tones, monitoring of oxygen tension, erythropoietin production (49), but increased ROS levels initiate an inherent risk to oxidative stress induced signaling and cell injury by irreversibly oxidizing macromolecules. Free radicals are generated at low levels under normal physiological conditions and play important roles in various signaling and metabolic pathways (50-51). But under stressed condition, large amount of free radicals are generated and they react with biomolecules viz proteins, lipids, carbohydrates and nucleic acids and destroy or alter their biological functions. These radicals aggravate numerous pathologies that ultimately leads to brain damage in different neurodegenerative diseases, aging and cancer.
So, level of ROS determines cellular fate. At low levels, ROS triggers autophagy with constant removal of damaged mitochondria and consequently cell survives. On the contrary high levels of ROS lead to cell death by promoting apoptotic cascade.
Apoptosis can be induced by both intrinsic and extrinsic signals through two major pathways viz. mitochondrial (intrinsic) or death receptor mediated (extrinsic). Intrinsic apoptotic cascade associates with the changes in the permeability of the outer mitochondrial membrane and ROS triggers this pathway by interacting with it. ROS regulated truncated form of Bid protein causes Bax/Bak oligomerization and creates mega-pore formation in the mitochondria. Next, apoptosome complex is formed in the cytosol by activating caspase 9 and then 3 to initiate apoptosis (52). Increased Fas receptor expression triggers mitochondrial permeability transition with the release of ROS which is the basic mechanism of apoptosis induction (53). Bcl2 blocks NO triggered apoptosis suggesting that mitochondria are the main target of apoptosis induction (54). Mitochondrial ROS are responsible for the full activation of caspase cascade. ROS, through disruption of mitochondrial membrane potential, generates mitochondrial pores and can contribute to cytochrome-C release (55). ROS can directly or indirectly increase the gating potential of the pores. In fact, H2O2 has the ability to induce apoptosis through initiation of caspase activation and mitochondrial cytochrome C release. However, at very high doses of H2O2 or other ROS, necrosis in brain tissue occurs (56).
Under normal physiological conditions, free radicals are produced continuously but counteracted by a number of antioxidants and endogenous enzyme systems viz. superoxide dismutase, catalase, vitamin C (ascorbic acid), glutathione, glutathione reductase, peroxidase and peroxiredoxin (57-59). As a result of oxidative stress, various macromolecules are oxidized viz.8-hydoxy-2-deoxyguanosine, an oxidized product of deoxyguanosine are used as biomarkers of in vivo oxidative DNA damage (60). Lipid peroxide generation causes alteration in cell membrane fluidity, increased membrane permeability and decreased membrane ATPase activity leads to severe cell injury after cerebral ischemic stroke. Lipid peroxidation has the most significant impact on plasma membrane permeability, structure and function and its damage leads to disruption of ionic gradients. The entry of Na+ and water leads to cell swelling. Malonaldehyde is formed as a result of decomposition of short lived and unstable lipid peroxides in cerebral stroke. 4-hydoxy-2-Noneneal, a derivative of w-6 family of polyunsaturated free fatty acids is also increased after stroke that causes cellular injury (61).
Development of hypoxia is one of the crucial events in cerebral ischemic stroke. In cerebral ischemia ROS and toxic metabolites are produced in the mitochondrial respiratory chain which are involved in the deregulation of cellular signaling pathways responsible for survival or death of neuronal cells. It is interesting to note that a full activation of HIF and NF-kB occurs by ROS in synergy with specific signaling pathway. Both hypoxia and ROS are involved in the maintenance of redox homeostasis and various cellular signaling pathways (fig3).
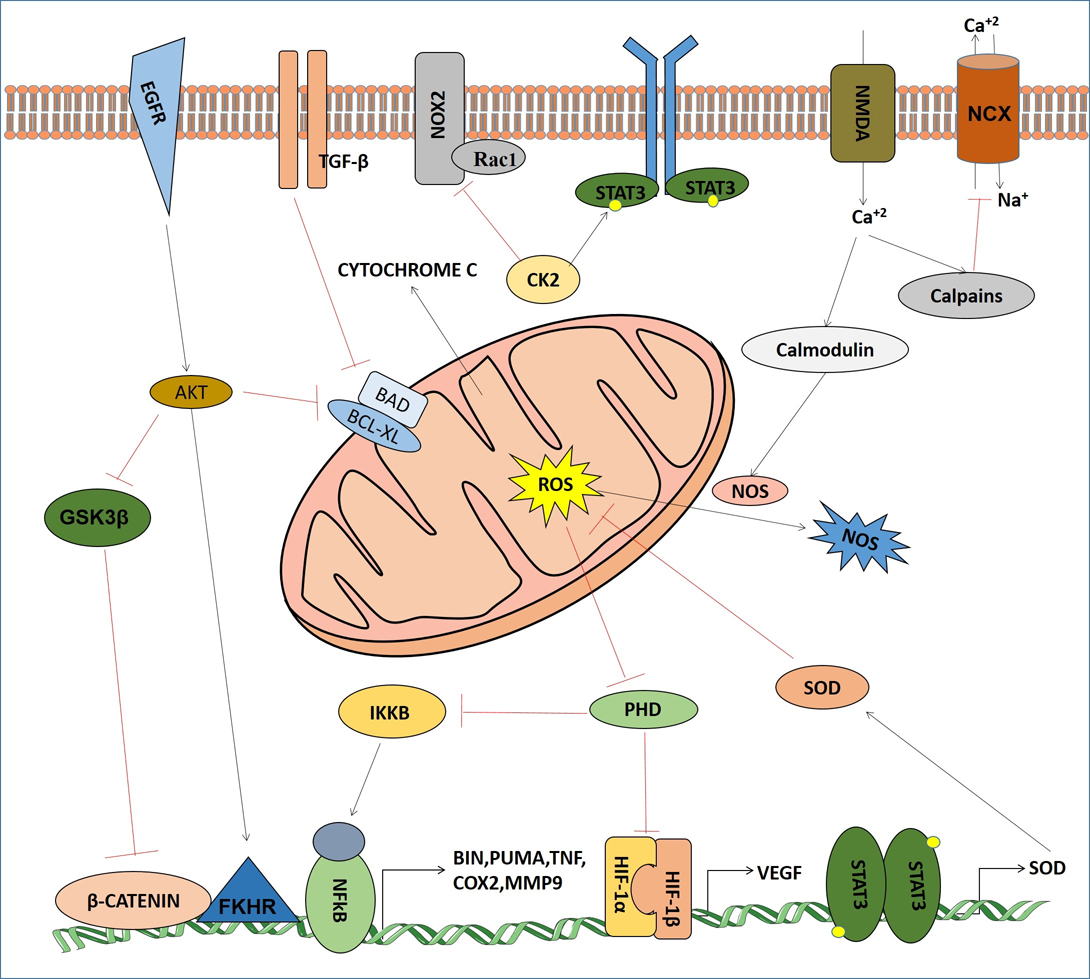 Figure 3
Figure 3Signaling crosstalks of mitochondrial ROS and NOS with major cell signaling components in cerebral ischemia.
ROS at low levels play important role in normal physiological signaling and metabolic pathways. But after cerebral ischemia, multiple detrimental processes such as overproduction of oxidants, inactivation of detoxification systems and consumption of antioxidants take place that results in mitochondrial dysfunction. Furthermore, accumulation of calcium and ROS due to excitotoxicity, disrupts natural anti-oxidative defense of the brain tissues. Various gene expressions are also deregulated by the participation of various signaling pathway based transcription factors. All of these events act synergistically to damage cellular proteins, lipids and DNA, which deteriorates cellular architecture and induces cell death by necrosis, apoptosis or both depending on the severity of mitochondrial dysfunction (3-4). They also contribute to numerous pathologies by alteration of the functions of the target molecules and damage the functions of organs.
HIF-1, a dimeric protein complex associates with angiogenesis in hypoxia. In ischemic condition HIF-1 plays dual role depending on the various functions of its target proteins in some specific types of brain cells. It has two subunits: HIF-1α and HIF-1β (62). The latter is insensitive to oxygen tension scenario and is constitutively expressed. HIF-1α is continuously synthesized and degraded in normal oxygen tension of the cells. Under hypoxic situation, there is an inhibition of HIF-1α degradation. Hence, it is rapidly accumulated and binds to hypoxia responsive elements (HRE), thereby activating its target genes (63). In the ischemic brain many of them have neuroprotective effect. In hypoxic region of ischemic brain HIF-1α is the principal gene involved in coordinating the shift from aerobic to anaerobic metabolism by inducing a variety of glycolytic enzymes and glucose transporters. HIF-1α regulates the expression of glucose transporter 3 (GLUT3) (64), the main transporter of glucose in neurons.
HIF-1α expression in cerebral ischemia is associated with cellular ROS level (9-10). However it has been shown by recent studies that increased ROS levels lead to reduced HIF-1α expression while other studies have shown that it has no effect. Under hypoxic condition mitochondrial complex III produces ROS that stabilizes HIF-1α. But in cytosol, ROS may be derived from NADPH oxidase, and it plays a major role on HIF-1α expression in normoxia than hypoxia. The complexity of relationships between ROS and HIF-1α in cerebral ischemia is primarily due to the diverse species of ROS generated by mitochondrial O2 metabolism and their properties, such as chemical nature, reactivity, specificity and half-life for their biological targets.
Casein kinase2 (CK2), a prominent oncogenic kinase plays a key role in ROS development in cerebral ischemia. It acts as a neuroprotectant via inhibition of NADPH oxidase (NOX) by Rec1 regulation (65). Observation from focal ischemia in rat showed that inhibition of CK2 in ischemic region causes PARP-1 accumulation resulting in cytochrome C and AIF release from mitochondria which activates a series of apoptotic events (66). Recent reports also show CK2 directly phosphorylates JAK as well as STAT3 for SOD2 transcription that detoxify ROS (67-68). Moreover, CK2 activates HIF-1α and NF-kB by phosphorylation for production of VEGF and other angiogenic proteins which are needed for angiogenesis and in escaping from hypoxia conditions.
Several reports describe the hyper activation of EGFR pathway in both ischemia and reperfusion conditions where transactivation of EGFR via phosphorylation leads to AKT activation (69). AKT plays most prominent role in neuroprotection by regulating forkhead transcription factor (FKHR) and glycogen synthase kinase 3 beta (GSK3β); BAD or caspase-9 or other apoptogenic components, for bypassing apoptosis (70-71). It is also reported that AKT, activated by ischemic ROS, provided hypothermic conditions to negatively regulate ROS production (72). Interestingly, AKT inhibits Raf so that ERK1/2 gets activated in ischemic condition but the scenario is reversed in reperfusion (73).
TGF-β is one of the cytokine mediated factor that highly express in cerebral ischemia. Mainly it regulates the expression of apoptotic (Bad) and anti-apoptotic (Bcl-2, Bcl-x1) proteins. It also crosstalks with MAPK pathway by transactivation of ERK1/2 for neuroprotection (74). Recent studies have shown that TGF-β1 save neurons from excitotoxicity by up-regulation of the type-1 plasminogen activator inhibitor (PAI-1) to nullify the t-PA-potentiated NMDA-induced neuronal death in ischemia (75).
NF-kB pathway plays an important role in cerebral ischemia (76). ROS activated NF-kB regulates Bim and Noxa (77), TNF (78), IL-1, IL-6 (79), iNOS, ICAM-1 and MMP9, cytosolic phospholipase A2, COX-2, and microsomal prostaglandin E synthase 1 (80). It can also directly regulates HIF-1α (81). All those downstream factors may cause apoptosis, cellular damage or even promote cell survival in a context dependent manner.
Another most important pathway found in cerebral ischemia is calcium signaling. Generally, Ca2+ signals are needed for cell-to-cell communication and survival, by playing role as secondary messenger, but Ca2+ overload in ischemic area leads to calpain mediated inhibition of sodium-calcium exchanger (NCX) (82) as well as caspase and Ca2+ dependent endonucleases activation which leads to apoptosis (83). It also helps in nitric oxide (NO) production via calmodulin-NOS pathway (84). Excessive NO can combine with other ROS to produce peroxynitrite (ONOO?), which causes damage to proteins, membrane lipids, and DNA (85).
Improved recognition of signs and symptoms in cerebral ischemic stroke has paralleled the development of multiple pharmacological strategies to reduce the mortality and morbidity. The reluctance to use any therapeutic strategy for the treatment of acute cerebral ischemia is because of the narrow therapeutic window of time. Neuroprotective strategies are intended to rescue the penumbral tissue cells and to extend the window of time for revascularization. However, under the present scenario, no neuroprotective agents have been shown to impact clinical outcomes in ischemic stroke. Heterogeneous class of drugs like antiplatelet agents (viz. aspirin, dipyridamole, ticlopidine and clopidogrel) (86) have been successfully used for more than 2 decades for secondary stroke prevention (fig4).
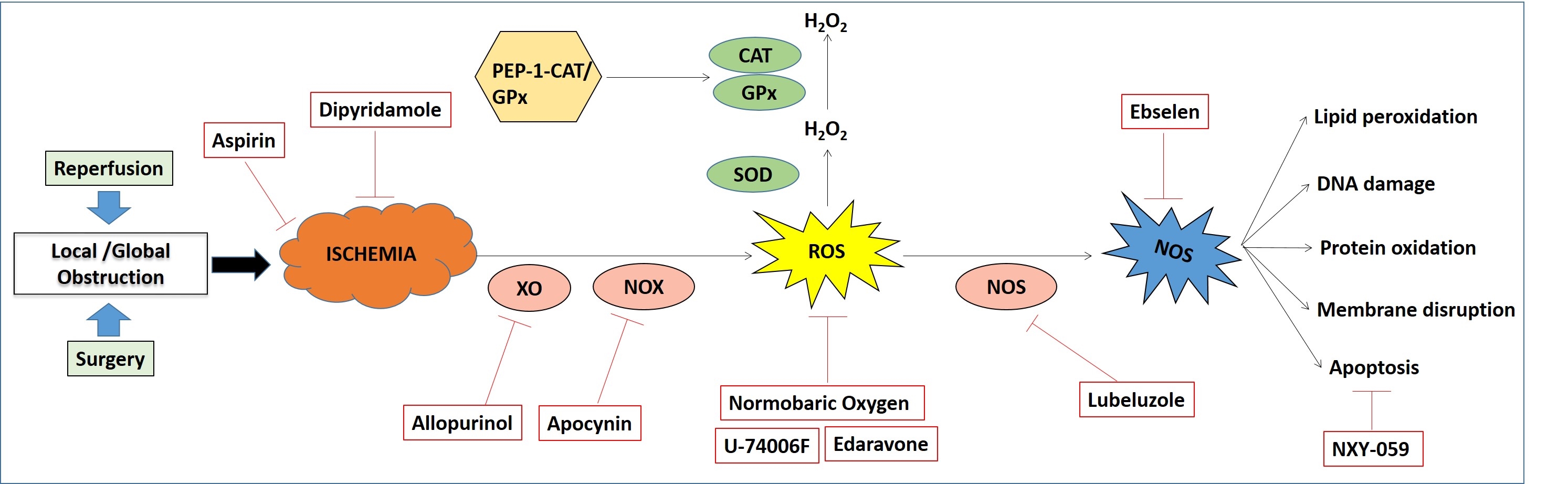 Figure 4
Figure 4Major therapeutic approaches for cerebral ischemia and their mode of actions.
In normal brain tissue, ROS are constantly generated during physiological procedures. These are however adjusted by endogenous defense mechanism carried by antioxidants. After Ischemic damage, free radical generation is incredibly expanded and causes redox disequilibrium in the characteristic endogenous antioxidant system. In this scenario detoxifications are inactivated and oxidants are overproduced. An expansion in ROS levels after ischemia brings about obvious oxidative anxiety and results in neuronal damage making free radicals as substantial helpful target, and much research has concentrated on evaluating the restorative impacts of antioxidants. There are three primary methodologies by which antioxidants work: (i) hindrance of free radical generation; (ii) scavenging of generated free radicals and (iii) escalating degradation of free radicals. Biological systems can either concentrate on the upregulation of endogenous antioxidants or on the conveyance of exogenous antioxidants.63
In this approach, the main source of ROS generation in diseased condition is targeted by some inhibitors of specific enzymes causing ROS generation. One fundamental source of ROS generation in ischemia reperfusion damage is the NADPH oxidases (NOXs). Hindrance of NADPH oxidase complexes with the pharmacological inhibitor apocynin (87) preceding reperfusion has shown reduction in ischemia in rodent models of stroke featuring the contributory part of NOX in brain damage.
Xanthine Oxidase (XO) is another protein that is associated with redox signaling pathways and is a vital source of ROS in the process of ischemic damage. Hindrance of XO is a potential restorative approach for the treatment of ischemia. Allopurinol is a normally utilized XO inhibitor that reduces levels of uric acid, as well as decreases the level of superoxide anion. Introductory trials with this medication are promising. Improvement in vascular and beneficial effects on inflammatory indices compared to placebo can be noticed in the patients treated with allopurinol (88).
Although pre-clinical to clinical trials has largely been disappointing, some compounds are capable of scavenging free radicals generated in ischemic stroke. Tirilazadmesylate (U-74006F) (89) is one of these compounds. It is an inhibitor of lipid peroxidation which has reported to reduce infarction size in rodents following transient focal ischemia. In the clinical trial of Tirilazad, when administered in 660 patients prior to focal ischemia showed maximum efficiency, whereas a decreasing efficiency can be observed with administration time from ischemic onset to thereafter (90). Another promising drug was NXY-059 which failed to show clinical efficacy in a clinical trial on 4000 patients (91). It is evident from different reports that NXY-pre059 has neuroprotective action, which causes neurological recovery in different stroke models in both rodents and non-human primates.
Edaravone (Radicava) is another free radical scavenger which is widely used by clinicians of Japan for the treatment of infarctions. The effectiveness of edaravone is still unclear, however it is known that it can scavenge peroxyl, hydroxyl and superoxide radicals (92).
Increasing levels of the antioxidant superoxide dismutase (SOD) is associated with ROS in lesion progression. Strategies are being aimed at reducing oxidative stress by targeting SOD. The overall efficacy of SOD is induced by its catalytic activity in the conversion of O2.- to less reactive H2O2. It is evident that catalase (CAT) and glutathione peroxidase (GPx) help to eliminate the by-product of H2O2. In vitro efficacy of CAT against ischemic injury was discerned when the overexpression of CAT by adenoviral vectors and transduction with PEP-1-CAT fusion protein demonstrated its neuroprotection ability (93).
Ebselen is an inhibitor of glutathione peroxidase-like activity which reacts with ONOO? and subsequently scavenges it (94). Another novel antioxidant approach is the inhalation of gases after or during ischemia. The use of inhaled hydrogen gas to selectively reduce the hydroxyl radical in a transient model of ischemia elicits beneficial effect. Other than this, normobaric oxygen (NBO) has been demonstrated in several studies to reduce infarct volume and neurological deficits in rodent models of focal ischemia. Moreover, combination therapy with NBO and ethanol was shown to have neuroprotective effects after ischemic injury in rodents (95).
Another approach to treat ischemic injury is the induction of a natural cell protection molecule NO, which is a vasoactive molecule produced by either endothelial NO synthase (eNOS), inducible NO synthase (iNOS) or neuronal NO synthase (nNOS). It is a short-lived, highly reactive intermediate that is receiving attention for its contribution to the cerebrovascular circulation during both physiological and pathological states. From a recent study it was evident that inhalation of NO could significantly reduce ischemic brain damage and improve neurological outcome in rodent stroke models. There are several advantages of gaseous treatment since it can rapidly penetrate biomembranes and diffuse into the cytosol, mitochondria and nucleus.
Lubeluzole causes reduction in NO levels and subsequent ONOO? production in hypoxic cells by inhibiting glutamate-mediated nitric oxide synthase pathway (96). A large clinical study with more than 1700 patients was initiated, but unfortunately no noticeable variation was observed between lubeluzole or placebo groups. Therefore, finding a prominent therapeutic modality is still now in hype (97).
Reperfusion injury occurs when blood supply returns to the tissue after lack of oxygen during ischemia. Scarcity of oxygen and nutrient supply from the obstruction of blood during ischemia creates a condition in which the restoration of circulation results in inflammation and oxidative damage with the induction of oxidative stress. Though reperfusion is essential to protect brain tissue, it also leads to secondary injury from influx of neutrophils. This often leads to the development of seizures. Cerebral reperfusion syndrome presents as a triad of ipsilateral headache, contralateral neurological deficits, intracerebral hemorrhage (ICH) and seizure. The symptoms arise can be from immediately after restoration of blood flow to up to one month since restoration. Acute stroke patients who receive reperfusion therapy with intravenous tissue plasminogen activator and intra-arterial therapy are at greater risk of developing seizures than patients who do not receive acute therapies (98-99)
In conclusion, this review illustrates our current knowledge of cerebral ischemic stroke, its cause, involvement of multiple signalling networks in connection with mitochondrial dysfunction, therapeutic strategies including clinical trials and its pros and cons. In fact, HIF1α, NF-kB, Stat3 and other signaling pathways are induced by ROS and, in turn, can regulate ROS production under cerebral ischemia. From a molecular point of view, transcription factors HIF1α, Stat3 and NF-kB may be considered as master regulators in this context. The combinational therapeutic strategies such as aspirin and streptokinase inhibitor may have adverse outcomes and thus should be carefully studied at preclinical level using appropriate animal models. The antiplatelet drugs have the ability to reduce platelet activity through a variety of mechanisms. Their biological activities go well beyond the platelet and include effects on other cell types throughout the body. The treatment of acute cerebral ischemia is dependent on clear understanding of the activity of the agents on the cerebrovasculature and within the brain parenchyma, especially on cerebral blood flow, the BBB, and neuronal cells. The most beneficial pharmacological regimen will minimize brain injury in cerebral ischemia.
This work was supported by grants received from Council of Scientific and Industrial Research (CSIR), DST (SERB: EMR/2017/000992/HS and Nanomission: SR/NM/NS-1058/2015) and DBT (Biocluster-Kolkata), Govt. of India and ICMR (WOS-DHR).
Abbreviations: ROS: Reactive oxygen species; VEGF: Vascular endothelial growth factor; TGF: Transforming growth factor; PDGF: Platelet derived growth factor; NO: Nitric oxide; ATP: Adenosine triphosphate ; HIF: Hypoxia-inducible factors; NADPH: Nicotinamide adenine dinucleotide phosphate; PHD: Prolyl Hydroxylase Domain; BBB: Blood brain barrier; NF-kB: Nuclear factor-Kappa B; MAPK: Mitogen-activated protein kinase; MMP: Matrix metalloproteinases; CBF: Cerebral blood flow; NMDA: N-Methyl-D-aspartic acid; AMPA: Aminomethylphosphonic acid; PMF: Proton motive force; ETC: Electron transport chain; FFAs: Free fatty acids; Bcl2: B-cell lymphoma 2; GLUT3: Glucose transporter 3; JAK: Janus kinase; NOX: NADPH oxidases; PARP-1: Poly (ADP-ribose) polymerase 1; STAT3: Signal transducer and activator of transcription 3; SOD2: Superoxide dismutase2; EGFR: Epidermal growth factor receptor; FKHR: Forkhead box protein O1; GSK3β: Glycogen synthase kinase 3 beta; ERK: Extracellular signal–regulated kinases; PAI-1: Plasminogen activator inhibitor-1; IKK: inhibitor of IK-B kinase; TNF: Tumor necrosis factor; COX-2: Cyclooxygenase-2; NCX: Sodium-calcium exchanger; XO: Xanthine Oxidase; CAT: Catalase; GPx: Glutathione peroxidase; NBO: Normobaric oxygen; eNOS: Endothelial NO synthase; ICH: Intracerebral hemorrhage