

 , Leandro Martins de Freitas 2,†
, Leandro Martins de Freitas 2,†1 Institute of Biomedical Sciences, Federal University of Rio de Janeiro, Rio de Janeiro, RJ 21941-902, Brazil
2 Multidisciplinary Institute of Health, Federal University of Bahia, Vitória da Conquista, BA 45029-094, Brazil
†These authors contributed equally.
Abstract
Chimeric antigen receptor–engineered macrophages (CAR-Ms) are emerging as a transformative frontier in immunotherapy and represent a promising strategy for pancreatic cancer. Unlike chimeric antigen receptor T-cell (CAR-T) or chimeric antigen receptor natural killer cell (CAR-NK) cells, which encounter limitations when applied to solid tumors, CAR-Ms combine innate tumor-homing capabilities with engineered antigen specificity to overcome these barriers. Recent studies have demonstrated encouraging antitumor activity in preclinical pancreatic ductal adenocarcinoma (PDAC) models; however, key questions remain regarding the persistence, safety, and scalability of these models. Nonetheless, despite these uncertainties, CAR-Ms may represent a paradigm shift from immune activation focused solely on direct cytotoxicity to one that also integrates immune orchestration and tumor microenvironmental reprogramming.
Keywords
- chimeric antigen receptor
- macrophages
- tumor microenvironment
- cellular therapy
Pancreatic cancer is one of the deadliest types of cancer, with around 511,000 new cases and 467,000 deaths reported in 2022. It accounts for nearly 5% of all cancer deaths and is the sixth leading cause of cancer mortality worldwide [1]. The most common and aggressive form is pancreatic ductal adenocarcinoma (PDAC), which is rising in incidence by about 0.5% to 1% per year [2]. By 2030, it is expected to become the second leading cause of cancer-related death [3]. Despite remarkable progress in precision oncology, PDAC continues evade to most therapeutic approaches, where the combination of late diagnosis, genetic heterogeneity, and dense desmoplastic stroma makes PDAC uniquely refractory to standard interventions.
Chemotherapy, despite being the main treatment option, offers modest benefit and limited durability. It includes neoadjuvant, adjuvant, and palliative care, with adjuvant chemotherapy the most commonly chosen option in clinical practice. However, only 8–32 % of patients responded to treatment with different chemotherapeutic agents [4, 5]. Targeted therapies directed against Kirsten rat sarcoma virus oncogene homolog (KRAS), MYC proto-oncogene (MYC), or DNA repair pathways have shown promise in molecular subsets. However, even immunotherapies that have revolutionized the treatment of other solid tumors, such as checkpoint inhibitors, have produced only minor responses in PDAC, largely due to both intrinsic and acquired resistance mechanisms. These therapeutic failures converge on a common biological barrier, an immunosuppressive and fibrotic tumor microenvironment (TME) that excludes effector immune cells and promotes tolerance rather than immunity.
In this context, the role of tumor-associated macrophages (TAMs) is receiving more attention in pancreatic cancer research. The interaction between tumor cells and TAMs induces a cyclical and prognostic immunosuppressive environment [6]. The presence of TAMs in the TME shapes an environment that supports tumor growth, angiogenesis, and immune invasion. The phenotypic diversity demonstrates the adaptability of macrophages to signals from the tumor microenvironment [7]. Understanding their phenotype is essential for the development of effective therapies. Additionally, unlike lymphocytes, macrophages naturally migrate toward hypoxic and necrotic tumor areas, even before their differentiation, as monocyte-derived macrophages are often found to infiltrate tumors in high numbers [8].
The emergence of Chimeric Antigen Receptor (CAR) technology has revolutionized the field of cellular immunotherapy, with CAR-T cells demonstrating remarkable success in treating hematologic malignancies [9]. However, the efficacy of CAR-T cell therapies remains limited when applied to solid tumors, underscoring the need for alternative strategies [10, 11]. To address this challenge, a conceptual pivot has emerged: instead of forcing lymphocytes into an inhospitable environment, why not reprogram the cells that already arrive there? This idea highlights why macrophages have become promising candidates for CAR-based engineering, due to their innate ability to infiltrate solid tumors and modulate the TME [12, 13]. The extracellular matrix is one of the key barriers that limit the efficacy of CAR-T and CAR-NK therapies, as these cells struggle to access the tumor site efficiently. Thus CAR-Macrophages (CAR-Ms) are engineered to combine the natural advantages of macrophages with the antigen-specific targeting capabilities of CAR technology, providing a novel approach to cancer treatment.
The design of chimeric-antigen receptor macrophages (CAR-M) shares several
similarities with CAR-T cells. It typically consists of an extracellular domain
(e.g., cluster of differentiation 19, CD19, cluster of differentiation 22, CD22,
or human epidermal growth factor receptor 2, HER2), a transmembrane domain (such
as CD8), and an intracellular signaling domain, including molecules like
CD3
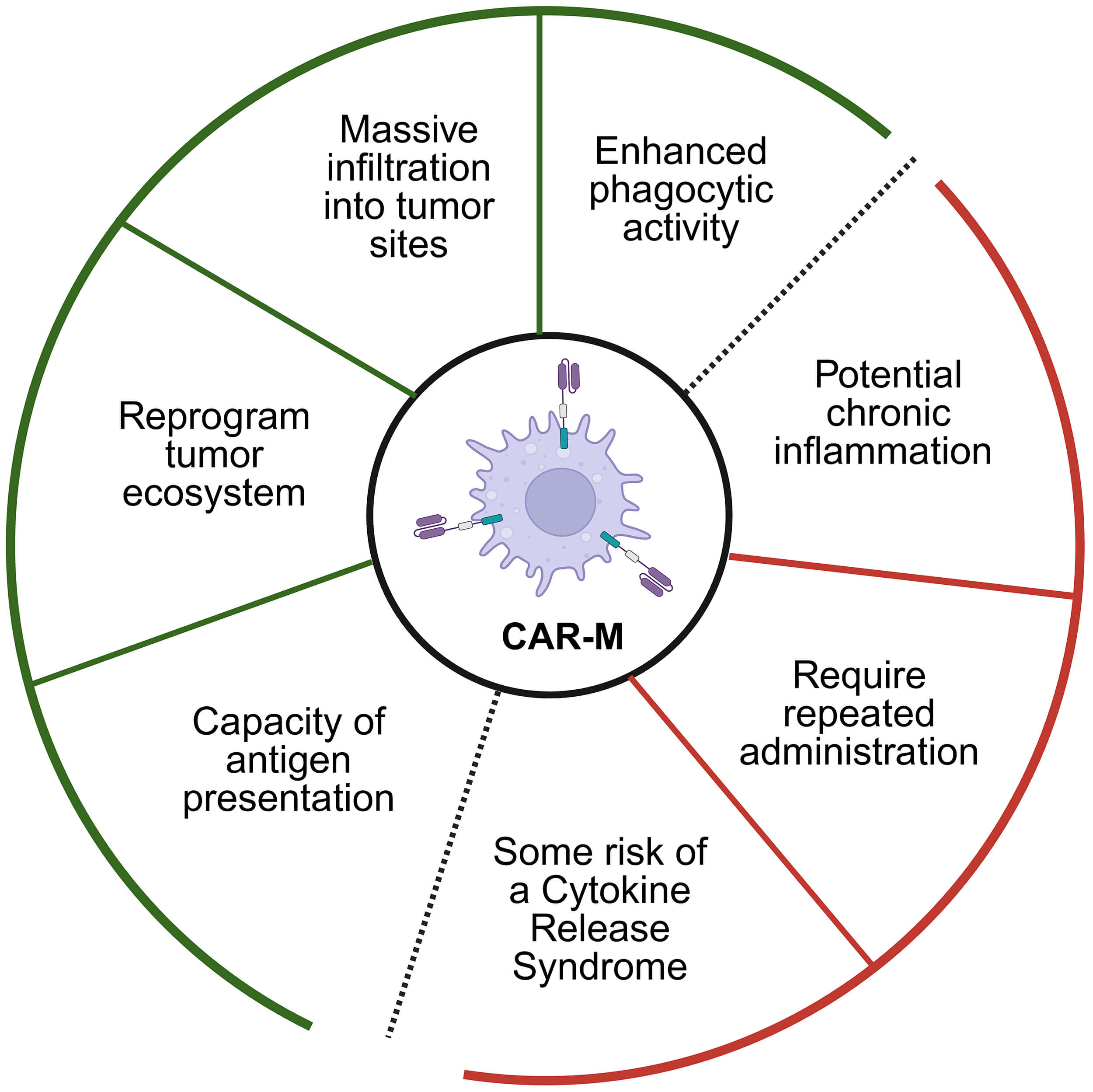 Fig. 1.
Fig. 1.
Functional advantages and potential risks of chimeric antigen receptor–engineered macrophage (CAR-M)-based approaches. CAR-M therapy provides multiple antitumor mechanisms, ranging from enhanced phagocytosis to tumor microenvironment remodeling, inside the green zone, while also posing concerns such as inflammatory toxicity, dosing frequency, and cytokine-mediated adverse events, inside the red zone.
Recent advancements in cellular immunotherapy have highlighted the significant potential of CAR-M as a promising therapeutic avenue for pancreatic cancer, explored through distinct but complementary strategies. One line of research has focused on targeting the c-MET oncogene, which is frequently overexpressed in pancreatic tumors and correlates with poorer patient outcomes, demonstrating that CAR-M targeting c-MET (CAR-M-c-MET) effectively suppresses pancreatic cancer progression and, notably, synergizes with cytotoxic chemotherapy to enhance its efficacy in preclinical models [27]. In a parallel effort, advancing a different therapeutic paradigm, another study developed an “off-the-shelf” allogeneic platform by generating CAR-M cells from human iPSCs that target the prostate stem cell antigen (PSCA), PSCA-targeted iPSC-derived CAR-Ms [28]. These CAR-iMacs exhibited potent, antigen-specific antitumor activity, resulting in reduced tumor burden and improved survival in pancreatic cancer mouse models, with the added advantage of being derived from a scalable and cryopreservable source of progenitors [28]. In another approach, Wang et al. (2025) [29] developed FAP-CAR macrophages, which are engineered in vivo to seek and destroy cancer-associated fibroblasts (CAFs), the cells responsible for creating the dense fibrotic barrier that makes pancreatic cancer impenetrable. By remodeling this protective wall, the FAP-CAR-M therapy effectively cleared the way for other treatments. This “barrier removal” strategy paves the way for direct-attack platforms such as c-MET or PSCA-CAR macrophages, as well as conventional chemotherapies, to gain access to the tumor core, significantly enhancing their therapeutic potential [29]. Together, these complementary paradigms demonstrate that CAR-M therapy can be strategically adapted: from direct cytotoxic targeting (c-MET, PSCA) to microenvironmental remodeling (FAP). It is interesting to note that these studies not only expand the repertoire of actionable targets in pancreatic cancer to include both c-MET and PSCA but also validate two powerful and distinct CAR-M platforms: one that can be integrated with standard chemotherapy and another that offers an “off-the-shelf”, allogeneic cell product for broader clinical application. The combination of the approaches could provide a more sophisticated therapeutic response to pancreatic cancer, enhancing the likelihood of achieving a better clinical outcome.
The CAR-M therapy presents several key advantages, one of them is the ability to modulate the complex and often immunosuppressive TME effectively. Unlike lymphocytes, CAR-M can remodel the TME into a more pro-inflammatory state that is hostile to cancer, a significant advantage in solid tumors. Moreover, this therapy leverages the natural dual functions of macrophages: phagocytosis and antigen presentation, where CAR-M not only directly engulfs and digests tumor cells but also processes and presents tumor antigens to the rest of the immune system. This antigen presentation enhances the activation of other immune cells, such as T-cells, thereby triggering a broader and more sustained systemic immune response against the cancer. Additionally, while risks such as Cytokine Release Syndrome (CRS) exist, macrophages possess intrinsic regulatory functions that may help control cytokine release more effectively than CAR-T cells, potentially mitigating this severe side effect (Fig. 1).
However, despite the significant advancements and therapeutic potential of CAR-M in pancreatic cancer, the field is characterized by numerous controversies and unanswered questions that demand further investigation. Before solving these controversies, the adoption of this approach is limited. Debates persist regarding the optimal strategies for engineering and administering CAR-M to maximize efficacy while minimizing risks, particularly in defining the ideal CAR construct configuration and delivery methods. Significant gaps remain in understanding the therapy’s fundamental mechanisms, such as the variability of phagocytic efficiency within the immunosuppressive TME and the most effective ways to combine CAR-M with other treatments to achieve synergistic effects. Furthermore, critical safety questions remain, including how risks documented in CAR-T therapy, like Cytokine Release Syndrome (CRS), translate to macrophage-based systems and whether the intrinsic regulatory functions of macrophages can be reliably controlled to prevent severe adverse events. The long-term implications of CAR-M treatment on the immune system, including potential chronic inflammation or dysregulation, are largely unknown, highlighting a pressing need for extensive long-term follow-up studies. Finally, as CAR-M approaches diversification, the field must establish comparative standards to evaluate efficacy and safety across different delivery systems, target antigens, and cellular sources, as well as resolve complex regulatory and ethical questions related to patient selection and consent. For CAR-Ms to transition from the bench to the bedside, these questions must be addressed to pave the way for CAR-M to transition from an experimental treatment to a safe and established option in the oncological arsenal.
As the field of cellular therapy continues to grow, it is becoming clear that the possibility of using a cell that combines innate characteristics, such as phagocytosis and antigen presentation, along with the capacity for TME remodeling, represents a promising approach. The most powerful aspect of this approach is its potential to reprogram the tumor ecosystem, transforming an immunosuppressive environment into one that supports immune activity. In our opinion, the next important step is to combine macrophage engineering with new tools from omics, such as spatial transcriptomics and single-cell RNA sequencing, and immunometabolism. These technologies could enable the understanding of the tumor immune microenvironment, facilitating the creation of CAR-Ms that adapt their behavior based on the local signals they will find inside the tumor, improving their persistence and safety. We also believe that progress in this field must be accompanied by caution. The enthusiasm for translating engineered macrophages into the clinic should not replace the need to understand how they behave in vivo, how long they survive, how they interact with healthy tissues, and how they influence the immune balance over time. To avoid repeating the mistakes seen with earlier immunotherapies, we need standardized models and long-term studies that can truly predict human outcomes. Even with these challenges, the future of CAR-M therapy remains promising. The combination of immunology, cell engineering, and bioinformatics can open new possibilities for safer and more adaptable cell therapies. With these interdisciplinary advances, CAR-Ms could evolve from a laboratory concept into a clinically reliable and personalized treatment for pancreatic cancer, giving patients a real chance at improved outcomes.
PPA and LMdeF designed the research study. PPA and LMdeF drafted the manuscript. Both authors contributed to critical revision of the manuscript for important intellectual content. Both authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.