




1 Department of Pharmacology, Faculty of Pharmacy, Dr. M.G.R. Educational and Research Institute, 600017 Chennai, Tamil Nadu, India
2 Pharmaco-Netinformatics Research Lab, Department of Psychiatry, Saveetha Medical College and Hospital, Saveetha Institute of Medical and Technical Sciences (SIMATS), Saveetha University, 602105 Chennai, Tamil Nadu, India
3 Department of Community Medicine, Sree Balaji Medical College and Hospital, 600044 Chennai, Tamil Nadu, India
4 Department of Community Medicine, Vinayaka Mission Medical College and Hospital, 609609 Karaikal, Pondicherry, India
5 Department of Genetic Engineering, School of Bioengineering, College of Engineering and Technology SRM Institute of Science and Technology, 603203 Kattankulathur, Tamil Nadu, India
Abstract
Excessive fructose consumption has emerged as a critical driver of obesity and metabolic dysfunction, with far-reaching implications for multiple organ systems. This review synthesizes current evidence on the biochemical and molecular pathways underlying fructose induced disease mechanisms, discussing how fructose metabolism activates the “survival switch”, promotes fat storage, and generates uric acid, mitochondrial dysfunction, and oxidative stress, thereby disrupting energy homeostasis. Key organ-specific consequences are explored, including hepatic steatosis and progression to non-alcoholic fatty liver disease, pancreatic β-cell dysfunction, renal fibrosis, intestinal barrier disruption with microbial dysbiosis, cardiometabolic impairment, pulmonary inflammation, and neurocognitive decline with relevance to Alzheimer’s disease. Moreover, mechanistic insights highlight the role of fructokinase C activation, adenosine triphosphate (ATP) depletion, leptin resistance, pro-inflammatory signaling (mechanistic target of rapamycin complex-1 (mTORC1), renin angiotensin system (RAS), Toll-like receptor 4 (TLR4)), and cross-talk between fructose metabolism and organ-specific pathophysiology. Animal and human studies consistently reinforce the central role of fructose overload in driving obesity and associated complications. Meanwhile, this review frames fructose not merely as a caloric contributor but as a metabolic disruptor, thereby underscoring the urgent need for public health interventions, dietary regulation, and mechanistic research to mitigate fructose-driven metabolic disease.
Keywords
- fructose metabolism
- obesity
- organ dysfunction
- oxidative stress
- metabolic signaling
- non-alcoholic fatty liver disease
Obesity is a major public health issue among children and adolescents worldwide.
According to the World Health Organization, one in eight people is classified as
obese; specifically, nearly 890 million adults and 160 million children
categorized as obese in 2022. Obesity is a chronic disease characterized by the
accumulation of excess body fat. Consequently, obesity increases the risk factor
for type 2 diabetes mellitus, non-alcoholic fatty liver disease, heart disease,
cancer and other related metabolic disorders. The etiology of obesity is
multifactorial involving diet, lifestyle, and genetic predisposition, with diets
rich in sugar, salt, and fat playing a major role in the overall development [1, 2]. Thus, the increased consumption of dietary sugars, particularly
fructose-containing sugars, is an emerging public health concern due to a strong
association with obesity and related metabolic disorders. Since fructose differs
from glucose, energy cannot be utilized directly by all cells in the body.
Instead, fructose is converted into glucose, fatty acids, or other metabolic
substrates, thereby creating an energy imbalance. Fructose also serves as a
substrate for de novo lipogenesis and contributes to increased adiposity
by impairing metabolic signaling pathways implicated in obesity. Animal models
have demonstrated the detrimental effects of excessive fructose intake, typically
defined as 30%–60% of total daily energy, which promotes rapid hepatic
steatosis and insulin resistance. In humans, “high” intake is characterized by
the consumption of at least 25% of the total energy from fructose-containing
sugars (often
This article presents a narrative literature review summarizing animal, mechanistic, and human evidence on fructose metabolism and multiorgan pathophysiology. Since this work is designed as a narrative overview rather than a systematic review or scoping review (ScR), the article does not follow PRISMA guidelines. A literature search was conducted in databases such as PubMed, Scopus, Web of Science, Google Scholar, ScienceDirect, SpringerLink, and Wiley Online Library using the search terms “fructose metabolism”, “high fructose corn syrup”, and “non-alcoholic fatty liver disease”. The search prioritized peer-reviewed studies from 2015 to 2025, while incorporating earlier seminal works to provide necessary conceptual context. Following a review of the identified titles and abstracts, 61 studies were selected for qualitative discussion based on the associated mechanistic relevance and clinical significance. Consistent with the narrative format, formal flow diagrams and systematic risk-of-bias tools were not utilized; instead, the literature was curated to provide a cohesive conceptual map of fructose-driven organ dysfunction.
Fructose is a simple sugar or monosaccharide naturally found in fruits, vegetables, and honey. Additionally, fructose is located in the disaccharide sucrose, which consists of fructose and glucose. The primary sources of fructose include sucrose derived from beetroot and sugarcane, as well as high-fructose corn syrup (HFCS). Common dietary sources of fructose-containing sugars include fruit juice, fruit drinks, sweetened cereal grains, honey, sweets, desserts and caloric sweeteners. Although fructose is naturally present in fruits and honey, fructose found in processed foods and beverages is typically consumed as sucrose-commonly known as table sugar or HFCS. In modern diet, HFCS is used as a sweetener in sugar-sweetened beverages (SSBs) [10], which are beverages sweetened with added sugars such as fructose, sucrose, brown sugar, lactose, malt syrup, HFCS, and molasses. Indeed, examples of SSBs include sodas, energy drinks, iced tea, fruit drinks, sports drinks, sweetened tea and coffee, and flavored waters. According to a survey across 187 nations, SSB consumption in adults was higher among middle-income countries than in high- or low-income countries [11, 12].
Fructose is used in many parts of the world as a caloric sweetener, meaning fructose is consumed regularly in modern diets. In addition to dietary sources, fructose is also generated in various tissues as a metabolic by products when glucose is converted to fructose by the rate-limiting enzyme aldose reductase. This process is referred to as the polyol pathway, and the aldose reductase enzyme is stimulated by hypoxia, stress and starvation. In diabetes, carbohydrate ingestion leads to elevated glucose concentrations, which in turn promote fructose metabolism. Under hyperglycemic conditions and in ischemic states, fructose is abundantly generated in the brain, heart, and kidneys [13, 14, 15]. Generally, weight regulation in animals involves a balance between nutrient intake and energy utilization through metabolic pathways. Energy utilization is primarily driven by mitochondria, which are known as the power house of the cell. Active energy is stored in cells as adenosine triphosphate (ATP), which is replenished either through nutrient intake or by oxidation of stored fat reserves. A high-calorie diet leads to excessive fat accumulation, which causes obesity, whereas a low-calorie diet leads to weight loss; however, ATP levels typically remain balanced in normal conditions. Notably, fructose metabolism disrupts ATP homeostasis; ATP-consuming pathways metabolize fructose but, do not stimulate compensatory ATP synthesis. Therefore, intracellular ATP concentrations decrease, leading to leptin resistance; even when ATP levels are replenished, excess caloric intake results in fat accumulation and storage [16, 17, 18, 19].
Conversely, hypercaloric fructose supplementation, which provides energy
surpluses of 25–35% above baseline, consistently elevates fasting
triglycerides, uric acid, and hepatic lipid deposition. These findings highlight
that fructose-related metabolic injury is primarily driven by excessive energy
intake rather than isocaloric substitution. Fructose metabolism in hepatocytes
rapidly consumes ATP via ketohexokinase-C (KHK-C), generating AMP, which is
subsequently degraded by AMP deaminase-2 (AMPD2) to inosine monophosphate and,
ultimately, uric acid [20, 21]. Elevated uric acid concentrations activate NADPH
oxidase, promoting oxidative stress, mitochondrial dysfunction, and endothelial
nitric oxide depletion. These effects impair insulin signaling pathways
(PI3K-Akt) and enhance hepatic de novo lipogenesis, establishing a
mechanistic link between fructose-induced hyperuricemia, insulin resistance, and
cardiometabolic disease [22]. Moreover, oxidative stress decreases the ATP
generation by inhibiting the aconitase enzymes in the Krebs cycle, as well as
carnitine palmitoyl transferase-1a (CPT1A) and enoyl CoA hydratase in
mitochondrial beta fatty acid oxidation. Simultaneously, oxidative
phosphorylation is suppressed, further reducing ATP synthesis. Thus, fructose
lowers the intracellular ATP levels. The prevailing hypothesis is that reduced
ATP activates a cellular survival response that disrupts energy homeostasis and
contributes to weight dysregulation. Meanwhile, ATP remains chronically depleted
due to sustained oxidative stress and mitochondrial dysfunction. Consequently,
leptin resistance increases hunger, stimulating high caloric intake and weight
gain. Fructose metabolism may indirectly reduce
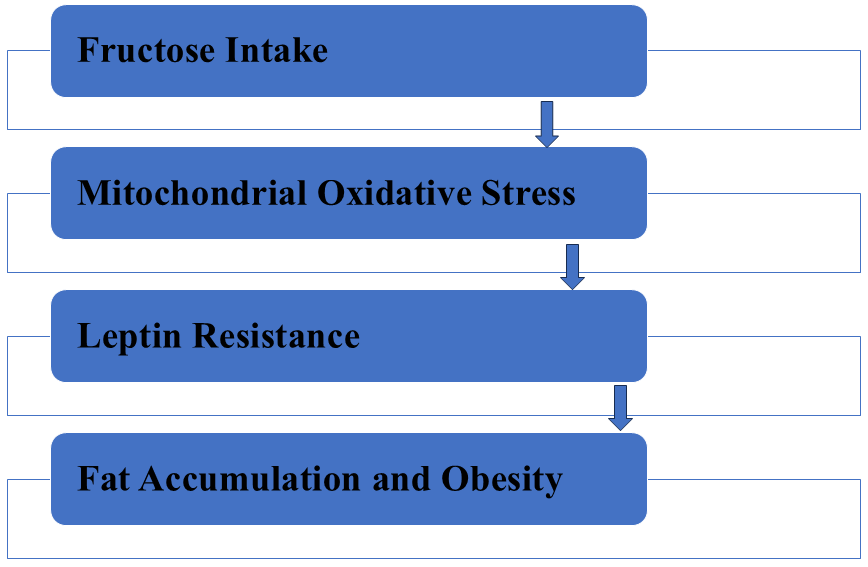 Fig. 1.
Fig. 1.
Fructose metabolism via fructokinase C promotes mitochondrial oxidative stress, leading to leptin resistance and ultimately contributing to fat accumulation and obesity.
Interpreting animal and human data together highlights a consistent theme: many animal models show organ-level injury under concentrated or supplemental fructose exposure (largely hypercaloric regimens), whereas human controlled trials demonstrate divergent effects depending on the energy context. Thus, mechanistic animal evidence can therefore plausibly explain why hypercaloric fructose increases hepatic lipogenesis and oxidative stress, whereas isocaloric replacement in humans largely avoids these effects. Hence, we emphasize that public health guidance should target reductions in energy intake resulting from added sugars, particularly from SSBs, rather than demonizing intrinsic fructose in whole fruits. To improve clarity and interpretability, animal and human evidence were tabulated separately (Table 1, Ref. [25, 26, 27, 28, 29] and Table 2, Ref. [30, 31, 32, 33, 34, 35, 36]). Each study entry includes an exposure context classification (isocaloric vs hypercaloric), which indicates whether fructose was provided as a calorie-matched substitution or as an additional caloric load; the study design/evidence type (experimental animal, randomized controlled trial (RCT), controlled feeding, cohort) is also documented to indicate the strength of evidence.
| Reference | Experimental model | Evidence type | Fructose exposure/dose | Exposure context | Major outcome findings |
| Cigliano et al. 2018 [25] | Male, Sprague-dawley rats | Experimental animal study | Fructose rich diet × 2 weeks | Hypercaloric | Oxidative stress + hippocampal inflammation |
| Yoo et al. 2017 [26] | Female, Sprague-dawley rats | Experimental animal study | 30% fructose diet/high fat diet/combined | Hypercaloric | ↑ endothelial dysfunction, dyslipidemia, ↑ aortic wall thickness |
| Vedova et al. 2019 [27] | Mice | Experimental animal study | 10% fructose water + 22% fat diet | Hypercaloric | Pulmonary oxidative stress + fibrosis |
| Nakayama et al. 2010 [28] | Sprague-dawley rats | Experimental animal study | 60% fructose feed × 6 weeks | Hypercaloric | Tubulointerstitial injury/tubular hyperplasia |
| Guo et al. 2021 [29] | Weaned piglets | Experimental animal study | Fructose supplementation × 35 days | Hypercaloric | Gut microbiota shift + intestinal barrier dysfunction |
Isocaloric: fructose replaced other carbohydrate energy with no net change in
total energy intake (calorie-matched substitution). Hypercaloric: fructose was
provided as additional calories above baseline/maintenance intake (via
supplemental or ad libitum concentrated diets), resulting in a net positive
energy balance. Classification was based on the exposure description and caloric
design reported in each study.
| Reference (Year) | Study design | Exposure context | Fructose/SSB Dose | Primary metabolic outcomes |
| Chiavaroli et al. 2015 [30]; Khan et al. 2016 [31] | Meta-analysis | Isocaloric (weight neutral) | Fructose replacing other carbohydrates | No significant effect on triglycerides, hepatic fat, or insulin sensitivity; modest HbA1c benefit |
| Jayalath et al. 2015 [32] | Prospective cohort | Hypercaloric (ad libitum) | High sugar-sweetened beverage (SSB) intake | Dose-response relationship with increased systolic blood pressure and incidence of hypertension |
| Imamura et al. 2015 [33] | Systematic review/Cohort | Hypercaloric | 13%–18% increased risk of type 2 diabetes (T2D), independent of adiposity | |
| Luo et al. 2015 [34] | Randomized crossover | Acute fructose vs. glucose | 75 g pure fructose drink | Increased hunger signaling and brain reactivity to food cues compared to glucose |
| Ma et al. 2016 [35] | Observational cohort | Long-term SSB intake | Significant association with increased visceral adipose tissue (VAT) and NAFLD risk. | |
| Stanhope et al. 2015 [36] | Controlled intervention | Hypercaloric (25% energy) | HFCS or fructose-sweetened beverages | Dose-dependent increase in LDL-C, ApoB, and postprandial triglycerides in as little as 2 weeks |
Isocaloric: fructose replaced other carbohydrate energy with no net change in total energy intake (calorie-matched substitution). Hypercaloric: fructose was provided as additional calories above baseline/maintenance intake (via supplemental or ad libitum concentrated diets), which results in a net positive energy balance. Classification was based on the exposure description and caloric design reported in each study. HbA1c, Hemoglobin A1c; T2D, Type 2 Diabetes; VAT, Visceral Adipose Tissue; NAFLD, Non-alcoholic Fatty Liver Disease; HFCS: High-Fructose Corn Syrup; LDL-C, Low-Density Lipoprotein Cholesterol; ApoB, Apolipoprotein B; SSB, Sugar-Sweetened Beverage.
In controlled feeding trials, isocaloric substitution of fructose for other carbohydrates generally does not reduce triglyceride levels, hepatic fat content, or insulin sensitivity. However, hypercaloric fructose supplementation with a net energy surplus of 25%–35% of total energy intake consistently increased fasting triglycerides and hepatic lipid accumulation. Large-scale prospective cohort studies provide quantitative evidence for the impact of SSBs. For example, a single daily serving (approximately 355 mL, providing 25–30 g of fructose) is associated with a 13%–18% increase in the risk of type 2 diabetes (relative risk (RR) = 1.13–1.18; 95% confidence interval (CI): 1.02–1.21). Similarly, habitual SSB intake is linked to a higher incidence of hypertension (RR = 1.12; 95% CI: 1.06–1.19) and significant increases in visceral adipose tissue over long-term follow-up. These data suggest that the metabolic risk associated with fructose is most pronounced when consumed in quantities that promote a positive energy balance [11].
Fructose, when consumed in moderate amounts from natural sources such as fruits
and vegetables, is generally well-tolerated and contributes to normal energy
metabolism. However, excessive intake of added fructose, especially from SSB s
and processed foods, can lead to significant metabolic and organ-specific
complications. Indeed, chronic fructose overconsumption has been linked to
oxidative stress, inflammation, mitochondrial dysfunction, and hormonal
imbalances, collectively disrupting energy homeostasis. These changes contribute
to obesity, insulin resistance, hepatic steatosis, renal injury, cardiovascular
abnormalities, pulmonary inflammation, and neurocognitive decline [37, 38]. Fig. 2
depicts multiorgan disorders that can be induced by high-fructose intake,
including neurodegenerative, cardiovascular, metabolic, hepatic, renal,
pulmonary, and gastrointestinal dysfunction. Furthermore, Fig. 2 illustrates the
organ-specific consequences of chronic fructose overload, including hepatic
steatosis and inflammation, pancreatic
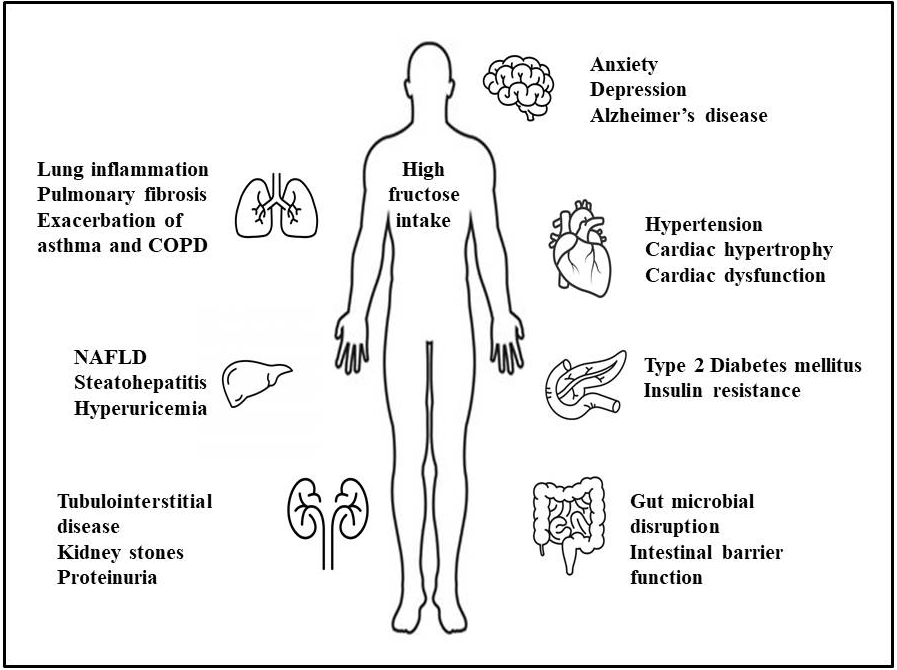 Fig. 2.
Fig. 2.
Systemic effects of excessive fructose intake on human health.
Fructose exerts various peripheral effects that may impair the physiological functions of visceral organs. Chronic intake is associated with increased fat accumulation and elevated pro-inflammatory markers. Meanwhile, emerging research suggests that disruption of cerebral metabolism, particularly in the hypothalamus and hippocampus, may be a potential etiology of neurological dysfunction, affecting cognition, appetite regulation, and motivation [9, 39].
Mechanistic studies, primarily in animal models, indicate that fructose may
influence cognitive function and emotional regulation by altering the
serotonergic system and inducing oxidative stress—processes implicated in
psychiatric and neurodegenerative conditions. In rat models, chronic fructose
administration has been shown to impair hippocampal mitochondrial bioenergetics
and intensify the effects of traumatic brain injury on synaptic plasticity. While
a significant role for fructose in Alzheimer’s disease (AD) has been proposed in
data from animal studies, this association remains largely hypothetical and is
predominantly supported by mechanistic speculation. Nonetheless, fructose-induced
insulin resistance is hypothesized to correlate with intracerebral biomarkers,
such as senile plaques and neurofibrillary tangles. For instance, the amyloid
beta (A
Specifically, fructose modulates molecular mechanisms and stimulates numerous
low-grade inflammatory pathways, thereby contributing to lung disorders.
Moreover, fructose intake promotes cellular metabolic stress, whereas uric acid
levels are elevated via an effector mechanism. Uric acid, in turn, activates
inflammatory signaling pathways such as the renin angiotensin system (RAS), the
mechanistic target of rapamycin complex-1 (mTORC1), and the androgen receptor
(AR). Furthermore, long-term fructose consumption alters protein expression and
maintains an inflammatory state. Hence, a detrimental feedback mechanism remains
a cause of sustained inflammation and epithelial and endothelial dysfunction in
lung diseases [43]. Epidemiological research indicates that dietary
fiber, as well as fruits and vegetables, is associated with a reduced risk of
chronic obstructive pulmonary disease (COPD). In contrast, increased fructose
consumption is linked to an increased risk of asthma and chronic bronchitis.
However, certain dietary components influence the risk of COPD by altering
oxidative stress and inflammatory responses. An animal study on the chronic
effects of fructose along with smoking cigarettes, observed an increase in
pulmonary emphysema in mice due to the lung parenchymal destruction and
alteration in the respiratory mechanisms. A possible mechanism for the
association between fructose intake and pulmonary emphysema is the activation of
an inflammatory response in the lungs, initiated at extrapulmonary sites [44].
The liver is the prime candidate for triggering fructose-induced inflammation.
Excessive fructose intake from food or beverages leads to increased formation of
advanced glycation end-products (AGEs) in the gut due to fructose malabsorption,
which enter the bloodstream and activate the receptor for advanced glycation
end-products (RAGEs). Inflammation triggered by RAGE activation can explain the
link between high fructose intake and chronic inflammatory diseases. The RAGE
expression in the lungs plays a significant role in the pathology of asthma.
Thus, high fructose intake may increase inflammation and exacerbate asthma states
[45]. High fructose levels were found to stimulate the fibrotic
phenotype of human lung epithelial cells by increasing oxidative stress, thereby
activated the latent transforming growth factor beta-1 (TGF-
Sugars that contain fructose can contribute to weight gain and elevate cardio metabolic risk factors and diseases, only in the instance of excess caloric intake. Nonetheless, there is growing evidence that consuming SSBs elevates cardiovascular risk through the onset of hypertension, dyslipidemia, inflammation, coronary heart disease, and stroke. A correlation with an unhealthy lifestyle probably influences the negative with impact of SSBs, as individuals who drink SSBs tend to consume additional calories, engage in less physical activity, smoke more frequently, and maintain poor dietary habits [38]. Evidence from a systematic review shows that consumption of SSBs is significantly linked to elevated blood pressure (BP) and higher occurrence of hypertension [39]. Despite several potential explanations, the exact mechanisms by which fructose increases BP remain incompletely understood. Nevertheless, current possibilities include decreased baroreflex activity, increased sympathetic nervous system and RAS activity, elevated circulating catechol amine levels, augmented sodium reabsorption, elevated uric acid production, and impaired endothelial function. Notably, current evidence that fructose directly raises BP remains limited. High fructose intake may be associated with increased caloric intake and weight gain, as well as insulin resistance, all of which are independently associated with elevated BP [46]. High fructose consumption leads to metabolic syndrome, hypertension, and obesity, which, in turn, contribute to cardiac hypertrophy and fibrosis [9]. Nevertheless, the specific mechanisms underlying cardiac inflammation induced by a high-fructose diet remain incompletely understood. Result shows that the effects of high-fructose diet on cardiomyocytes in adult mice and found a significant increase in cardiomyocyte size and in the relative wall thickness of the left ventricle (LV). Echocardiographic assessments of cardiac performance also demonstrated a notable decrease in ejection fraction percentage (EF%) and fractional shortening percentage (FS%) 12 weeks after consuming a diet containing 60% fructose. The mRNA and protein levels of monocyte chemoattractant protein-1 (MCP-1) were significantly elevated in HL-1 cells and primary cardiomyocytes treated with high fructose. The research findings indicate that a high-fructose diet induces cardiac inflammation by recruiting macrophages into cardiomyocytes, thereby adversely affecting cardiac function. Fructose-induced dyslipidemia and hypertension are key mediators of cardiometabolic risk. Population-based interventions that reduce SSB consumption could prevent new cases of hypertension and diabetes [47, 48].
Studies indicate that excessive fructose consumption may lead to pancreatic beta cells dysfunction, which can reduce in insulin secretion and may contribute to the development of insulin resistance. High fructose intake has also been shown to lower ATP levels in the liver, which may lead to decreased insulin binding at the cellular level, a potential reduction in insulin receptor numbers, and the development of insulin resistance [49]. Additionally, fructose stimulates the hepatic de novo lipogenesis while inhibiting fatty acid oxidation, thereby promoting hepatic fat accumulation, which in turn triggers inflammation and insulin resistance. Increased hepatic insulin resistance promotes insulin secretion by pancreatic beta-cells, thereby contributing to the progressive dysfunction of these cells. Gradually, a decline in beta-cell function can lead to insufficient insulin production, thereby intensifying fructose-induced inflammation and oxidative stress, further exacerbating hepatic insulin resistance. The effect of fructose on diabetes and glycemic control remains controversial. A meta-analysis of controlled feeding trials indicated that replacing carbohydrates with isocaloric fructose led to notable improvements in glycemic control, with a 0.53% decrease in HbA1c, without a significant effect on insulin levels in individuals with diabetes. The current level of fructose in diabetic diets appears to be within recommended limits. Since fructose has a low glycemic index, fructose may serve as an alternative sweetener for diabetic patients, although these patients are prone to postprandial hyperglycemia. In individuals with mild non-insulin-dependent diabetes mellitus, fructose lowers postprandial glucose and insulin levels, compared to most other carbohydrate sources. Clinical research has shown that fructose has either enhanced metabolic control in patients with diabetes or has not produced significant changes. Patients who are prone to hypertriglyceridemia should avoid high doses of fructose due to the associated potential of fructose to increase triglyceride levels. However, there are few long-term controlled trials of fructose use in patients with diabetes [50].
Approximately 70% of fructose in humans is metabolized by the liver. Thus, high fructose intake leads the liver to increase de novo fatty acid synthesis and the subsequent accumulation of triglycerides. Consequently, fructose has been suggested as a significant contributor to the onset of MASLD. When the amount of fructose exceeds the capacity for intestinal clearance, fructose enters the portal vein; the subsequent increase in blood fructose rapidly triggers transport to the liver, the primary site of fructose metabolism [51]. Thus, fructose is metabolized in the liver and is more likely to promote lipogenesis than glucose. High-fructose diets increase metabolic flux through aldolase B by accumulating fructose-1-phosphate. Subsequently, triokinase promotes the phosphorylation of D-glyceraldehyde to generate pyruvate and acetyl-CoA, thereby encouraging lipid dysregulation. Fructose is a major dietary contributor to hepatic de novo lipogenesis and the progression of MASLD, particularly when consumed in excess; however, fructose does not form the sole determinant of hepatic fat accumulation. MASLD is a condition characterized by excessive accumulation of fat in the liver, occurring in the absence of alcohol use, autoimmune diseases, or viral infection. The etiology of MASLD includes obesity, high sugar and fat consumption, and a sedentary lifestyle. If left untreated, MASLD may advance to metabolic dysfunction-associated steatohepatitis (MASH), which involves not only fat accumulation but also inflammation and mild scarring, ultimately progressing to severe fibrosis, cirrhosis, and possibly liver cancer, representing the final stage of the disease. Importantly, fructose has been identified as a key factor in the development of MASLD, as numerous studies have shown a strong link between fructose intake and the severity of inflammation and fibrosis. The liver damage caused by fructose is largely mediated by activation of lipogenesis and inflammatory signaling pathways, which subsequently lead to fibrosis and hepatocellular carcinoma (HCC). Additionally, the overproduction of free radicals and uric acid resulting from high fructose intake significantly contributes to the progression of fatty liver, inflammation, fibrosis, and HCC through multiple signaling pathways [52, 53]. High consumption of SSBs elevates serum uric acid levels and causes hyperuricemia. Increase in uric acid levels due to fructose may lead to negative health effects. Excessive fructose intake promotes hepatic de novo lipogenesis and steatosis, particularly under hypercaloric conditions. Hence, limiting added sugars and SSBs is an effective strategy for reducing the risk of MASLD [54].
The absorption of fructose and glucose in the small intestine has been characterized and is significantly different. Glucose is primarily absorbed into enterocytes via an active process, along with sodium, via sodium-dependent glucose transporters (SGLTs). When luminal glucose levels are elevated, glucose transporter-2 (GLUT2) is rapidly and transiently recruited to the apical membrane of enterocytes, indicating that GLUT2 contributes to glucose uptake [48]. Fructose absorption in the small intestine occurs primarily through GLUT5, a facilitative, sodium-independent fructose transporter located on the apical membrane of enterocytes. Therefore, under conditions of high luminal fructose load, GLUT2 may transiently translocate to the apical membrane to augment fructose uptake [55]. Increased fructose consumption may alter gut microbial composition, leading to dysbiosis. In addition, high fructose impairs intestinal barrier function, including reduced tight junction protein levels, thereby increasing permeability to pathogen-associated molecular patterns (PAMPs). The enhanced movement of PAMPs, including lipopolysaccharides, into the portal vein activates Toll-like receptor 4 (TLR4) signaling pathways in the liver, potentially contributing to the onset of hepatic insulin resistance. Furthermore, fructose metabolism may influence mucosal nitric oxide homeostasis. The amino acids L-arginine and L-citrulline may help mitigate the reduction in arginase activity and, consequently, reduce fructose-related intestinal barrier dysfunction. Nitric oxide modifications and decreased arginase activity may disrupt tight junction proteins in the small intestine. Additionally, certain probiotics may help delay the onset of fructose-induced intestinal barrier dysfunction through unknown mechanisms. Fructose exacerbates symptoms in individuals with irritable bowel syndrome (IBS), a condition marked by abdominal discomfort, diarrhea, and bloating. Research involving IBS patients indicated a significant prevalence of fructose malabsorption, reaching as high. Notably, even patients who tested negative for fructose metabolism disorders experienced abdominal symptoms after fructose consumption, indicating that fructose intolerance is relatively common. In some beverages sweetened with HFCS-55 (55% fructose), a 500 mL serving provides 27.5 g of fructose. Evidence from human studies indicates that this level of fructose in beverages and food products may contribute to the development of inflammatory diseases [56].
Fructose is metabolized in the proximal tubules in the renal system. In cases of high consumption, fructose can cause adverse effects, potentially due to an excess of uric acid, a byproduct of fructose metabolism. The activation of glycolysis by fructose metabolism promotes other pathways, such as lipogenesis, the pentose phosphate pathway, and the hexosamine pathway, and supplies biosynthetic precursors that support energy production in renal inflammation and fibrosis [53]. Animal studies have revealed that healthy rats fed a high-fructose diet exhibited mild tubulointerstitial damage, accompanied by inflammation and fibrosis. Meanwhile, in rats with chronic kidney disease, fructose exacerbated tubular damage and increased interstitial inflammation and fibrosis. Fructose administration in rats causes renal hypertrophy, tubulointerstitial disease, glomerulosclerosis, and proteinuria in existing renal disease. Fructose is freely filtered at the glomerulus and partially reabsorbed in the proximal tubule, where fructose is metabolized by fructokinase (KHK-C). Excessive fructose intake promotes uric acid production and oxidative stress, thereby contributing to tubulointerstitial injury. The formation of uric acid by xanthine oxidoreductase induces oxidative stress and inflammation. Thus, an increase in uric acid levels leads to an afferent arteriolopathy, which in turn causes glomerular hypertension. High fructose consumption elevates uric acid and induces oxidative renal stress, contributing to early kidney injury. Dietary reduction of SSBs can lower hyperuricemia and CKD progression risks. Furthermore, SSBs enhance the excretion of elements associated with the risk of kidney stones, including oxalate, uric acid, and calcium. Research has indicated that fructose consumption is independently associated with an increased risk of kidney stones. The intake of fructose, particularly from sucrose and high-fructose corn syrup, has increased substantially in recent decades and is independently associated with an elevated risk of nephrolithiasis [57, 58].
Excessive caloric intake contributes substantially to obesity and metabolic dysfunction, whereas fructose exerts distinct organ-specific effects through distinct metabolic pathways. Unlike glucose, fructose bypasses key glycolytic control points, leading to rapid ATP depletion, uric acid formation, and mitochondrial stress, which promote lipogenesis and insulin resistance [59].
Historically, low-purine diets for hyperuricemia have often inadvertently increased carbohydrate and fructose intake, reducing uric acid levels. In contrast, the dietary approaches to stop hypertension (DASH) and Mediterranean diets improve insulin sensitivity, reduce uric acid levels, and lower inflammation. While reducing added sugars remains the cornerstone of prevention, modern clinical management of obesity and metabolic dysfunction has evolved to include pharmacological and surgical interventions. Indeed, semaglutide (a GLP-1 receptor agonist) has demonstrated significant efficacy in reducing visceral adiposity and improving hepatic insulin sensitivity, potentially counteracting the proinflammatory signaling induced by chronic fructose consumption. Furthermore, bariatric surgery provides a potent metabolic reset, rapidly improving glycemic control and reversing MASLD by fundamentally altering the hormonal response to nutrient intake. Overall, while animal models help define the molecular pathways of fructose-driven injury, human evidence confirms these risks at the population level. Thus, adopting nutrient-dense diets, such as the DASH diet, can substantially reduce the burden of gout and cardiovascular disease. However, for individuals with advanced obesity or established metabolic injury, the integration of semaglutide or bariatric surgery may be necessary to achieve the weight loss and metabolic stabilization required to halt disease progression. Combining these advanced clinical interventions with public policies, such as SSB taxation and nutrition education, offers a comprehensive approach to reducing the global burden of fructose-mediated metabolic disease [60, 61, 62, 63].
In conclusion, while moderate fructose metabolism via KHK-C contributes to normal energy homeostasis, the evidence summarized in this review demonstrates that excessive intake triggers ATP depletion, uric acid generation, and oxidative stress. This suggests that metabolic harm results from overconsumption that exceeds the processing capacity of the liver. Unlike glucose, fructose bypasses key regulatory steps, leading to rapid mitochondrial dysfunction and lipogenesis. While caloric overload is a broad contributor to metabolic disease, the organ-specific pathways identified in this review—specifically the synergistic effect of fructose on insulin resistance and multiorgan injury—distinguish fructose as a unique metabolic disruptor. Naturally occurring fructose in whole fruits and vegetables represents a small fraction of total intake and is associated with essential fiber and micronutrients, posing no significant risk. However, the high density of added fructose in processed foods and beverages provides a clear mechanistic link to the progression of diabetes and cardiovascular disease. Therefore, based on the mechanistic pathways delineated in this review, reducing the intake of concentrated added fructose remains a primary strategy for mitigating metabolic dysfunction. This synthesis highlights how cross-organ pathways drive disease progression, providing a biological basis for dietary modifications emphasizing whole-food consumption and reducing refined sugars.
ATP, Adenosine Triphosphate; AMPD2, AMP Deaminase-2; CPT1A, Carnitine Palmitoyltransferase-1A; HFCS, High-Fructose Corn Syrup; IR, Insulin Resistance; KHK-C, Ketohexokinase C; MASLD, Metabolic dysfunction-associated steatotic liver disease; MASH, Metabolic dysfunction-associated steatohepatitis; NAFLD, Non-Alcoholic Fatty Liver Disease; NASH, Non-Alcoholic Steatohepatitis; RAGE, Receptor for Advanced Glycation End Products; RAS, Renin-Angiotensin System; ROS, Reactive Oxygen Species; SSB, Sugar Sweetened Beverage; TG, Triglycerides.
ESS: Conceptualization, Literature Review, Writing—Original Draft; NG: Supervision, interpretation of data, Writing—Review & Editing, Project Administration; MN: Data Collection, Visualization, Methodology; APM: Literature Search, Figures Preparation; AR: Data Analysis, Technical Support; SS: Conceptualization, Supervision, Funding Acquisition, Writing—Review & Editing, Corresponding Author. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The author(s) would like to thank their university for providing technical support throughout the course of the study.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.