






 , Letícia Coelho Montagna 1, Laíza Manfroi 1, Yan de Oliveira Laaf 1, Luigi Ferrazza Maiochi 1, Marcus Adonai Castro da Silva 1, André Oliveira de Souza Lima 1,*
, Letícia Coelho Montagna 1, Laíza Manfroi 1, Yan de Oliveira Laaf 1, Luigi Ferrazza Maiochi 1, Marcus Adonai Castro da Silva 1, André Oliveira de Souza Lima 1,*
1 Polytechnic School, University of Vale do Itajaí (Univali), Itajaí, SC 88302-202, Brazil
Abstract
Enhanced biological phosphorus removal (EBPR) systems utilize phosphorus-accumulating organisms (PAOs) to remove phosphorus from wastewater since excessive phosphorus in water bodies can lead to eutrophication. This study aimed to characterize a newly isolated PAO strain for its potential application in EBPR systems and to screen for additional biotechnological potential. Here, sequencing allowed for genomic analysis, identifying the genes and molecules involved, and exploring other potentials. Additionally, assessing the phosphorus removal performance of the PAO strain in common effluents is essential for its potential application in large-scale systems.
A strain designated LAMA1607 was isolated from activated sludge and selected based on its ability to remove total phosphate from the culture medium. Genomic DNA was extracted and sequenced using the Illumina NovaSeq 6000 platform. Assembly and annotation were performed using CLC Genomics Workbench v.24.0 (QIAGEN®) and Rapid Annotation using Subsystem Technology (RAST)/Pathosystems Resource Integration Center (PATRIC) server tools. Functional prediction of uncharacterized proteins was completed using PHYRE2, and secondary metabolite identification was performed using antiSMASH. Further, additional enzymes with biotechnological applications were manually curated through the Association of Manufacturers and Formulators of Enzyme Products (AMFEP) list. The phosphorus removal capability was assessed in domestic and fishery effluents under enriched and unenriched conditions, where pH, microbial growth, and total phosphorus were monitored over 48 hours.
The genome sequence comprised 5,234,874 bp divided into 20 contigs, 5540 coding sequences, and a GC content of 38.0%; subsequently, LAMA1607 was identified through Basic Local Alignment Search Tool (BLAST) analysis as Priestia megaterium. Genome annotation revealed 27 genes potentially involved in phosphorus removal, including eight encoding transport proteins, three regulatory proteins, twelve enzymes, and others related to phosphorus incorporation and polyphosphate (polyP) granule formation. Moreover, other enzymes of interest were identified, such as hydrolases, lipases, proteases, and amylases, alongside secondary metabolite gene clusters, such as Non-ribosomal peptide synthetase-independent siderophore. P. megaterium LAMA1607 effectively removed up to 70% of the total phosphorus from the fishery effluent.
Genomic analysis suggests that P. megaterium LAMA1607 possesses the mechanistic functions for phosphorus uptake, transport, and storage while also identifying additional biotechnologically relevant enzymes and capabilities. Meanwhile, tests on the effluent demonstrated significant phosphorus removal. These findings support the biotechnological potential and application of P. megaterium LAMA1607 in EBPR systems.
Graphical Abstract
Keywords
- eutrophication mitigation
- microbial phosphorus metabolism
- genomic analysis
- polyphosphate accumulation
- effluent bioremediation
Eutrophication is the excessive growth of primary producers and aquatic plants in a body of water at levels considered to cause interference with the desired use [1]. Eutrophication results from nutrient oversupply in water due to human activity, whereby the nutrients, mainly phosphorus and nitrogen, subsidize the growth of primary producers; when these nutrients are not limiting factors, the excessive growth of the producers is triggered. Consequently, algal, cyanobacterial, and aquatic macrophytes bloom, which has effects on human health, elevates the concentration of organic matter, restricts fishing and recreational activities, causes changes in marine biodiversity and alterations in the composition of species, and promotes variations in water salinity [2].
The enhanced phosphorus biological removal (EBPR) utilizes phosphorus-accumulating organisms (PAOs), microorganisms that store polyphosphate granules during water treatment when this nutrient is in a surplus [3]. In effluents, phosphorus is found in both organic and inorganic forms, with the latter being divided into polyphosphates and orthophosphates. The simplest form for microbial metabolism to assimilate is orthophosphate.
To understand the phosphorus removal capacity of the organism and other characteristics of interest, it is useful to determine the genome and the genes of the organism that are involved in the process. Thus, genomic analysis and gene annotation are the basis of prospecting molecules and metabolic pathways. When the prospecting and testing phases are completed, the obtained genetic information, including the mechanisms of action and removal rates, can guide future research and industrial use. Moreover, investigating other metabolic capabilities is useful for determining further biotechnological uses, addressing secondary metabolite production, and other industrially relevant enzymes.
Considering the addressed topics, this manuscript aimed to isolate and identify the microorganism of interest, LAMA1607, and the proteins and genes related to the phosphorus removal processes, thereby distinguishing the mechanisms through which LAMA1607 functions. Additionally, experiments were performed to obtain practical results in two different effluents: domestic and fishing industries. Thus, the results are an extensive analysis of the potential of microorganisms for EBPR use in wastewater treatments. Annotations of biosynthetic gene clusters and manual curation of biotechnologically relevant enzymes were also performed to explore the genomic potential of LAMA1607 further.
This study identifies and characterizes a newly isolated strain of Priestia megaterium with significant biotechnological potential for EBPR and provides a detailed genomic analysis revealing novel genes and metabolic pathways involved in phosphorus removal. The innovation of this work lies in the combination of practical testing in different effluents with the exploration of the organism’s genomic potential, thereby creating new possibilities for industrial applications in wastewater treatment systems.
Microbial strains were isolated from activated sludge samples collected at two distinct wastewater treatment plants. The first sample originated from domestic sewage treatment and was collected after complete wastewater treatment. The second sample was retrieved directly from the effluent treatment system of a textile industry before the clarification tank. Both samples were collected using sterilized vials and refrigerated until processing. The samples underwent decimal serial dilution with peptone water (0.1%) (Merck, Darmstadt, Germany) to a final concentration of 10-5 cells for microbial isolation. The samples were then inoculated in duplicate onto R-2A agar plates (Difco, Detroit, MI, USA) and incubated at 30 °C for 5 days. Colonies exhibiting distinct morphologies from the initial plates were selected for further replication, resulting in pure cultures.
The previously isolated microorganisms were cultivated in 10 mL of liquid R2A
medium for 48 hours to analyze phosphate granule production. The R2A medium used
in this study was composed of (per liter) 0.5 g yeast extract, 0.5 g proteose
peptone, 0.5 g casamino acids, 0.5 g glucose, 0.3 g dipotassium phosphate
(K2HPO4), 0.3 g magnesium sulfate (MgSO4
The effluent samples used for phosphorus removal testing were collected from two distinct origins: a local fish-processing industry and domestic wastewater treatment. Samples were refrigerated until filtration through 47 mm glass microfiber filters (Millipore, Burlington, MA, USA) and subsequent sterilization. A pre-culture medium and inoculum of the previously isolated LAMA1607 strain were prepared for the experiments. Pre-culture vials were prepared in 125 mL Erlenmeyer flasks containing 25 mL of nutrient broth and grown at 30 °C for 48 hours. The effluent samples (300 mL each) were divided into two duplicates: one enriched with glucose (0.5%) and another non-enriched. Each replicate was inoculated with 1 mL of the pre-culture medium and incubated at 30 °C with agitation at 150 rpm. A control vial containing 100 mL of sterile effluent sample was also prepared. During growth, pH, microbial growth, and orthophosphate concentration were monitored at 0 h, 24 h, and 48 h intervals.
Phosphorus was quantified using colorimetric analysis of vanadomolybdophosphoric acid, following Standard Methods 4500-P [1]. Solutions A and B were prepared as follows. For solution A, 25 g of ammonium molybdate was dissolved in 300 mL of deionized water. Solution B was prepared by dissolving 1.25 mg of ammonium metavanadate in deionized water by boiling, cooling, and then adding 330 mL of HCl. Solutions A and B were then mixed and diluted to 1 L. The pH was adjusted to 7.0 according to the Standard Methods [1]. A standard phosphate solution was prepared by dissolving 2196 mg of KH2PO4 in 1 L of deionized water. A calibration curve was constructed using five different concentrations of the standard phosphate solution (4, 7, 13, 16, and 18 µM). Absorbance readings were measured at a wavelength of 460 nm (see Supplementary Material for details). Sample replicates were prepared for analysis by mixing 35 mL of the sample with 10 mL of the colorimetric reagent and adjusting the final volume to 50 mL using deionized water. Absorbance was measured at 460 nm. Microbial growth was monitored by measuring absorbance at 600 nm. A calibrated pH meter probe (Mettler Toledo, Zurich, Switzerland) was used for all pH measurements. All readings were performed at the same predetermined time intervals. Experimental graphics were generated using the ggplot2 v.3.5.1 package in R (R Project for Statistical Computing, Vienna, Austria; https://ggplot2.tidyverse.org) [2].
Genomic DNA was extracted from the isolated bacteria using the commercial DNeasy Blood and Tissue kit (QIAGEN®, Hilden, North Rhine-Westphalia, Germany) following the manufacturer’s instructions. The 16S rRNA gene was amplified by polymerase chain reaction (PCR) using the universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) under standard amplification conditions (Tm 55 °C, 35 cycles). The amplified PCR products were purified and sequenced at Macrogen sequencing company (Seoul, Korea) using the ABI 3100 automated sequencer (Applied Biosystems, Foster City, CA, USA) with BigDye Terminator kit v. 3.1 (Applied Biosystems, Foster City, CA, USA). The obtained sequences were edited using CLC Genomics Workbench v.24.0 (QIAGEN®, Redwood City, CA, USA; https://digitalinsights.qiagen.com) and compared with 16S ribosomal RNA sequences (Bacteria and Archaea) deposited in the National Center for Biotechnology Information (NCBI) database through BLASTn searches (version 2.13.0+, National Center for Biotechnology Information, Bethesda, MD, USA; https://blast.ncbi.nlm.nih.gov). This comparison aimed to identify sequences of type strains most closely related to the isolated bacteria’s 16S rRNA gene sequence. Operational taxonomic units (OTUs) are groups of closely related sequences with high similarity. This study used a 97% sequence similarity threshold to define OTUs. Based on the results of the BLASTn search, the genus Priestia was chosen for phylogenetic tree construction. Reference 16S rRNA sequences were retrieved from the NCBI RefSeq database and aligned using ClustalW (version 2.1, European Bioinformatics Institute, Hinxton, UK; https://www.ebi.ac.uk) [5]. The phylogenetic tree was then constructed using the maximum likelihood method (HKY+G model) with 500 bootstrap replications in MEGA 11 (Molecular Evolutionary Genetics Analysis software, version 11, Tokyo, Japan; https://www.megasoftware.net) [6]. Halobacillus alkaliphila was employed as an outgroup to root the tree. The 16S rRNA gene and whole-genome sequences obtained in this study will be deposited in the NCBI GenBank upon publication under BioProject accession no. PRJNA1112526.
The complete genome of LAMA1607 was sequenced using the NovaSeq® 6000 sequencing platform (Illumina, San Diego, CA, USA) at Macrogen, Seoul, South Korea. The sequencing employed the shotgun sequencing approach with 151 bp paired-end reads. An iterative optimization process was used to generate the most accurate assembly possible. Paired-end reads from the filtered sequencing were evaluated based on quality scores (minimum Q20 or Q40) and read length (minimum 70 bp). Multiple de novo assemblies were performed using CLC Genomics Workbench v.24.0 (QIAGEN®, Redwood City, CA, USA; https://digitalinsights.qiagen.com) with varying word size parameter values (21 to 61). Only contigs exceeding 4000 bp were retained after each assembly. The reads were then mapped back to the contigs generated in the assembly process. Different length fraction (0.5 to 0.9) combinations and similarity fraction (0.6 to 0.9) parameters were tested during mapping. All resulting assemblies were compared based on the number of contigs and the total data volume. The assembly with the optimal balance of these factors was chosen for subsequent annotation.
The selected assembly was annotated using two automated pipelines: Pathosystems Resource Integration Center (PATRIC) [7] and Rapid Annotation using Subsystem Technology (RAST) [8] with the ClassicRAST annotation module. The resulting annotations were screened for the presence of “hypothetical proteins”. These proteins were submitted to the PHYRE2 server [9] for further annotation based on predicted protein structure homology. Proteins with a confidence score exceeding 60% were considered reliable for assigning additional functional annotations, following established protocols [10], and screening manually for other enzymes with industrial applications and usefulness in effluent treatment. Furthermore, secondary metabolite biosynthetic gene clusters were identified and annotated by antiSMASH version 7.0 (EMBL-EBI, Hinxton, UK; https://antismash.secondarymetabolites.org) using strictness ‘relaxed’ [11]. Finally, the entire protein list underwent manual curation using the NCBI Conserved Domain Search and SmartBLAST tools to confirm the additional annotations. The tbl2asn program was then used to prepare the annotated genome for submission to GenBank.
Following genome annotation, a set of genes encoding enzymes and other proteins potentially involved in LAMA1607’s phosphorus removal mechanisms and polyphosphate (PolyP) granule formation were analyzed. This analysis focused on genes associated with various aspects of phosphorus metabolism, including phosphorus transport, which facilitates the uptake of this essential nutrient across the cell membrane; phosphorus incorporation, encompassing genes involved in the intracellular storage of phosphorus; phosphatases and other enzymes, playing crucial roles in the breakdown or modification of phosphorus-containing compounds; regulatory genes, which control the expression of different genes in the pathway, ensuring a coordinated response to cellular phosphorus requirements. By analyzing these functional categories of genes, a conceptual model for the phosphorus removal mechanisms of LAMA1607 was constructed based on the available genomic data.
Sixty microorganisms exhibiting diverse morphologies were isolated from the two inoculated wastewater samples (domestic and textile industry) onto R2A agar plates. These samples were split equally between the two sources (30 isolates each). Following initial isolation, 17 colonies were excluded due to contamination. The remaining 43 isolates (23 from domestic wastewater and 20 from textile wastewater) were evaluated using Neisser’s staining to assess the presence of polyphosphate inclusions. This analysis revealed that 65% of isolates from domestic wastewater and 85% from textile wastewater stained positive for polyphosphate (Fig. 1A).
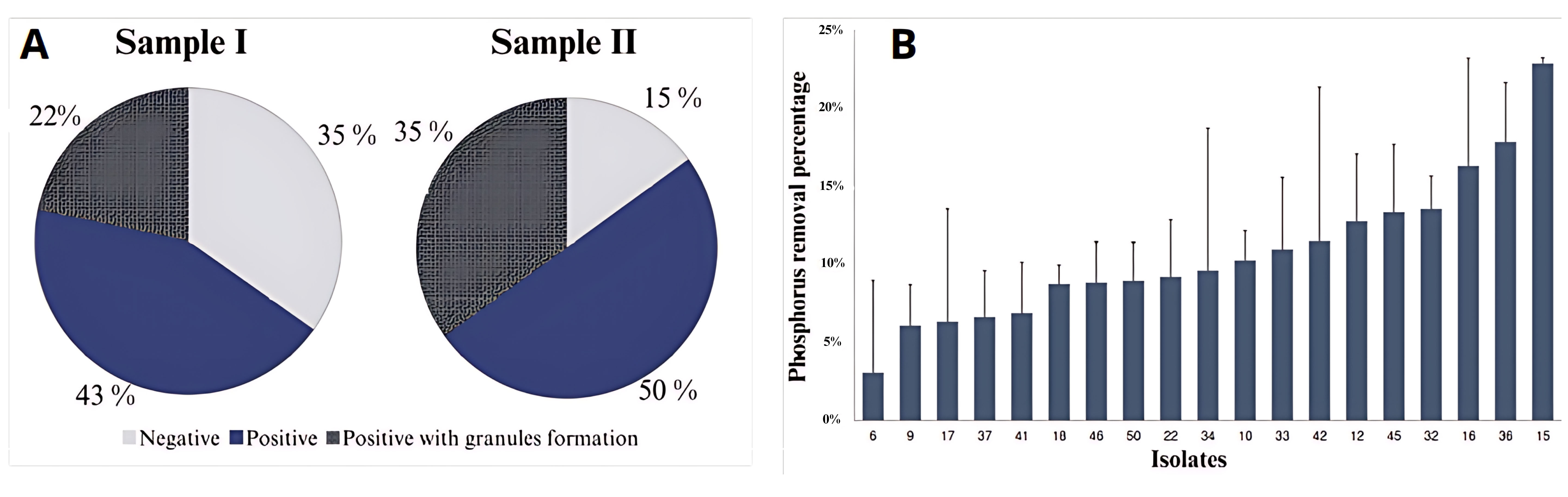 Fig. 1.
Fig. 1.
Preliminary phosphorus removal results. (A) Percentages of total isolates based on polyphosphate accumulation and the presence of polyphosphate granules, as determined by Neisser’s staining method, in domestic (Sample I) and textile industry effluents (Sample II). (B) Isolates can remove phosphorus and their respective phosphorus removal percentages after 48 hours of incubation.
Subsequently, the inorganic phosphorus removal capacity of isolates stained positive for polyphosphate inclusions was assessed. Among the 43 isolates tested, 19 exhibited significant phosphorus removal activity. Notably, isolates from textile industry wastewater displayed a higher removal rate. In total, 10 of the 17 polyphosphate-positive isolates from this source were capable of phosphate removal, whereas only 8 of the 15 positive isolates from domestic wastewater demonstrated the same ability (Fig. 1B). Interestingly, the remaining 13 organisms displayed increased phosphate concentrations after the 48-hour incubation period, suggesting that not all isolates with detectable polyphosphate inclusions possessed phosphate accumulation capabilities. For further characterization, isolate 15 from domestic wastewater was chosen due to its demonstrably high phosphate removal efficiency (Fig. 1B). This isolate was subsequently designated LAMA1607 and served as the target organism for the remainder of this study.
Regarding total phosphate removal, the domestic effluent experiment did not achieve significant removal after 48 hours, with no variations observed between the enriched and unenriched sets (Fig. 2). Conversely, the enriched fish effluent experiment displayed a 70% reduction in initial phosphate concentration by 48 hours, while the non-enriched set exhibited a 30% reduction (Fig. 2A). Microbial growth patterns differed between enriched and unenriched sets across both effluent types (Fig. 2B). In the domestic effluent experiment, the enriched set exhibited a rapid growth spurt in the first 24 hours, followed by a decline (decay phase), which suggests the utilization of readily available carbon. In the fish effluent experiment, the enriched set displayed the most pronounced growth during the first 30 hours (exponential phase), followed by a plateau (stationary phase). Conversely, the non-enriched set in both experiments exhibited a lag phase initially, followed by a slower growth phase (exponential phase). The pH of the culture medium differed between enriched and unenriched sets across both effluent types (Fig. 2C). In the domestic effluent experiment, the enriched set exhibited minimal variations throughout the incubation period. The non-enriched set also displayed minimal variations, with a slight increase in pH after 24 hours. Conversely, the fish effluent experiment revealed a significant decrease in pH in the enriched set, dropping from an initial value of 7.0 to 3.78 by the end of the experiment. The non-enriched fish effluent set exhibited a slight increase in pH (Fig. 2C).
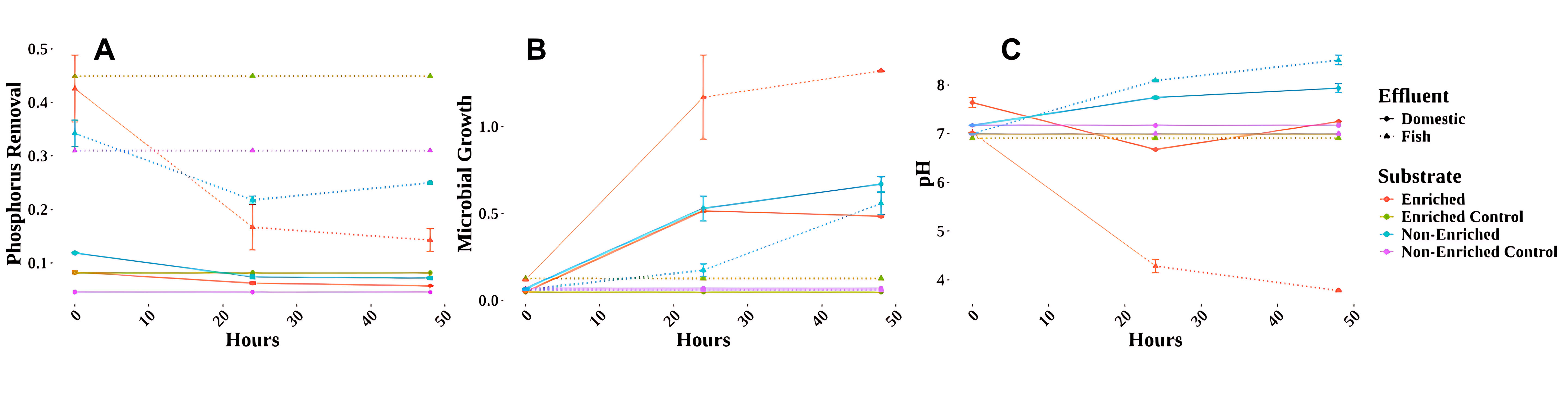 Fig. 2.
Fig. 2.
Phosphorus removal, microbial growth, and pH results for fish and domestic effluent. Measurements were performed at 0 h, 24 h, and 48 h while considering a glucose-enriched substrate and a non-enriched substrate, as well as a control for both variables. Samples were divided between the effluent source (domestic sewage treatment and fish industry wastewater). (A) Phosphorus removal: Absorbance measured at a wavelength of 460 nm. (B) Microbial growth: Absorbance measured at a wavelength of 600 nm. (C) pH measurements.
By analyzing the 16S rRNA BLAST sequences and the phylogenetic tree (Fig. 3), LAMA1607 was identified as Priestia megaterium since this species showed the highest sequence similarity to LAMA1607 in the analysis.
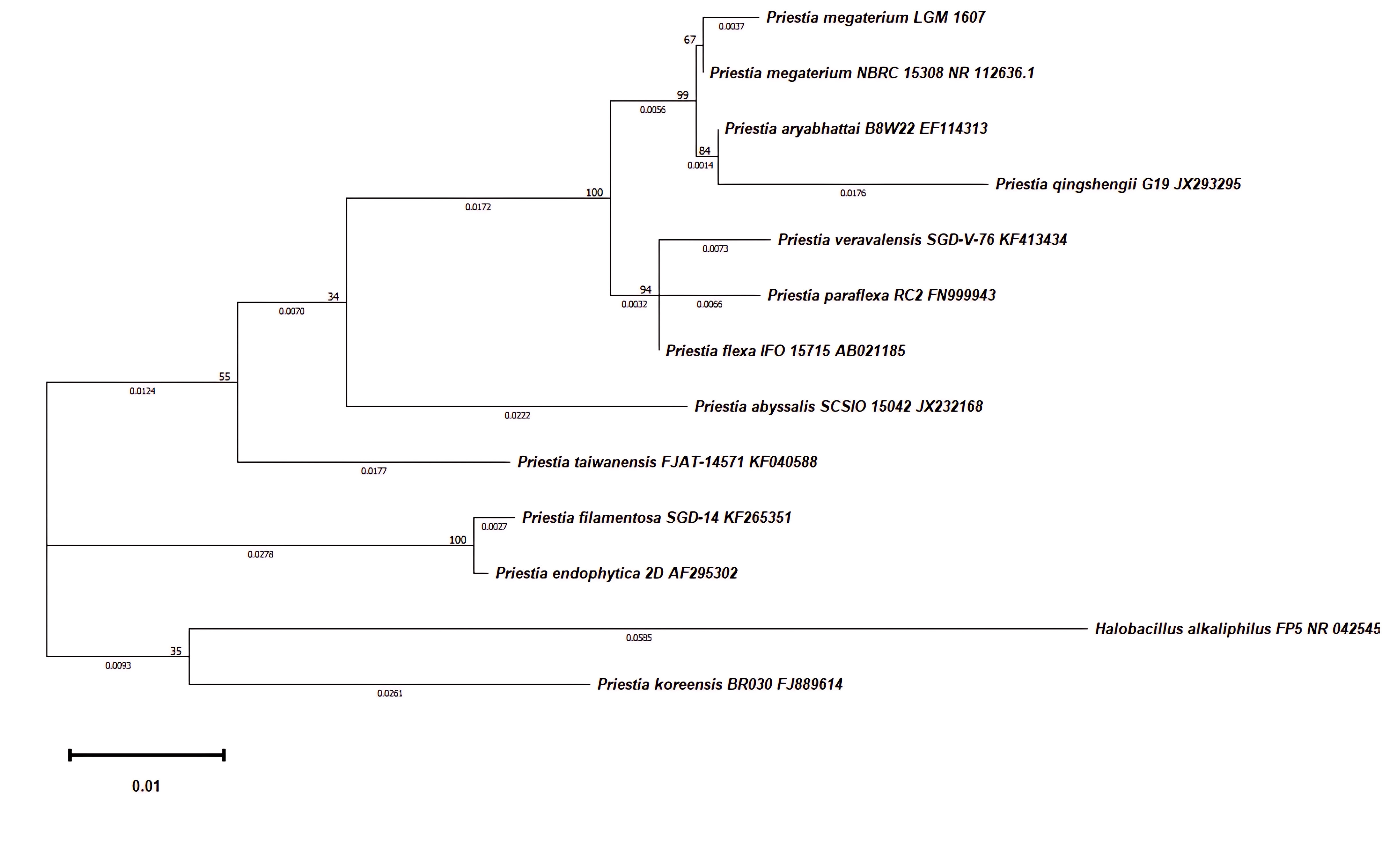 Fig. 3.
Fig. 3.
Phylogenetic tree of Priestia megaterium LAMA1607. Maximum-likelihood tree showing the phylogenetic relationships of 16S rRNA gene sequences of P. megaterium LAMA1607. Bootstrap 500 replicates.
Genome assembly using CLC Genomics Workbench resulted in 5,234,874 bp, divided into 20 contigs, with an L50 of 3, an N50 of 1,017,811, and a GC content of 38.0%, summarized in Table 1. The RAST software presents the genome annotation divided into subsystems and pathways, which are already known by their role (Fig. 4). In total, 5540 coding sequences (CDs) were identified, forming 325 subsystems and 79 RNAs. A portion of 76% of the genes are not associated with them, while 24% are. The most common subsystems were amino acids and derivatives (360), carbohydrates (313), protein metabolism (195), cofactors, vitamins, prosthetic groups, pigments (160), and nucleosides and nucleotides (109). A total of 19 CDs were associated with the phosphorus metabolism subsystem.
| Item | Description |
| Sequencing platform | NovaSeq® 6000 (Illumina) |
| Genome status | WGS |
| Genome quality | Good |
| Coarse consistency | 99.6 |
| Fine consistency | 98.1 |
| Contigs | 20 |
| Assembly | RAST and PATRIC |
| Size (bp) | 5,234,874 |
| GC content (%) | 38.0 |
| Number of coding sequences | 5540 |
| Number of RNAs | 79 |
RAST, Rapid Annotation using Subsystem Technology; PATRIC, Pathosystems Resource Integration Center; WGS, whole-genome sequencing.
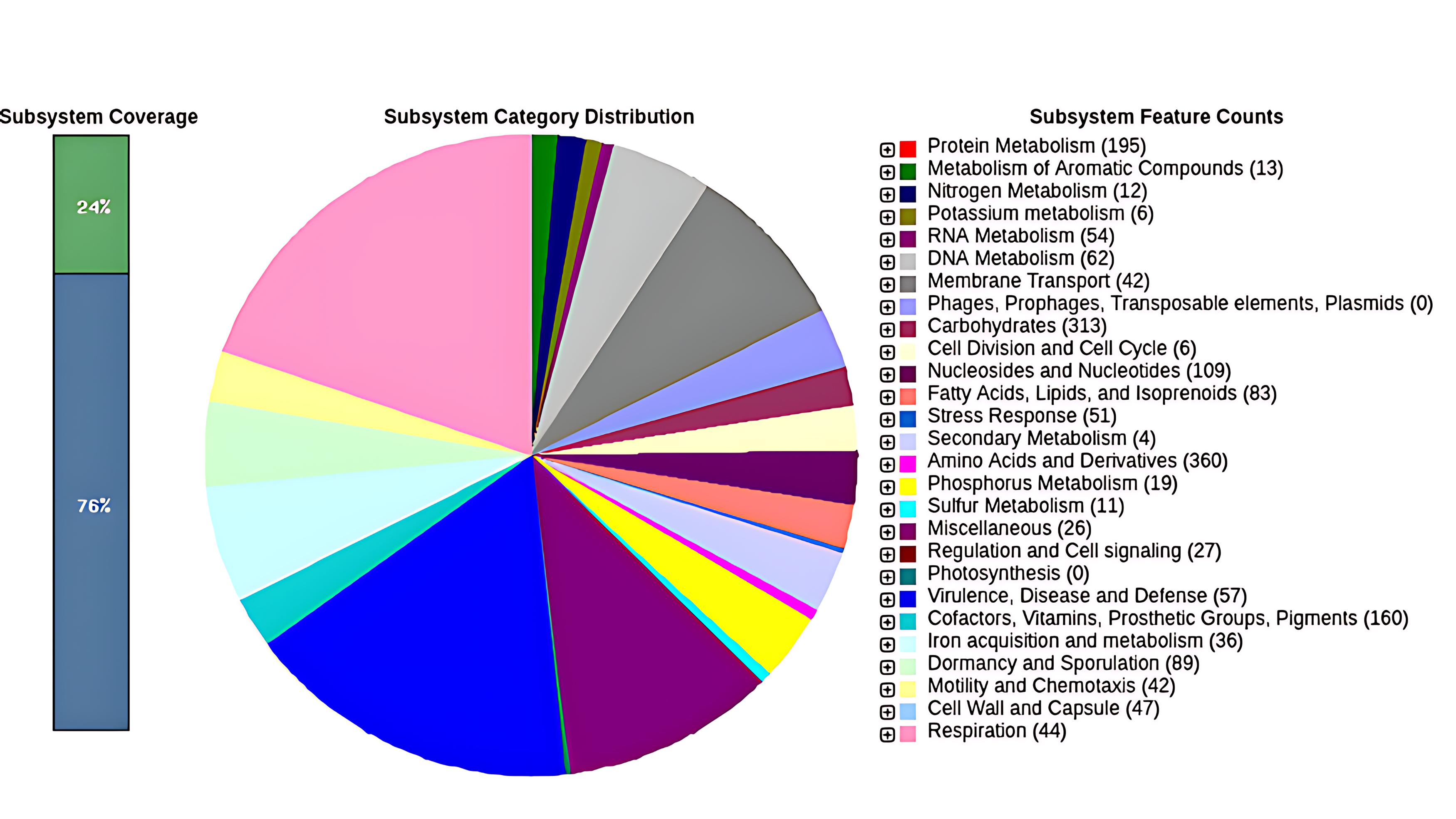 Fig. 4.
Fig. 4.
Subsystems identified in Priestia megaterium LAMA1607. Overview of the subsystem categories assigned to the P. megaterium LAMA1607 genome. A pie chart was generated using the Rapid Annotation using Subsystem Technology (RAST) server (blue); 76% of genes are related to the identified subsystems features, while 24% are not (green).
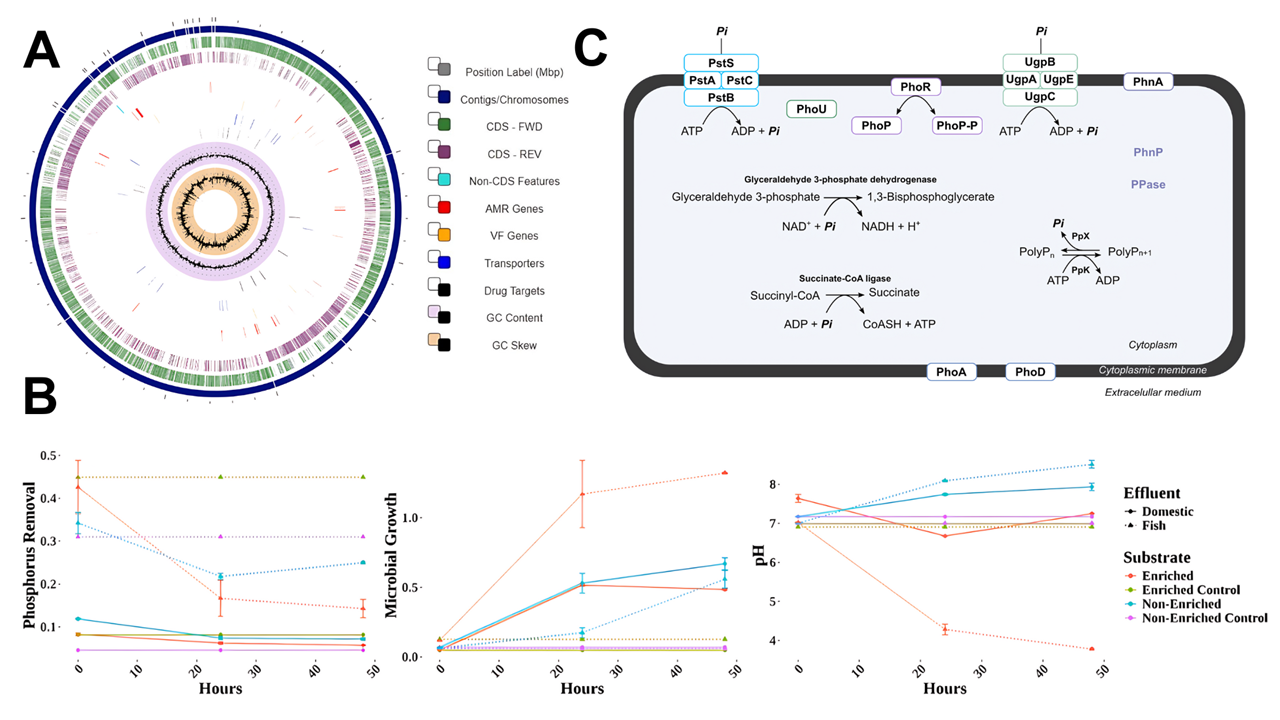
Additionally, the antiSMASH server [11] annotation identified eight biosynthetic gene clusters responsible for secondary metabolites, including NI-siderophore, phosphonate, RRE-element containing, type III PKS, and tRNA-dependent cyclodipeptide synthase (Fig. 5B). Table 2 summarizes the 15 enzymes found after manual curation using the Enzyme Commission (EC) number, based on the list of Association of Manufacturers and Formulators of Enzyme Products (AMFEP), which lists the main enzymes with biotechnological applications.
| Enzyme | Enzyme commission | Size in aa | Applications in | ||
|---|---|---|---|---|---|
| Food | Feed | Technical | |||
| Alpha-acetolactate decarboxylase | 4.1.1.5 | 257 | Yes | ||
| Alpha-amylase | 3.2.1.1 | 633 | Yes | Yes | Yes |
| Alpha-glucosidase | 3.2.1.20 | 648 | Yes | ||
| Aryldialkylphosphatase | 3.1.8.1 | 293 | Yes | ||
| ATP-dependent protease La type I | 3.4.21.53 | 657 | Yes | Yes | Yes |
| Beta-amylase | 3.2.1.2 | 548 | Yes | ||
| Carboxyl-terminal protease | 3.4.21.102 | 477 | Yes | Yes | Yes |
| Catalase KatE | 1.11.1.6 | 541 | Yes | Yes | |
| Catalase KatE-intracellular protease | 1.11.1.6 | 581 | Yes | Yes | |
| Glutaminase | 3.5.1.2 | 333 | Yes | ||
| Glycogen branching enzyme | 2.4.1.18 | 592 | Yes | ||
| Lysophospholipase | 3.1.1.5 | 228 | Yes | Yes | |
| Manganese catalase | 1.11.1.6 | 218 | Yes | Yes | |
| Protein-glutamine gamma-glutamyltransferase | 2.3.2.13 | 246 | Yes | ||
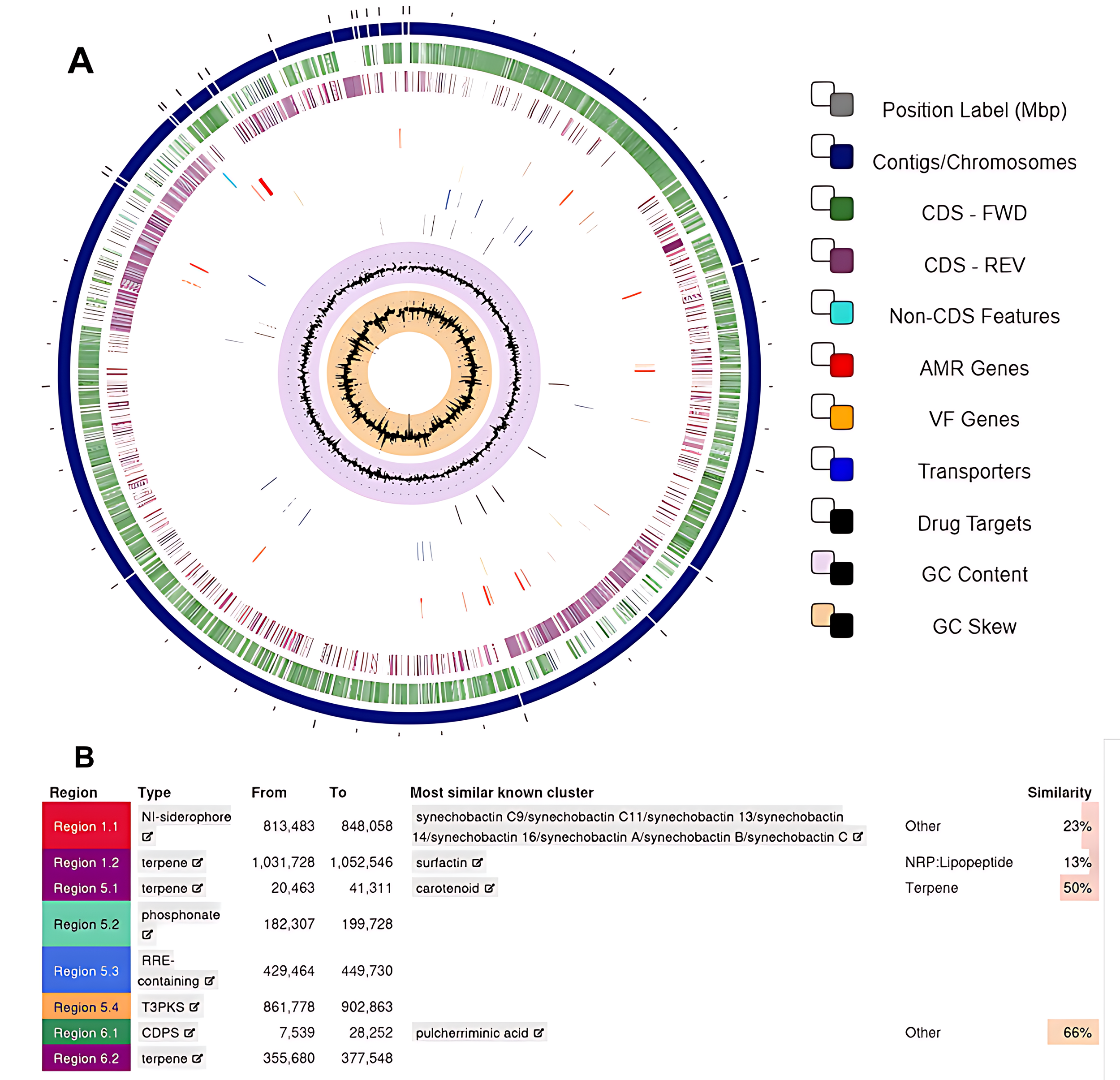 Fig. 5.
Fig. 5.
Circular view of the genome and biosynthetic gene clusters. (A) A circular view of the genome assembly was generated using PATRIC software. Data from outermost to innermost: contigs (dark blue); forward coding sequences (green); reverse coding sequences (purple); antimicrobial genes (red); virulence factors genes (orange); transporters (blue); drug targets (black); GC content (lilac); GC skew (orange). (B) Gene clusters responsible for synthesizing secondary metabolites were identified using the antiSMASH server. CDS-FWD, Coding Sequences - Forward Strand; CDS-REV, Coding Sequences - Reverse Strand; AMR, Antimicrobial Resistance Genes; VF, Virulence Factors.
The PATRIC server annotation also illustrated 5540 CDs, divided into 3814 proteins with functional assignments, 1726 hypothetical proteins, and 79 RNAs. From these 3814 proteins, 1448 were assigned to subsystems, and 885 were assigned to pathways. The most common subsystems were ribosomal proteins, single-copy (42), ribosome large subunit (LSU), bacterial (36), flagellum (34), cell division initiation-related clusters (28), spore germinant receptors (27), and glycerolipids and glycerophospholipids (27). Furthermore, 95 infectious disease-related genes were identified (Fig. 5A).
In total, 27 phosphorus removal-related genes were found in the P. megaterium LAMA1607 genome; of these, eight encode transport proteins, three encode regulatory proteins, twelve encode enzymes, two encode enzymes related to phosphorus incorporation, and two encode enzymes related to polyphosphate (polyP) granules (Table 3).
| Gene | Function | Description |
|---|---|---|
| PhnA | Enzyme | Alkylphosphonate utilization operon protein |
| PhoA | Alkaline phosphatase A | |
| PhoD | Phosphodiesterase/alkaline phosphatase D | |
| PhnP | Phosphodiesterase | |
| PpaC | Pyrophosphate phospho-hydrolase Ppase | |
| PpK | PolyP granules | Polyphosphate kinase |
| PpX | Exopolyphosphatase | |
| Gap | Pi incorporation | Glyceraldehyde 3-phosphate dehydrogenase |
| SucC | Succinyl-CoA ligase | |
| PstA | Transport | Phosphate ATP-binding cassette (ABC) transporter, permease protein |
| PstB | Phosphate ABC transporter, ATP-binding protein | |
| PstC | Phosphate ABC transporter, permease protein | |
| PstS | Phosphate ABC transporter, substrate-binding protein | |
| UgpA | Glycerol-3-phosphate ABC transporter, permease protein | |
| UgpB | Glycerol-3-phosphate ABC transporter, substrate-binding protein | |
| UgpC | SN-glycerol-3-phosphate transport ATP-binding protein | |
| UgpE | Glycerol-3-phosphate ABC transporter, permease protein | |
| PhoP | Regulatory | Transcriptional regulatory protein PhoP |
| PhoR | Phosphate regulon sensor protein | |
| PhoU | Phosphate transport system regulatory protein |
When comparing the two sets of replicates across effluent samples (enriched and non-enriched), the enriched sample obtained better phosphorus removal rates in both cases. However, this can be easily explained by the metabolic phosphate demand during substrate utilization. These phosphorus removal results also correlate with the genetic findings of P. megaterium LAMA1607.
Further investigation also showed that phosphate utilization was higher in the effluent from the fish processing industry. This evidence is supported by the presence of volatile fatty acids (VFAs), an essential nutrient for phosphate utilization in EBPR systems [12, 13]. Considering the amino acid-rich substrate provided by the fish effluent, the drastic drop in pH also suggests a possible metabolic utilization alongside glucose. There was a minor increase in pH in the non-enriched set, indicating a potential uptake of carbonic acids, such as acetate, in the absence of glucose. This is supported by earlier evidence showing the importance of VFAs in EBPR systems [14, 15]. Furthermore, the low phosphate removal and low content of VFAs in the domestic effluent might have affected these results. However, despite low phosphate removal, significant growth can still be observed in the enriched and domestic effluent samples. This can be explained by the presence of genes responsible for adaptability to low phosphorus availability in the substrate, detected through gene annotation. The non-enriched set registered higher pH readings after 24 hours than the fish effluent samples, which is further evidence of the utilization of carbonic acids, as mentioned before.
Gene annotation has elucidated that P. megaterium LAMA1607 can interact with phosphorus in different ways and displayed other biotechnologically relevant enzymes for effluent treatment and industrial use, including hydrolytic enzymes, lipases, proteases and amylases (Table 3), alongside biosynthetic gene clusters of interest, such as NI-siderophore (Fig. 5B). These findings demonstrate a genetic repertory of products of interest for biotechnological applications, available for cloning and further use in other works [10, 16] as they are not only significant for the existence of the organism itself but also useful for effluent treatment. Moreover, the genome size and characteristics of LAMA1607 resonate with previous bacterial genomes that possessed biotechnological potential.
Among the regulatory genes detected (Table 3), three were connected to the phoPR system: a two-component signal transduction system that consists of a histidine kinase, which is a sensory protein (phoR) located in the cytoplasmic membrane, and a cognate response regulator (phoP). The third component is a membrane-associated metal binding protein (phoU) that does not control phoR activity but helps to regulate intracellular phosphate metabolism. Furthermore, a previous study proposed that phoU can inhibit phoR when the pst complex actively transports phosphate, operating as a mediator between the systems [17]. This system controls the Pho regulon and mediates the adaptation under phosphate limitation [18]. The most common members controlled by the phoPR are extracellular enzymes, Pi-specific transporters (such as pstSCAB and ugpBAEC), and enzymes involved in storing and saving the nutrient [19].
Among the transport proteins, four were related to the pstSCAB ABC-type transporter. These consist of a periplasmic protein that binds the phosphate and leads it to the membrane channel (pstS), two transmembrane proteins that establish the transport channel (pstA and pstC), and a dimer whose function is to provide adenosine diphosphate (ADP) for the transport (pstB) [18]. Similar to the pstSCAB operon, the ugpABCE genes encode an ABC transport system for glycerol-3-phosphate and glycerophosphodiester, consisting of two ABC permease transporters (ugpA and ugpE), a periplasmic binding protein (ugpB) and an ATPase (ugpC). The regulation of ugp is mainly phoBR-dependent [20]. Both pstSCAB and ugpBAEC are high-affinity transporters. Thus, in effluents, P. megaterium LAMA1607 can continue to obtain phosphorus even at low exogenous concentrations, growing increasingly compared to other microorganisms without these transporters.
Regarding the enzymes, nine genes were related to alkaline phosphatases, monomeric enzymes that catalyze the hydrolysis of both phosphomonoesters and phosphodiesters [21]. Specifically, two were identified as alkaline phosphatases A and D (PhoA and PhoD). These enzymes are regulated by P availability via the two-component regulatory system phoBR. Phosphatase is considered a key enzyme for utilizing dissolved organic phosphate [22]. In addition to the alkaline phosphatases, an inorganic pyrophosphatase (Ppase) was discovered, which catalyzes pyrophosphate into two orthophosphate molecules [23]. Due to a lack of available orthophosphate in waters, the production of these two groups of phosphatases plays a crucial role in the effluent treatment by breaking complex phosphorus molecules and making Pi ready for the bacteria to assimilate.
Phosphonoacetate hydrolase (PhnA) was also found, homologous to alkaline phosphatases with the exception that it hydrolyzes a carbon–phosphorus bond, converting phosphonoacetate to acetate and inorganic phosphate [24]. Hence, It can be considered an extra source of inorganic phosphorus acquisition for incorporation by the microorganism.
Still among the enzymes, PhnP, a phosphodiesterase with specificity towards ribonucleoside 2′,3′-cyclic phosphates, was found. The phn operon encodes a multienzyme system that enables bacteria to metabolize organophosphonates when the inorganic phosphate is scarce [25]. However, the other operon components were not found in this research, although these genes may not have been present in the part analyzed during the sequencing/procedure. Another explanation could be that it is just an enzyme with a very similar structure to the one of interest (PhnP); therefore, experimental evidence is required to confirm the presence of the desired enzyme in the genome. Nevertheless, this protein can be part of a system that allows the bacteria to assimilate phosphate from organic molecules.
Two enzymes associated with polyphosphate granules were found. One is a polyphosphate kinase (PPK), related to the formation of the polyP granules by catalyzing the conversion from ATP into polyP and ADP [26], and the other one is an exopolyphosphatase (PpX), responsible for the polyP hydrolysis and Pi release [27]. These two enzymes are important for effluent treatment as they are considered crucial enzymes for EBPR from wastewater; they are also closely related to anaerobic phosphorus release and oxic phosphorus uptake [28]. For the storage of Pi, most bacteria induce the expression of PpK, which can accumulate polyphosphate as a Pi reservoir and enable its reuse when necessary [29].
Concerning the proteins related to the incorporation of phosphorus, the enzyme glyceraldehyde 3-phosphate dehydrogenase converts glyceraldehyde 3-phosphate and Pi to 1,3-bisphosphoglycerate, in the first step of glycolysis that leads to ATP formation. The last enzyme targeted was succinyl-coA ligase, which catalyzes succinyl-CoA conversion to succinate. In this reaction, a phosphoryl group is transferred to ADP (or Guanosine diphosphate (GDP)) to form ATP (or Guanosine triphosphate (GTP)) in the citric acid cycle [30]. In these reactions, phosphorus is incorporated by P. megaterium LAMA1607 through the formation and utilization of ATP, providing a function to the inorganic phosphate that has been removed from the effluent. From this analysis of the genomic data, a model has been constructed that contains all the identified mechanisms that may be important for effluent phosphorus removal (Fig. 6).
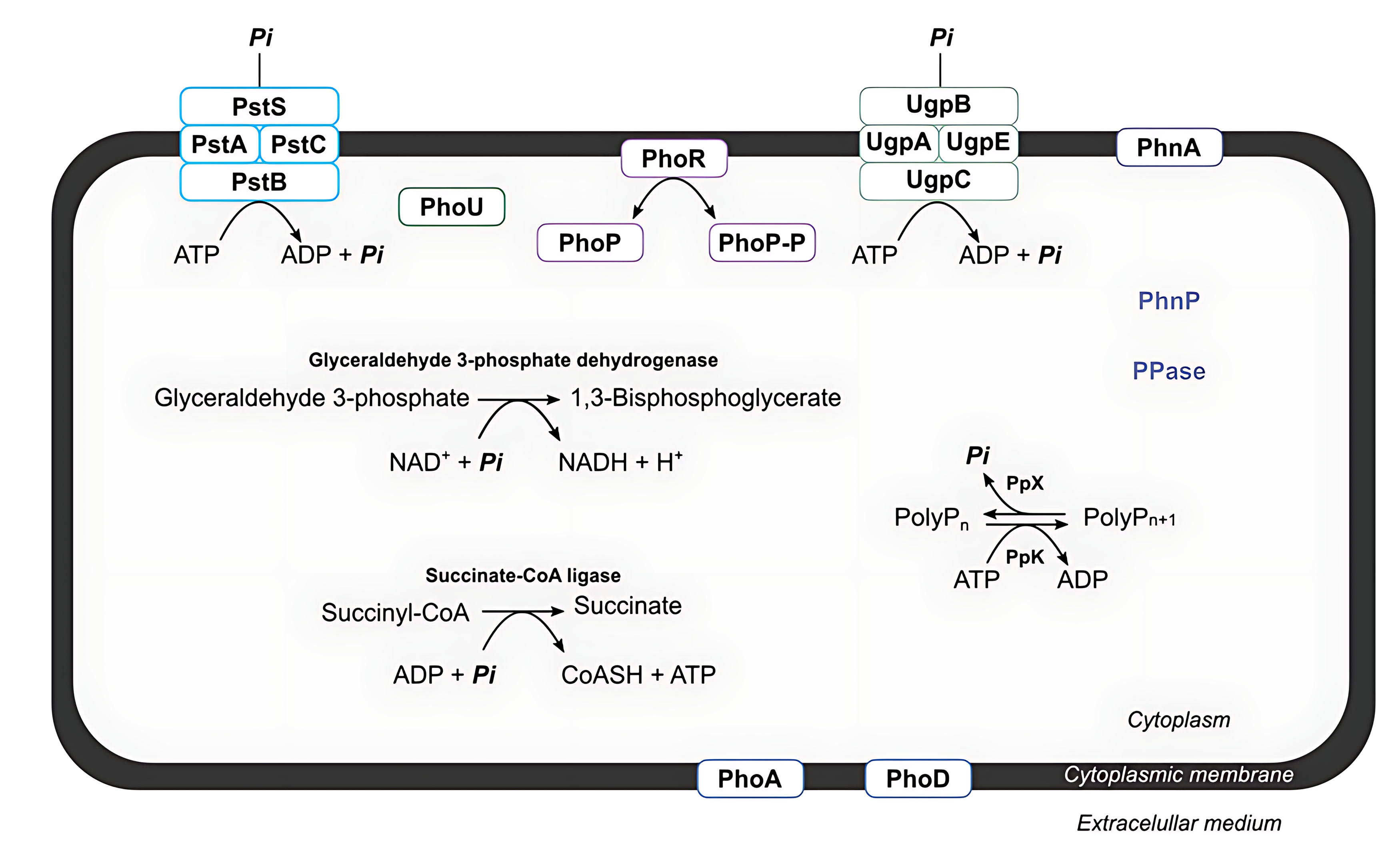 Fig. 6.
Fig. 6.
Proposed model for transport, accumulation, and incorporation of phosphorus in Priestia megaterium LAMA1607. The entire phosphorus metabolism is controlled by the regulatory system PhoPR, which recognizes the presence of phosphorus through interaction with PhoU and activates the transcription of genes that encode proteins responsible for the phosphorus removal mechanisms. Phosphorus is transported from the extracellular medium to the cytoplasm through the PstABCS and UgpABCE transporters. Alkaline phosphatases (PhoA and PhoD) and other enzymes (PhnA, PhnP, and PPase) break down different phosphorus-containing molecules, in turn contributing to the increase in inorganic phosphorus (Pi) in the intracellular environment. The organism incorporates the free Pi in the cytosol with the assistance of the enzymes glyceraldehyde 3-phosphate dehydrogenase and succinyl-CoA ligase. Two-component regulatory system PhoPR (PhoPR), phosphate transport system regulatory protein PhoU (PhoU), alkaline phosphatase A (PhoA), alkaline phosphatase D (PhoD), phosphate ABC transporter PstABCS (PstABCS), glycerol-3-phosphate ABC transporter UgpABCE (UgpABCE), phosphonoacetate hydrolase PhnA (PhnA), phosphodiesterase PhnP (PhnP), inorganic pyrophosphatase Ppase (Ppase), nicotinamide adenine dinucleotide (NAD+), reduced nicotinamide adenine dinucleotide (NADH) and coenzyme A (CoASH).
The genes found in the genome indicate that mechanisms for phosphorus catalysis, transport, accumulation, and incorporation are present in P. megaterium LAMA1607, which may be useful for effluent treatment. Moreover, these results are consistent with the experimental analysis. The observed phosphorus removal rates in different effluents, particularly the higher efficiency in amino acid-rich effluents such as those from the fishing industry, directly correlate with the identified genomic capabilities. This alignment between genomic potential and experimental performance provides strong evidence supporting the biotechnological application of P. megaterium LAMA1607 in EBPR systems. Additionally, other features of biotechnological relevance for industrial purposes and improvement in effluent treatment, such as hydrolases, were identified. Thus, these results demonstrate that P. megaterium LAMA1607 offers potential for future application in EBPR systems and other biotechnological uses.
PAOS, phosphorus accumulating organisms; EBPR, enhanced biological phosphorus removal; PATRIC, pathosystems resource integration center; RAST, rapid annotation using subsystem technology; polyP, polyphosphate; VFAs, volatile fatty acids; ATP, adenosine triphosphate; ADP, adenosine diphosphate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; PpX, exopolyphosphatase; PPase, pyrophosphatase; PpK, polyphosphate kinase; PhnP, phosphodiesterase; PhnA, phosphonoacetate hydrolase; PhoA and PhoD, alkaline phosphatase; EC, Enzyme Commission.
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JBEAFA000000000. The version described in this paper is version JBEAFA010000000. The raw sequencing data have also been deposited in the Sequence Read Archive (SRA) under the accession code SRR29086236. The metabolic model figure is included in the manuscript and will be publicly available as part of the publication. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
MN was responsible for manuscript writing, phosphorus experimental testing and data analysis. LCM conducted DNA extraction protocols, being also involved in manuscript writing and data analysis. LM was responsible for bacterial isolation and pre-selection of organisms with potential for phosphorus accumulation. YL provided the phylogenetic tree analysis, BLAST manual curation and helped with manuscript writing. LFM performed genome assembly and annotation. MS designed the research study and provided proposed model for phosphate utilization. AL generated and analyzed the genomic data and designed the research study. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The microbial strain used in this study is registered in accordance with Brazilian regulations under the Sistema Nacional de Gestão do Patrimônio Genético (SisGen), registration number AAC6A3B. No additional ethical approval is required.
Not applicable.
The authors acknowledge funding from the National Council for Scientific and Technological Development (CNPq) for the support of the Brazilian National Institute of Science and Technology - INCT-Mar COI (Process 400551/2014-4), as well as from the Foundation for Research and Innovation Support of the State of Santa Catarina (FAPESC, Process 2020TR1448). We also wish to thank CNPq for the support provided to AL (Process 312363/2018-4) and the Government of the State of Santa Catarina for the scholarships (Article 170 - IC) provided to MN, LCM, and LM.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbe1604037.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.