










1 Department of Chemistry and Center for Photochemical Sciences, Bowling Green State University, Bowling Green, OH 43403, USA
†These authors contributed equally.
Abstract
While monoclonal antibodies have shown success in cancer immunotherapy, their limitations prompt exploration of alternative approaches such as aptamers and peptides targeting programmed death ligand 1 (PD-L1). Despite the significance of these biotechnological tools, a comprehensive review encompassing both aptamers and peptides for PD-L1 targeting is lacking. Addressing this gap is crucial for consolidating recent advancements and insights in this field. Biotechnological advances leveraging aptamers and peptides represent a cutting-edge approach in refining the targeting proteins. Our review aims to provide valuable guidance for researchers and clinicians, highlighting the biotechnological advances utilizing aptamers and peptides refining PD-L1 targeting.
Keywords
- PD-L1
- peptide
- aptamer
- multiple presentation
In cells, intracellular protein-protein interactions (PPIs) play vital roles. They facilitate the formation of protein-protein complexes that aid signal transduction and facilitate the binding of transcription factors to promoters and enhancers. Pharmacological strategies to inhibit intracellular PPIs have utilized small molecules with molecular weights less than 500 Da and biologics based on proteins with molecular weights greater than 5000 Da. Small molecules are efficient candidates in terms of cell permeability and inhibition of intracellular proteins [1, 2]. But their activity is hindered when a single mutation occurs at a target site, and cancer cells can easily acquire resistance against these drug candidates. Moreover, although the surface interaction between two proteins could be too large to be covered by small molecule candidates [3], there are currently several small molecule inhibitors, targeting the programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) interaction, undergoing clinical trials [4, 5]. On the other hand, large molecules such as antibodies can achieve high selectivity and affinity by binding to a large protein surface, but they face challenges when it comes to cell permeability [6]. Biotechnological advances leveraging aptamers and peptides represent a cutting-edge approach in refining the targeting PPIs.
Aptamers, short single-stranded DNA or RNA molecules [7, 8, 9], and peptides offer precise targeting capabilities due to their high specificity and affinity for proteins. By exploiting these biotechnological tools, researchers can design novel therapeutics capable of modulating the protein-based pathway with enhanced precision and efficacy. Furthermore, the versatility of aptamers and peptides allows for the development of multifunctional agents capable of synergistically targeting proteins while simultaneously delivering payloads such as drugs or imaging agents. In this review, we aim to provide a synthesis of the most recent advancements in biotechnological approaches employing aptamers and peptides for targeting Programmed Death Ligand 1 (PD-L1). These innovative strategies hold significant potential for not only enhancing personalized cancer diagnostics but also revolutionizing treatment strategies. By examining the latest progress in this field, we endeavor to shed light on the promising convergence of biotechnology and immunotherapy. Immunotherapy has become a significant research focus [10]. Presently, various approaches are employed in cancer treatment, including radiotherapy, chemotherapy, and surgery, each adhering to its own set of medication administration standards. However, these methods are constrained by their respective limitations. In contrast, immunotherapy stands out for its notable specificity, binding affinity, and low toxicity in tumor treatment. Significant research efforts have been dedicated to this burgeoning field.
A recent breakthrough in cancer immunotherapy is the immunologic checkpoint blockade [11], among which the programmed cell death protein 1 (PD-1) has been most actively studied [12]. PD-1 is expressed on the surface of activated T cells, B cells and myeloid cells [13, 14]. PD-1 has two natural ligands, PD-L1 and PD-L2 [15, 16]. Engagement of PD-1 with either ligand suppresses immune responses and promotes self-tolerance [15, 16, 17]. PD-L1 and PD-L2 only share 37% sequence identity, but have similar functions and expression profiles [18, 19]. PD-L1 is expressed in immune cells such as T and B cells, dendritic cells, and macrophages, as well as in many different tumor types [20]. PD-L2 expression has been reported to be more restricted to antigen-presenting cells [21]. The overexpression of PD-L1 and/or PD-L2 in cancer cells reduces the body’s immune responses, enabling cancer cells to evade killing mediated by T cells [22, 23]. PD-L1 is expressed more widely by tumor cells than PD-L2, and the blockade of the PD-1/PD-L1 interaction is more frequently targeted by therapeutic agents. The 2018 Nobel Prize in Medicine, to James P. Allison and Tasuku Honjo, acknowledged the research behind this technique [24]. To date, several therapeutic antibodies, targeting PD-1 or PD-L1, have been approved by the FDA, and a few others are currently undergoing clinical trials [25, 26].
While monoclonal antibodies have achieved significant success in clinical trials, it is important to acknowledge that the overall pooled response rates of anti-PD-1/PD-L1 antibodies have been reported to be below 25% [27, 28]. In additional, monoclonal antibodies are expensive with some other shortcomings, such as lower oral bioavailability and poor tumor penetration [29, 30]. Although small-molecule compounds have significant advantages in dealing with these problems compared to antibodies, it is very difficult to design small molecules targeting the PD-1/PD-L1 interaction [18, 31]. The main reason is, due to the large, flat, and hydrophobic areas of the PD-1/PD-L1 interaction surface [32], small molecules usually cannot offer large surface area to break the interaction. Until now, only very limited small molecules show moderate blockade activity of PD-1/PD-L1 binding [33, 34, 35]. On the other hand, nucleic acid aptamers and peptides, which are larger than small molecules but smaller than antibodies, are promising alternatives to target the PD-1/PD-L1 interaction [36]. Numerous endeavors have been undertaken thus far to develop aptamers or peptides as highly sought-after alternatives to antibodies, particularly as PD-1 or PD-L1 inhibitors. PD-1 is expressed in immune cells and PD-L1 is overexpressed in cancer cells; therefore, inhibitors targeting PD-1 may have side effects resulting in immune disorders, while PD-L1 inhibitors may reduce these problems [37, 38]. In addition, therapeutic PD-L1 antibodies have been reported to be more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling [39]. In this review, we will focus on discussing aptamers and peptides against PD-L1.
DNA and RNA aptamers, both classes of nucleic acid molecules, exhibit distinct characteristics that influence their applications in research and therapeutics. This review predominantly focuses on DNA aptamers, with only one RNA aptamer targeting PD-L1 discussed, highlighting the potential challenges in obtaining RNA aptamers. These challenges may stem from factors such as nuclease susceptibility and structural stability. Compared to RNA, DNA aptamers are inherently more stable in biological environments. The absence of the 2′-OH group in DNA makes it less susceptible to hydrolysis and self-cleavage. They are also typically easier and more cost-effective to synthesize, increasing their accessibility for researchers developing novel therapeutics or diagnostic tools. Conversely, RNA aptamers often exhibit higher binding affinity to targets due to the presence of 2′-hydroxyl groups, which can form additional hydrogen bonds with target molecules. This increased affinity can be advantageous in applications where strong target recognition is crucial. RNA aptamers also offer a broader range of target specificity owing to their ability to fold into complex secondary and tertiary structures, potentially allowing for recognition of a wider array of target molecules. The choice between DNA and RNA aptamers ultimately depends on the specific requirements of the intended application, with each class offering unique advantages and considerations. Notably, the lack of reliable 3D structure prediction for single-stranded DNA (ssDNA) led to the modeling of aptPDL-1 as an RNA aptamer using RNAcomposer [40], underscoring the practical differences between working with DNA and RNA aptamers. However, modern techniques are increasingly addressing these challenges, narrowing the gap between these two classes of aptamers.
The evolution of aptamer selection techniques has addressed several challenges in targeted therapeutics development. Traditional Systematic Evolution of Ligands by Exponential Enrichment (SELEX) methods often struggle with achieving high specificity and affinity, leading to the development of innovative variants. Modular-SELEX combines multiple SELEX types to overcome limitations in affinity, specificity, and compatibility [41]. X-Aptamer libraries incorporate modified nucleotides, enhancing chemical diversity and interaction robustness [42]. These approaches help address the fundamental challenge of aptamer degradation by nucleases in the body. To combat nuclease-mediated degradation and improve in vivo stability, researchers have incorporated non-canonical nucleotides into aptamers. For instance, 2’-fluoropyrimidine-RNA aptamers have been developed for triple-negative breast cancer (TNBC) cell recognition, potentially enhancing their stability in biological environments [43]. Similarly, the use of modified dU in X-Aptamer libraries not only improves binding affinity but also contributes to increased nuclease resistance [42]. Bispecific aptamers like Ap3-7c demonstrate how structural modifications can enhance both functionality and stability [44]. REase-SELEX introduces mutations that may contribute to improved aptamer stability and affinity [45]. The loss-gain cell-SELEX strategy focuses on membrane protein targeting, potentially reducing off-target interactions and improving specificity [46]. These advancements in SELEX techniques and aptamer modifications are crucial steps toward developing stable, highly specific aptamers capable of withstanding physiological conditions, thereby increasing their potential for diagnostic and therapeutic applications.
We first explore the landscape of anti-PD-L1 aptamers, detailing their discovery and discussing strategies for enhancing their efficacy, which underscores the promising potential of aptamer-based therapeutics in cancer immunotherapy. A novel DNA aptamer called aptPD-L1 was reported by Lai et al. [40] in 2016 for the first time against PD-L1 protein (Fig. 1, Ref. [40]). The aptamer showed promising results in in vitro lymphocyte proliferation and suppressed in vivo tumor growth without noticeable liver or renal toxicity. The study findings demonstrated the effectiveness of restoring T-cell function and altering the microenvironment of tumors. The aptamer is 45 nucleotides long, which specifically binds to the target PD-L1 with a dissociation constant (Kd) of 4.7 nmol/L compared to bovine serum albumin (BSA) and blocks 58% of the interaction between PD-1/PD-L1. The aptPD-L1 sequence was 5′-ACG GGC CAC ATC AAC TCA TTG ATA GAC AAT GCG TCC ACT GCC CGT-3′ and since then it has been used in different research [40].
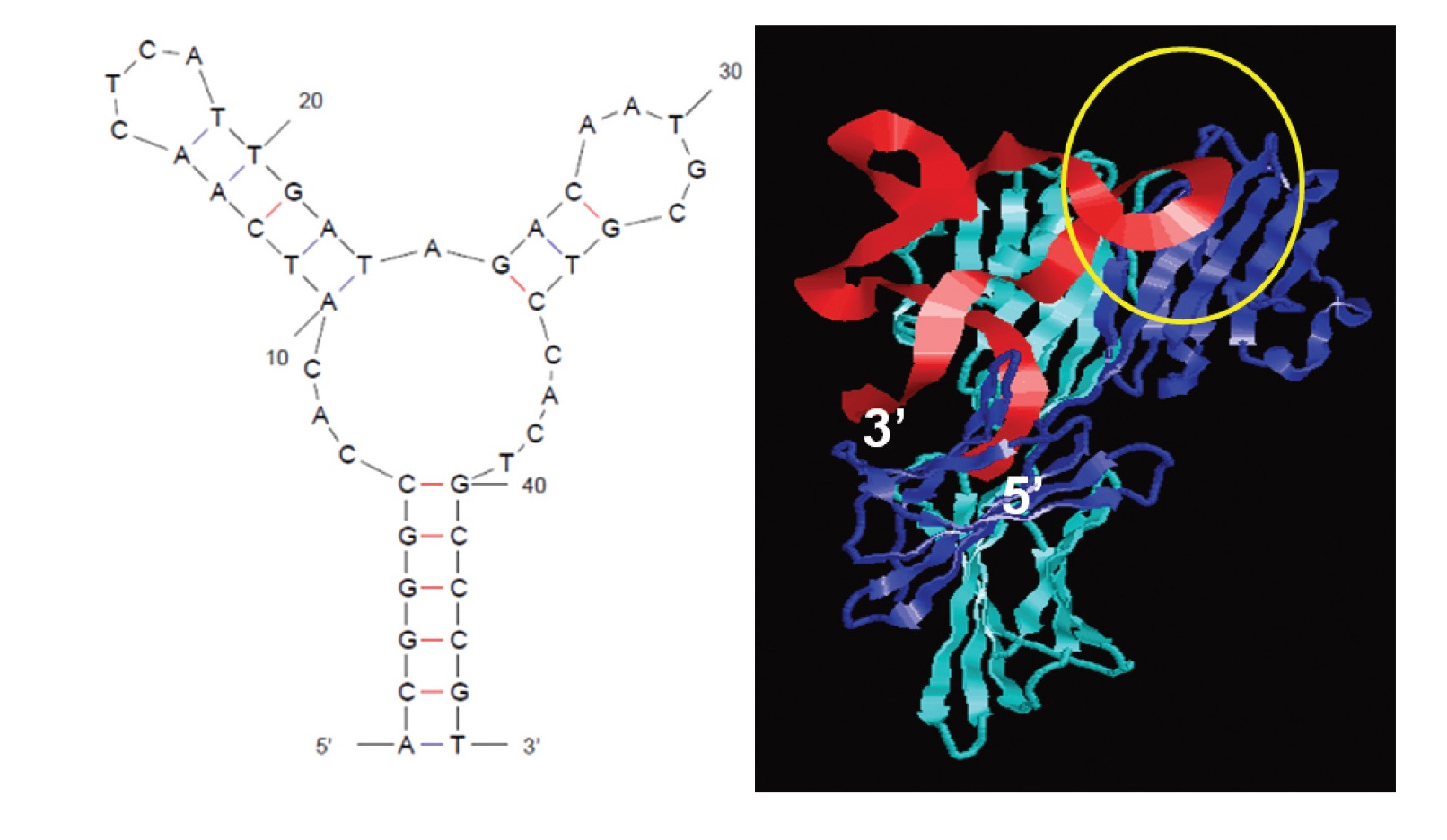 Fig. 1.
Fig. 1.
The secondary structure of PD-L1 antagonizing DNA aptamer (aptPD-L1) was predicted, and docking simulations were performed to visualize its interaction with the PD-L1 (programmed death ligand 1) protein (aptPD-L1 in red and PD-L1 in blue). The yellow circle highlights the binding region between PD-L1 and PD-1 [40].
A year later, in 2017, the protein SELEX method using magnetic beads was
performed, and two possible aptamers (Apt 5 and Apt 33) targeted PD-L1 was
reported [47]. Of these, Apt 5 showed high affinity and specificity with a Kd of
64.77 nM compared to the other candidate and was examined further. The selected
aptamer was labelled with ATTO 647N to determine its dissociation constant. Flow
cytometry data showed a better binding affinity for Apt 5 with a fluorescence log
intensity of 3.89
Similarly, another anti-PD-L1 aptamer has been reported to interfere with the
interaction between PD-1/PD-L1 and its antitumor effect. Cell SELEX was performed
using engineered PD-L1 expressing cells on a target. This work resulted in a
highly specific binding aptamer PL1 with Kd value of 95.73
The aptamer used in the homogeneous, low-volume, efficient, and sensitive
exosomal programmed death-ligand 1 (HOLMES-ExoPD-L1) technique is MJ5C [49],
a short single-stranded DNA molecule engineered specifically to efficiently
target PD-L1. MJ5C exhibits significant affinity for PD-L1 as evidenced by its
binding Kd of 91
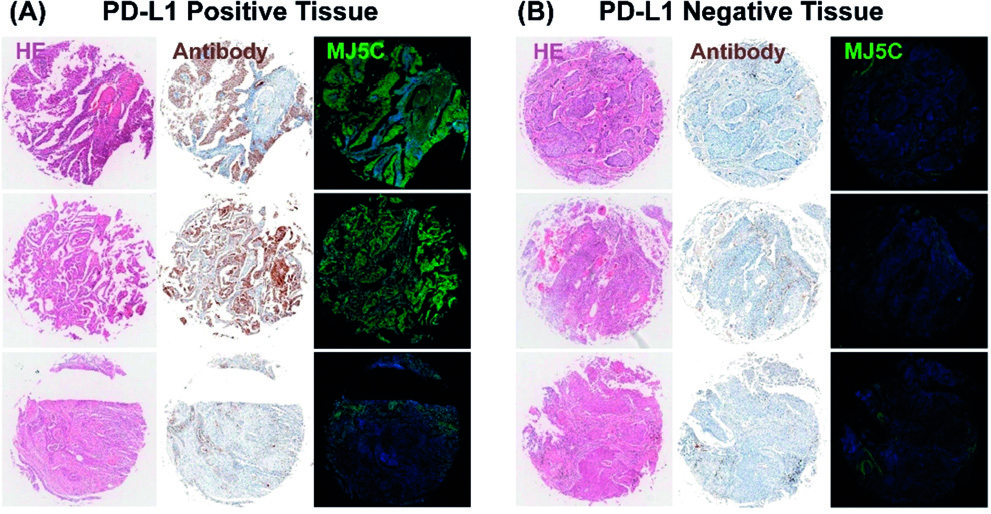 Fig. 2.
Fig. 2.
Fluorescence staining using the MJ5C aptamer and immunohistochemical staining with a PD-L1 antibody were performed on PD-L1 positive (A) and negative (B) tissues. Reproduced with permission from Huang et al. [49], Angewandte Chemie (International Ed. in English); published by Wiley-VCH, 2020.
Through the use of Modular-SELEX, clon-3 aptamer was identified [41]. High
compatibility and specificity was demonstrated using clon-3 in identifying PD-L1
expression in tumor samples. Aptamer dissociation constant (Kd) values were
obtained using the waveguide sensor at 70.1
In another research, aptamers A6 and B10 were studied with their high affinity
or specificity against PD-1 and PD-L1, respectively [50]. For PD-1, aptamer A6
had a Kd value of 47.84
When non-natural or modified nucleotides are incorporated into the aptamer sequence, it allows for improved binding qualities and a wider range of functions than conventional aptamers. This is referred to as “Xeno” in X-Aptamers. X-Aptamers XA-PD1-78 and XA-PD-L1-82 were selected using a bead-based library of X-Aptamers with various changes, including amino acid functional groups linked to dU at position 5. In pancreatic tumor tissue, XA-PD-L1-82 showed binding capacity comparable to PD-L1 antibodies, suggesting its potential for therapeutic use. Strong binding affinities were observed for the binding of XA-PD1-78 and XA-PDL1-82 (Kd = 56.72 nM and 20.18 nM, respectively) to cells expressing PD-L1. These X-Aptamers provide a synthetic replacement for antibodies in PD-L1 research, diagnostics, and possible therapeutic treatment [42].
As shown in Fig. 3 (Ref. [51]), researchers discovered two threose nucleic acid (TNA) aptamers, N5 and S42, that effectively inhibited the PD-1/PD-L1 interaction through an in vitro selection procedure. These TNA aptamers showed more than 50% inhibition of PD-1/PD-L1 interaction in enzyme-linked immunosorbent assay (ELISA) experiments (Fig. 3) and their binding affinities for PD-L1 protein were approximately 400 nM. Crucially, TNA aptamers also showed their efficacy in a more physiologically realistic setting by disrupting the PD-1/PD-L1 junction on the cell surface. Moreover, TNA aptamer N5 with reported truncated sequencing 5′-GAT TGA GTA GAT AGT GGT TCT GTA CGT AGT GAA AGA GTG G-3′ selectively accumulated at the tumor site and dramatically suppressed tumor growth in mouse tumor xenograft experiments, all without apparent damage. These aptamers differ from normal DNA or RNA aptamers due to their TNA composition, which confers increased resistance to nucleases and may increase their therapeutic value as immune checkpoint inhibitors for cancer immunotherapy [51].
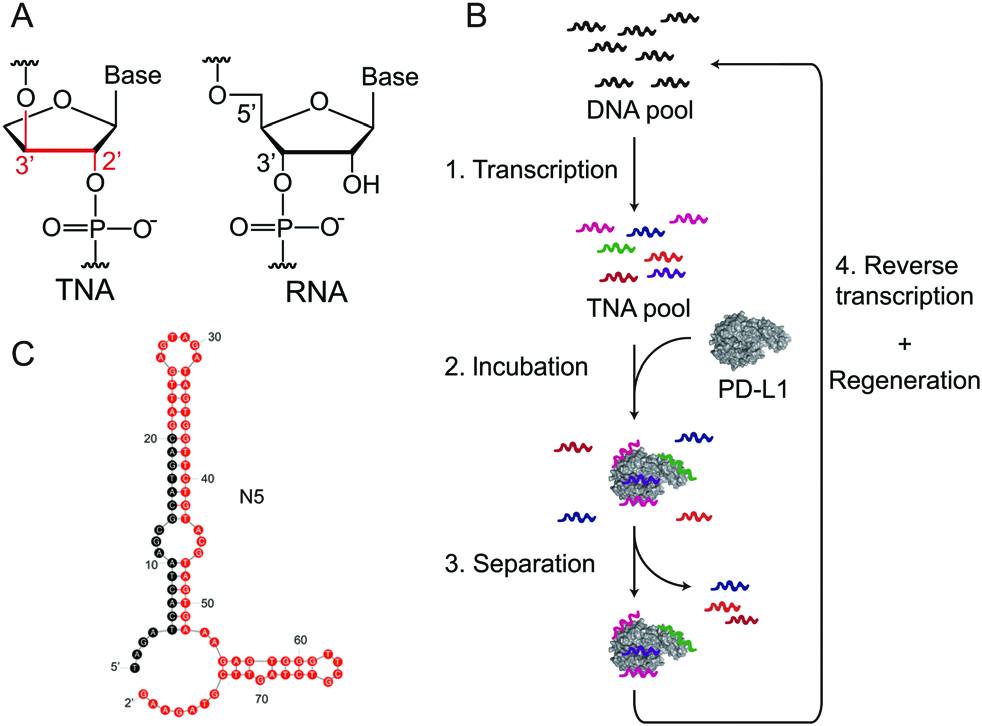 Fig. 3.
Fig. 3.
In vitro selection of PD-L1-binding Threose nucleic acid (TNA) aptamers includes several key steps. (A) The constitutional structures of TNA and RNA are compared, where each TNA repeating unit contains a threofuranose ring and is linked by a 20–30 phosphodiester bond. (B) A schematic illustration depicts the in vitro selection strategy employed. (C) The predicted secondary structures of the TNA aptamer N5 were generated using the RNA backbone in Mfold. Reproduced with permission from Li et al. [51], Chemical Communications (Cambridge, England); published by Royal Society of Chemistry, 2020.
In another study, DNA aptamer Ap3 was found and characterized. It binds preferentially to PD-L1 on tumor cells, with a Kd value of 169.0 nM for the protein and 204.2 nM for cells expressing PD-L1, respectively. The research team then combined Ap3 with an anti-PD1 aptamer to create a bispecific aptamer called Ap3-7c. They also invented a “recognize then conjugate” method based on click chemistry to produce a covalently attached bispecific aptamer called D-Ap3-7c. This innovative approach showed potent anticancer effects in vivo, demonstrating the unique ability of aptamers to target multiple immunological checkpoints simultaneously and enhance immunotherapy [44].
Using a novel SELEX approach called REase-SELEX, the type II restriction enzyme Alu I was critical in aptamer isolation. Alu I cleaved the non-binding material and caused massive variation in the DNA library, greatly expanding its spectrum. As demonstrated by many experiments, aptamer 8–60 showed high affinity (Kd = 1.4 nM) and specificity towards PD-L1. Notably, 8–60 outperformed a previously identified aptamer in reliably detecting PD-L1 expression levels in tissue sections from malignant melanoma, non-small cell lung cancer, and normal human tonsils. They concluded that REase-SELEX can isolate highly specific aptamers by producing targeted mutations during the selection process [45].
Recently, using a novel cell loss-gain-SELEX approach, the DNA aptamer (XQ-P3) was identified, which binds to PD-L1 with high affinity (Kd ~15 nM) and specificity. In triple-negative breast cancer cells that overexpress PD-L1, the XQ-P3 aptamer was able to improve the cellular uptake and cytotoxicity of the conjugated chemotherapy drug paclitaxel, as well as suppress the PD-1/PD-L1 interaction and restore T cell activity. The researchers used a variety of methodologies such as pull-down assays, competitive binding, and cell-based assays to verify the specificity of XQ-P3 for PD-L1 with a sequence of 5′-ACC GAC CGT GCT GGA CTC ATC TCG CTT TTT TCA CGG TCC ACA CTA CTA TGA GCG AGC CTG GCG-3′. These results identified the potential of the PD-L1 aptamer for immunomodulation and targeted treatment of triple-negative breast cancer [46].
In this section, we have thoroughly explored the field of PD-L1 aptamers, discussing their discovery and strategies for enhancing their effectiveness through conjugation with other aptamers or alternative approaches. We emphasize the promising potential of aptamer-based therapeutics in cancer immunotherapy. Table 1 (Ref. [40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]) provides a summary of the general information regarding these anti-PD-L1 aptamers.
| Researchers (Published year) | Aptamer names | Sequences (5′-3′) | Binding Affinity (Kd) |
| Lai et al. 2016 | aptPD-L1 [40] | ACGGGCCACATCAACTCATTGATAGACAATGCGTCCACTGCCCGT | 4.7 nM |
| Yazdian-Robati et al. 2017 | Apt 5 [47] | GCTGTGTGACTCCTGCAAGACGGACCAGCCTTGCCGCAAGACGGACCAGGGATTCAAACGAGCAGCTGTATCTTGTCTCC | 64.77 nM |
| Wang et al. 2018 | XA-PD-L1-82 [42] | N/A | 20.18 nM |
| Huang et al. 2020 | MJ5C [49] | TACAGGTTCTGGGGGGTGGGTGGGGAACCTGTT | 91 |
| Wu et al. 2020 | XQ-P3 [46] | ACCGACCGTGCTGGACTCATCTCGCTTTTTTCACGGTCCACACTACTATGAGCGAGCCTGGCG | |
| Li et al. 2021 | Clon-3 [41] | CCCTCCTCCTAACTGTTCCTACGAAACGAGCTTATGCGTAATGATGACTGTCGTAGTTCG | 8.6 |
| Li et al. 2021 | B10 [50] | AACAACGTATAACAATGCCCACGTCACCAGAGTACTATGG | 59.72 |
| Li et al. 2020 | N5 [51] | GATTGAGTAGATAGTGGTTCTGTACGTAGTGAAAGAGTGG | |
| Gao et al. 2021 | PL1 [48] | ATACCAGCTTATTCAATTGTAGAGTATAAAAAGAGTGATGATCTTTTGTAGGTTTTTTAGATAGTAAGTGCAATCT | 95.73 |
| Ren et al. 2021 | 8-60 [45] | N/A | 1.4 nM |
| Camorani et al. 2020 | TN145 [43] | CCUCAGCGCGCAACUUCCCUCCGUUCCCUGCCACGCGUCA | 37.36 |
| Sun et al. 2022 | Ap3 [44] | N/A | |
| Hu et al. 2023 | PD-L1-GEMs [52] | TTTACAGGTTCTGGGGGGTGGGTGGGGAACCTGTTTT | N/A |
In addressing the limitations of small molecules and biologicals, peptides that interfere with PPIs with a molecular weight ranging between 500 and 5000 have been developed [53]. They are easy to synthesize, have lower immunogenicity, better pharmacokinetics, and lack Fc-receptor-related side effects [54, 55, 56, 57]. Due to this unique property, they can act as a binding bridge between monoclonal antibodies and small molecules. Moreover, the peptides-based inhibitor can be easily modified for better stability and to penetrate tumors and effectively block the interaction between PD-1 and PD-L1 in both near and distant areas of the tumor’s blood vessels. The most common modification involves macrocyclization and stapling of peptides. Macrocylization of peptide enhances the rigidity in structure which help in improving binding specificity and resistance in proteolysis. They are resistant to degradation, target protein-protein interactions, and show promise in therapeutic development for disease like cancer. Recent advancements have expanded their potential in drug discovery [58]. On the other hand, stapling of peptides makes it more constrained, which supports further stability and bioavailability enhancing their efficacy as treatments. This technique involves reinforcing peptides by introducing a chemical “staple”, thereby making them more resistant to degradation within the body. This method is instrumental in developing drugs that specifically target protein interactions and has demonstrated potential in inhibiting disease-associated proteins [59]. Apart from macrocyclic peptide and stapled peptide, there is another class of peptide molecule known as miniproteins. These molecules are a diverse group of small-sized protein frameworks (1–10 kDa) known for their stability and effectiveness in drug-related functions. These miniproteins are mainly derived from natural sources and have been widely used in pharmaceutical development. Despite their different structures and functions, the methods for their application show more similarities among themselves than with more commonly used modalities such as antibodies or small molecules [60]. Peptide-based therapeutics also provide options like nano-scale drug delivery with easy conjugation to a targeting ligand or encapsulated system. Because of these advantages, peptide-based inhibitors have a future as remarkable candidates for cancer immunotherapy [61, 62]. Extensive studies have been carried out in various scientific research institutions to inhibit PD-L1 by using various display technologies like phage display, bacterial display, and mRNA display and by computational design peptide mimetic. These techniques are used to study protein interactions and identify peptides with high affinity for specific targets. Phage display involves expressing peptides on the surface of bacteriophages, which allows for easy manipulation and amplification of the peptide library [63]. Similarly, bacterial display involves expressing peptides on the surface of bacteria, often E. coli, which offers advantages in terms of speed and cost due to the fast growth and simple culture conditions of bacteria [64]. Furthermore, mRNA display links the peptide to its encoding mRNA via a covalent bond, allowing for the direct selection of peptides with desired properties from vast libraries and facilitating rapid evolution of high-affinity binders [65]. On the other hand, the Computational design of peptide mimetics leverages advanced algorithms and molecular modeling to design peptides that mimic the structure and function of natural proteins, providing a powerful approach to creating novel therapeutics and enhancing drug discovery processes [66]. The peptides discovered from these technologies are synthesized and purified by different way. One of common way is Solid-phase peptide synthesis (SPPS) that allows for the step-by-step addition of amino acids on a solid resin support, making purification easier and increasing yield [67]. Liquid-phase peptide synthesis (LPPS) is another technique that is often used for longer peptides because it is easy to handle and purify intermediates [68]. Recombinant DNA technology is widely used for large-scale production, where peptides are expressed in microbial systems such as E. coli or yeast [69]. Enzymatic synthesis, which uses proteases and peptidases, offers a biocatalytic route to peptide production with high specificity and mild reaction conditions [70]. Each method has its own advantages, and the choice depends on factors such as peptide length, complexity, and application requirements. Herein, we review the recent advances on peptide-based inhibitors targeting PD-L1.
To date, many peptide-based PD-L1 inhibitors have been reported. In 2014, Bristol-Myers Squibb [71] introduced a significant category of macrocyclic peptides derived from peptidomimetics, aiming at the PD-1/PD-L1 pathway. They disclosed three cyclic peptides Bristol-Myers Squibb (BMS) 57, BMS 71, and BMS 99—comprising 15, 14, and 13 amino acids, respectively. Magiera-Mularz et al. [72] conducted a study on the interactions between these peptides and PD-L1, elucidating the binding mechanisms along with certain aspects of their biological activity. The reported IC50 values for BMS-57 and BMS-71 were found to be 9 nM and 7 nM, respectively. Various techniques such as nuclear magnetic resonance (NMR)titration, differential scanning fluorimetry (DSF), and cell-based PD-1/PD-L1 blockade bioassay were employed to corroborate the affinities of BMS-57 and BMS-71 towards PD-L1 [72]. NMR titration analysis demonstrated that these two macrocyclic peptides lack activity towards PD-1, whereas they exhibited binding affinities with PD-L1, with Kd values significantly below 0.1 µM. These findings were corroborated by DSF tests and further validated through TCR experiments. Additionally, cell-line experiments revealed EC50 values of 566 nM and 293 nM for BMS-57 and BMS-71, respectively [73, 74, 75].
In a separate study, Chang et al. [76] identified the peptide sequence FPNWSLRPMNQM through phage display peptide screening, which exhibited a strong binding affinity for the PD-L1 protein. Subsequent investigations revealed that the peptide degraded rapidly in blood plasma. To address this stability issue, they developed an enzymolysis-resistant D-peptide by combining techniques involving chemical synthesis of D-proteins and mirror-image phage display (Fig. 4, Ref. [76]). The resulting peptide sequence NYSKPTDRQYHF (Fig. 5A) demonstrated a binding affinity of 0.51 µM and significant inhibition of tumor growth in a CT26 transplantation model, with no observed loss of body mass.
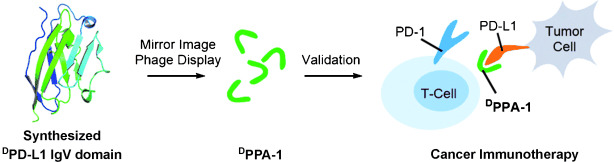 Fig. 4.
Fig. 4.
Researchers integrated protein chemical synthesis and mirror-image phage display to develop a proteolysis-resistant D-peptide antagonist named D-peptide antagonist-1 (DPPA-1), which targets the immune checkpoint protein programmed death ligand 1 (PD-L1). DPPA-1 has shown the ability to inhibit the PD-1/PD-L1 protein-protein interaction at the cellular level. Reproduced with permission from Chang et al. [76], Angewandte Chemie (International Ed. in English); published by Wiley-VCH, 2015.
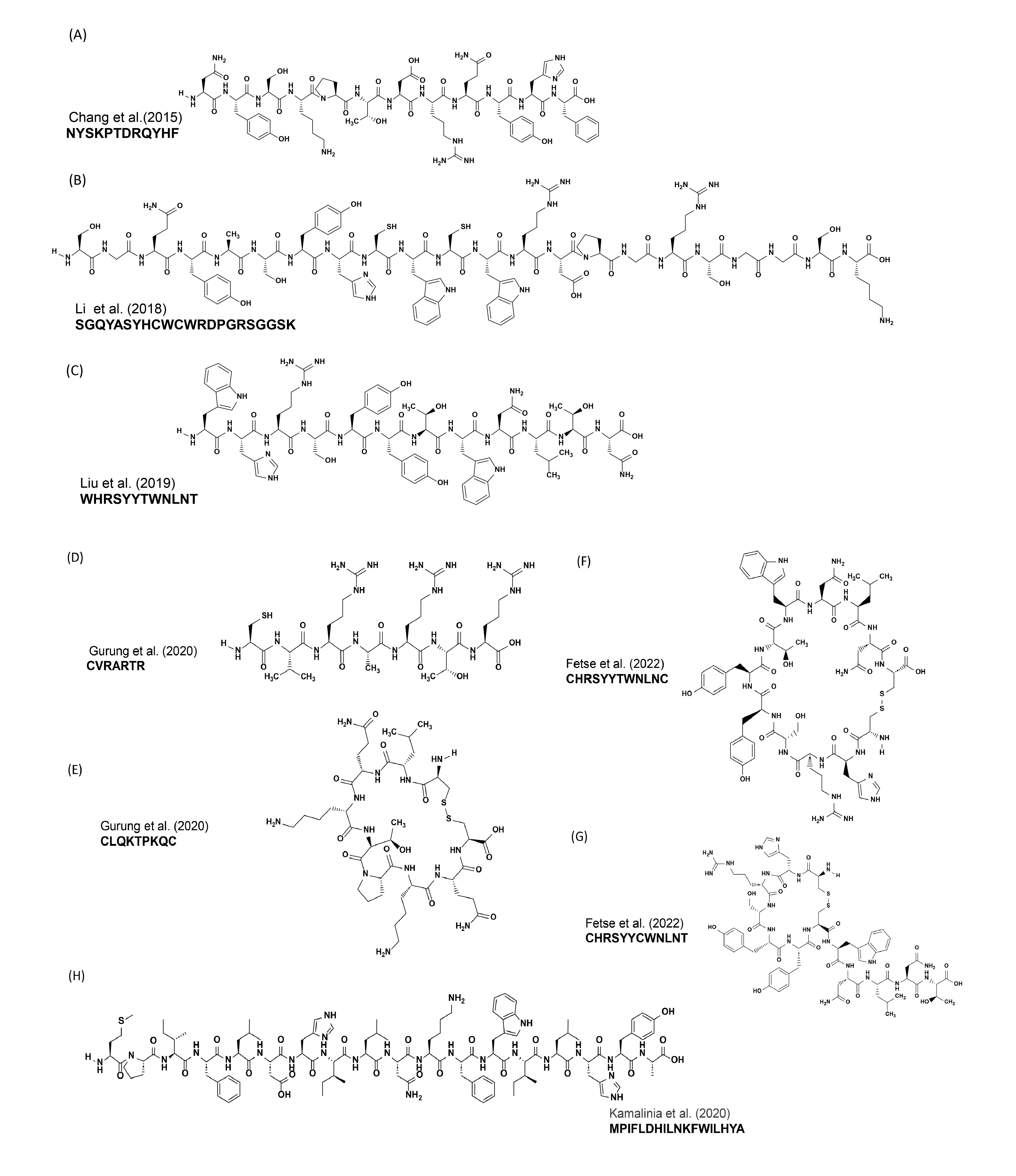 Fig. 5.
Fig. 5.
Structures and sequences of some anti-PD-L1 peptides. (A) NYSKPTDRQYHF [76], (B) SGQYASYHCWCWRDPGRSGGSK [77], (C) WHRSYYTWNLNT [78], (D) CVRARTR [79], (E) CLQKTPKQC [79], (F) CHRSYYTWNLNC [81], (G) CHRSYYCWNLNT [81], (H) MPIFLDHILNKFWILHYA [82].
In 2018, Li et al. [77] reported on a 22 amino acid residue peptide named targeting PD-L1 peptide (TPP-1), which targeted PD-L1. Utilizing the bacterial surface display method, they identified the peptide with the amino acid sequence SGQYASYHCWCWRDPGRSGGSK (Fig. 5B), possessing a binding affinity of 94 nM. The interference of TPP-1 peptide with the interaction between PD-1/PD-L1 was confirmed through a T-cell activation assay and mixed lymphocyte reaction. To evaluate TPP-1’s inhibitory effect on tumor growth in vivo, a xenograft mouse model was established using H460 cells. Mice treated with TPP-1 or PD-L1 antibody exhibited a 56% or 71% reduction in tumor mass growth rate, respectively, compared to those treated with a control peptide. These findings suggest that TPP-1 has the potential to inhibit or at least decelerate tumor growth [77].
In 2019, Liu et al. [78] developed a novel phage display strategy to identify a peptide capable of specifically binding to hPD-L1 residues responsible for the PD-1/PD-L1 interaction. This approach was further validated through docking studies. They identified four peptides (CLP001, CLP002, CLP003, CLP004), among which CLP002, with the sequence WHRSYYTWNLNT (Fig. 5C), exhibited the highest blocking efficiency (85%) and a binding affinity of 366 nM for PD-1/PD-L1 interaction. Additionally, CLP002 demonstrated an IC50 of 1.43 µM against DU-145, a prostate cancer cell line, and 2.22 µM against hPD-L1. Further investigations revealed that CLP002 was capable of restoring T-cell proliferation and preventing apoptosis during co-culture with cancer cells. Moreover, it displayed superior tumor penetration in a 3D tumor spheroid model compared to Cy5-labeled anti-PD-L1 antibody. Furthermore, CLP002 inhibited the growth of CT26 colorectal tumors and increased the survival of tumor-bearing mice [78].
Next, Gurung et al. [79] identified two PD-L1 binding peptides, PD-L1
pep-1 and PD-L1 pep-2, using a T7 phage library containing CX7C peptide.
Bio-panning was conducted on transfected HEK293T cells, with counter-selection
performed on non-transfected HEK293T cells. The peptide sequences CLQKTPKQC and
CVRARTR (Fig. 5D,E) exhibited selective binding affinities of 373 nM and 281 nM,
respectively, toward high PD-L1-expressing cells such as MDA-MB231 and CT26 tumor
cells, over low PD-L1-expressing cells like MCF7 tumor cells. This binding was
further augmented by treating cells with IFN-
In 2021, Yin et al. [80] utilized computational design to create a miniprotein mimetic aimed at blocking the PD-1/PD-L1 interaction by imitating the interface of an optimized PD-1. Initially, attempts to mimic the native structure of PD-1 resulted in the MNPD-1 peptide, which displayed no activity despite mimicking nine of the 22 hotspot residues from the interaction surface. However, with the optimized PD-1, they identified the mimetic peptide Mimetic of Optimized PD-1 (MOPD-1) (IQIREYKRCGQDEERVRRECKERGERQNCHYVIHKEGNCYVCGIICL), which exhibited a binding affinity of 0.3 µM for PD-L1. Remarkably, the designed peptide inhibited the interaction with an IC50 of 1–2 µM, irrespective of whether PD-1 or PD-L1 was immobilized. Further characterization using NMR spectroscopy and X-ray crystallography revealed that the binding residues from the modified PD-1 were crucial for the bioactivity of MOPD-1. Moreover, the mimetic peptides were found to be highly stable, with serum half-lives exceeding 24 hours without requiring any modifications such as cyclization. In vivo studies demonstrated that MOPD-1 suppressed tumor growth by revitalizing the immune system in a CD8+ T cell-dependent manner [80].
Recently, Fetse et al. [81] employed a side chain macrocyclization strategy with two cysteine residues scanning on the anti-PD-L1 linear peptide CLP-002 to discover a cyclic peptide with improved inhibition and binding affinity. Among 22 scans, they found that peptides C7 (CHRSYYTWNLNC, Fig. 5F) and C12 (CHRSYYCWNLNT, Fig. 5G) exhibited superior binding affinity compared to the unmodified CLP-002 peptide. Notably, the C7 peptide demonstrated an IC50 of 180 nM, which was 34 times better than the parent CLP-002 peptide’s IC50 of 6073 nM against mouse PD-1/PD-L1 interaction. The efficacy of C7 and C12 peptides was further supported by serum stability experiments. While the CLP-002 linear peptide displayed a half-life of 18.8 minutes and was nearly completely degraded after 3 hours in 50% human serum, the cyclic peptides C7 and C12 exhibited improved serum stability, with half-lives of 38 and 70 minutes, respectively. Among these, the C12 peptide showed the best serum stability, with approximately 25% of the peptide remaining intact after 3 hours. Furthermore, both cyclic peptides demonstrated superior in vivo anti-tumor efficacy even at a dose of 0.5 mg/kg compared to the parent peptide CLP-002 at 2 mg/kg [81].
Kamalinia et al. [82], in 2020, engineered an 18-residue linear peptide called SPAM using mRNA display, which binds to human PD-L1. The SPAM peptide has sequence MPIFLDHILNKFWILHYA (Fig. 5H), which shows different characteristics compared to known PD-L1 binding peptides and mAbs. It exhibits high selectivity for human PD-L1, with dissociation constants of 119 and 67 nM for unglycosylated and glycosylated human PD-L1, respectively. The SPAM peptide does not significantly bind to mouse PD-L1 or human PD-L2. Additionally, competition binding assays suggest that the SPAM peptide’s binding site overlaps with the binding site of PD-1 and therapeutic anti-PD-L1 antibodies. The peptide shows serum stability for up to 5 hours with no change in peptide retention time in 25% human serum [82].
In 2023, Shi et al. [83] discovered a peptide sequence that interacts
with the protein acyltransferase ZDHHC3, responsible for modifying the PD-L1
protein. Utilizing artificial intelligence, they successfully pinpointed the
precise peptide sequences to target ZDHHC3. Subsequently, these sequences were
modified through strategic stapling at optimal positions to enhance stability and
effectiveness. This stapling technique significantly improved the peptides’
stability, binding capacity to the target, and ability to penetrate cells. The
resulting stapled peptide was conjugated with linker GSGS, and E3 ligase peptide
ALAPYIP, named SP-PROTAC, was employed as a peptide-based PROTAC, effectively
reducing PD-L1 expression to less than 50% at 0.1 µM in both C33A and HeLa
cervical cancer cell lines. Furthermore, the expression of ZDHHC3 decreased in a
dose and time-dependent manner. In a C33A and T cell co-culture model, the use of
the peptide led to a dose-dependent increase in IFN-
BMS986189, a macrocyclic peptide that entered the clinical trial, provided
favorable safety and pharmacokinetic data. The results from a Phase I clinical
trial (ClinicalTrials.gov
NCT02739373; https://clinicaltrials.gov/ct2/show/NCT02739373) completed in
December 2016 led to the development of an analog of the original candidate,
BMS986189, for which a new Phase I clinical trial is ongoing in 2022 (ISRCTN
Registry ISRCTN17572332, https://www.isrctn.com/ISRCTN17572332). Inspired from
BMS986189, Rodriguez et al. [84] did structural and biological
characterization of pAC65 macrocyclic peptide and found its potency equivalent to
the current FDA-approved mAbs and BMS986189. The verification of its potency is
based on various assay. In Antagonist Induced Dissociation Assay (AIDA)-NMR experiments, they found that pAC57 is capable
of interfering with both the PD-1/PD-L1 and PD-L1/CD80 complexes formation. To
further support its effectiveness, they did a commercially available Homogeneous
Time Resolved Fluorescence (HTRF) assay which shows affinity of pAC65 for PD-L1
and dissociate PD-1/PD-L1 complex with an IC50 value of 1.80
In this section, we have extensively explored the realm of PD-L1 peptides, detailing their discovery and discussing strategies for enhancing their efficacy through conjugation with other molecules. We underscore the promising prospects of peptide-based therapeutics in cancer immunotherapy. Fig. 5 depicts the structures of several anti-PD-L1 peptides elucidated in this review. Moreover, Table 2 (Ref. [77, 78, 79, 80, 81, 82, 83]) summarizes the general information pertaining to these anti-PD-L1 peptides.
| Names | Sequences | Biological evaluation | Discovery Technologies | References |
| TPP-1 | SGQYASYHCWCWRDPGRSGGSK | Kd = 94 nM | Bacterial display | [77] |
| CLP-002 | WHRSYYTWNLNT | Kd = 366 nM | Phage display | [78] |
| IC50 = 1.43 µM | ||||
| Pep-1 | CLQKTPKQC | Kd = 373 nM | Phage display | [79] |
| Pep-2 | CVRARTR | Kd = 281 nM | Phage display | [79] |
| MOPD-1 | IQIREYKRCGQDEERVRRECKERGERQNCHYVIHKEGNCYVCGIICL | Kd = 0.3 µM | Peptide mimetic | [80] |
| IC50 = 1–2 µM | ||||
| SPAM | MPIFLDHILNKFWILHYA | Kd = 119 nM (unglycosylated) | mRNA display | [82] |
| Kd = 67 nM (glycosylated) | ||||
| C7 | CHRSYYTWNLNC | IC50 = 180 nM | Phage display and peptide macrocyclization | [81] |
| C12 | CHRSYYCWNLNT | IC50 = 440 nM | Phage display and peptide macrocyclization | [81] |
| SP-PROTAC | GIQDT(R8)NSKKQS(S5)DTH-GSGS-ALAPYIP | IC50 = 0.1 µM | Artificial Intelligence | [83] |
TPP-1, Targeting PD-L1 peptide; CLP-002, Checkpoint Ligand Peptide 002; MOPD-1, Mimetic of Optimized PD-1; SPAM, Signal Peptide based Affinity Maturated ligand; SP-PROTAC, stapled peptide-based proteolysis-targeting chimera.
Both dimerization and multiple presentations serve as valuable tools in aptamer or peptide development, providing avenues to enhance binding properties and potentially incorporate supplementary functionalities. The specific approach chosen depends on the desired outcome and the characteristics of the target molecule. In sections 4.1 and 4.2, we will examine recent developments in dimerization of anti-PD-L1 aptamers and peptides, respectively. In the following section 4.3, we will delve into their multiple presentations.
Bispecific inhibitors capable of concurrently blocking PD-1 and PD-L1 have emerged as promising candidates for enhancing the efficacy of immune checkpoint blockade therapy. These inhibitors work by releasing the brakes on T-cell antitumor activity, thus bolstering the immune system’s ability to combat cancer. In a groundbreaking study, Sun et al. [44] identified a novel DNA aptamer, termed Ap3, with the remarkable ability to specifically recognize PD-L1 expressed on tumor cells. Importantly, Ap3 competes with the binding of PD-1, thereby disrupting the PD-1/PD-L1 interaction crucial for immune evasion by cancer cells. Leveraging this finding, the researchers engineered a bispecific aptamer by integrating Ap3 with an anti-PD-1 aptamer known as 7c. This bispecific aptamer dimer demonstrated remarkable efficacy in preclinical studies, inducing a potent immunological response against tumors in vivo [44]. These findings represent a significant advancement in the development of targeted immunotherapies and highlight the potential of bispecific inhibitors against PD-1 and PD-L2 simultaneously, in revolutionizing cancer treatment strategies. Moreover, this work underscores the pivotal role of aptamers in facilitating precise and multifaceted targeting of immune checkpoints, paving the way for more effective and personalized therapeutic interventions in oncology.
Natural killer (NK) cells have emerged as a compelling alternative to T cells for adoptive cell therapy, given their robust killing capacity, off-the-shelf availability, and minimal toxicity. Despite their rapid and potent immune effects, NK cells face challenges related to inadequate infiltration into tumor sites and the immunosuppressive tumor microenvironment, limiting their therapeutic efficacy. Addressing these limitations, Zheng et al. [85] have introduced a groundbreaking approach utilizing a highly stable CD16/PD-L1 bispecific aptamer dimer characterized by exceptional affinity and selectivity. This innovative aptamer dimer orchestrates a profound antitumor immune response by recruiting CD16-positive NK cells to directly engage with PD-L1-overexpressing tumor cells (Fig. 6, Ref. [85]). Moreover, the aptamer-mediated upregulation of PD-L1 on tumor cells, often observed as an adaptive response to various immunotherapeutic strategies, can be counteracted by its specific binding to PD-L1, mitigating the negative impact of PD-L1 overexpression on therapeutic efficacy. Importantly, this aptamer-based immunotherapy offers a straightforward yet highly efficient approach with minimal side effects, thereby holding considerable promise for clinical translation. This strategy not only underscores the versatility of aptamers in modulating immune checkpoints, but also highlights their potential to enhance the effectiveness of NK cell-based therapies in combating cancer.
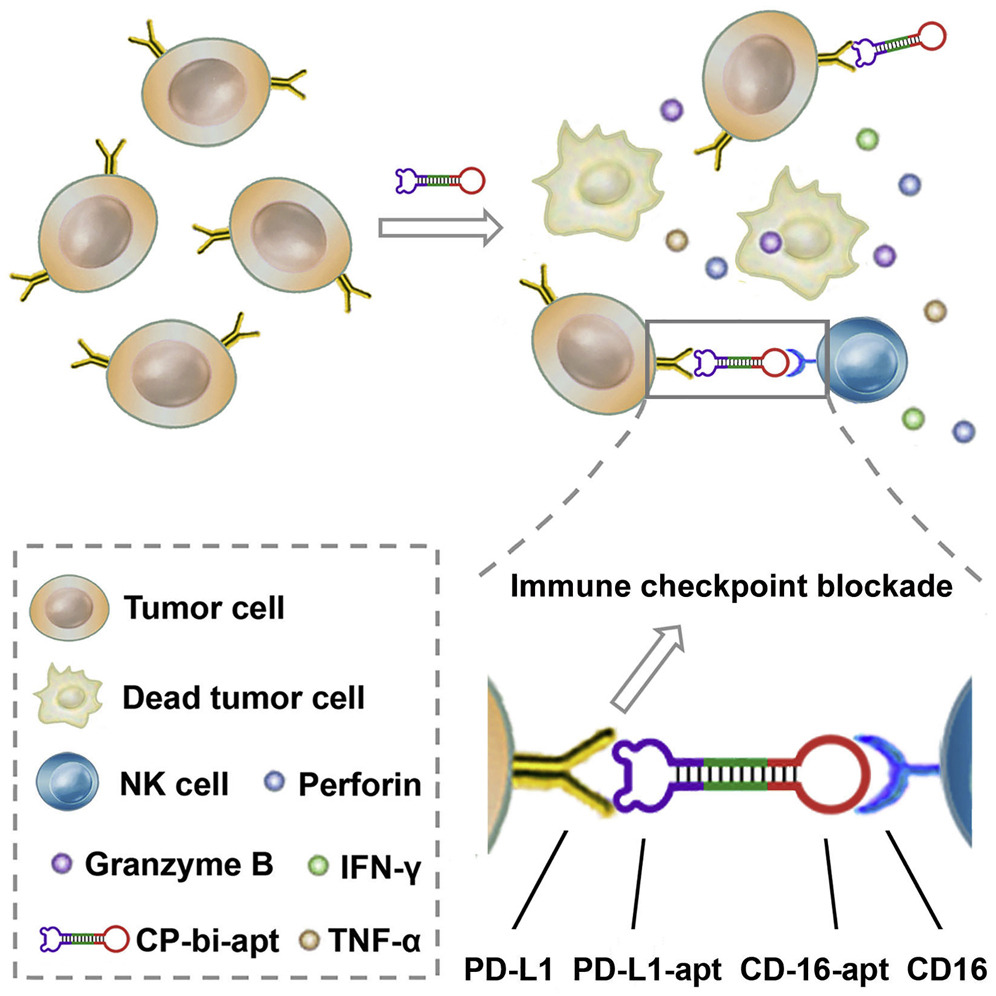 Fig. 6.
Fig. 6.
A schematic illustration of CD16/PD-L1 bi-specific aptamer (CP-bi-apt) shows its role in enhancing antitumor immunotherapy. Adoptive NK cells can induce a robust immune response with the engagement of CP-bi-apt. Additionally, CP-bi-apt enhances the antitumor effect through PD-L1 immune checkpoint blockade [85].
In a recent publication, Hu et al. [86] introduce a novel chimeric
peptide, Pal-DMPOP, designed to enhance anticancer efficacy. This innovative
peptide amalgamates the smallest fragment of the D-peptide inhibitor of
PD-1/PD-L1 (OPBP-1) with the optimized peptide inhibitor of CD47/SIRP
In another work, Shen et al. [87] identified the CD24/Siglec-10 blocking peptide (CSBP), utilizing a multifaceted approach involving cell-based phage display biopanning and D-amino acid modification strategy. Remarkably, CSBP was found to possess hydrolysis-resistant properties and demonstrated the ability to block the interaction of CD24/Siglec-10 and PD-1/PD-L1. Moreover, CSBP induced the phagocytosis of tumor cells by both macrophages and monocytic myeloid-derived suppressor cells (M-MDSCs), consequently activating CD8+ T cells. Notably, a synergistic reduction in tumor growth and alteration of the tumor microenvironment were observed upon combining radiotherapy with CSBP in both anti-PD-1-responsive MC38 and anti-PD-1-resistant 4T1 tumor models. This study introduces the first CD24/Siglec-10 blocking peptide capable of simultaneously blocking PD-1/PD-L1 interaction. Its mechanism involves enhancing tumor cell phagocytosis by macrophages and M-MDSCs, thereby augmenting CD8+ T cell activity for cancer immunotherapy.
Jacquot et al. [88] delved into the engineering of a long bispecific peptide through the fusion of anti-epidermal growth factor receptor (EGFR) and anti-programmed cell death ligand 1 (PD-L1) peptide modules. Despite reducing the affinity of the peptide modules for their individual targets, this fusion strategy enables the simultaneous engagement of EGFR and PD-L1. As a result, the long bispecific peptide exhibits selective binding to tumor cells that co-express EGFR and PD-L1 exclusively. Their study demonstrated that this affinity-attenuated bispecific peptide induces PD-L1 blockade solely in an EGFR-directed manner. These findings underscore the potential of this approach to enhance the selectivity and safety of PD-L1 checkpoint inhibition, offering a promising avenue for refined cancer immunotherapy strategies.
Next, we will outline recent advancements in multiple presentations for the enhanced targeting of PD-L1 with aptamers or peptides. For example, Li et al. [89] delved into the exploration of DNA nanostructures as enhancers of PD-L1 aptamer function. They ingeniously integrated four PD-L1 aptamers (Apt) into a Holliday Junction (HJ) framework, resulting in the formation of a tetravalent DNA nanostructure known as Apt-HJ. Notably, the average size of Apt-HJ measured 13.22 nm, surpassing the renal clearance threshold. Additionally, Apt-HJ underwent partial phosphorothioate modification, enhancing its resistance to nucleases. Comparative analysis revealed that the tetravalent Apt-HJ exhibited augmented affinity toward CT26 colon cancer cells in comparison to monovalent PD-L1 aptamers. Impressively, Apt-HJ demonstrated a substantial enhancement in antitumor efficacy in vivo compared to free PD-L1 aptamers, without concurrent elevation of systemic toxicity. These findings underscore the potential of employing multiple aptamers tethered to a DNA nanostructure to significantly enhance PD-L1 aptamer function in vivo.
To circumvent renal clearance of the aptamer, An et al. [90] embarked on conjugating the PD-L1 aptamer with albumin, forming a larger complex denoted as BSA-Apt, and subsequently assessed its potential to enhance in vivo antitumor efficacy. The PD-L1 aptamer underwent thiol modification and was conjugated to the amino group of BSA via an SMCC linker. With an average size of 11.65 nm, BSA-Apt exceeded the renal clearance threshold. Functionally, BSA-Apt preserved the PD-L1 aptamer’s capacity to bind with PD-L1-expressing tumor cells. Moreover, both the free aptamer and BSA-Apt amplified PBMC-induced antitumor cytotoxicity in vitro (Fig. 7, Ref. [90]). Notably, BSA-Apt elicited significantly stronger antitumor efficacy than the free PD-L1 aptamer in vivo, without concomitant elevation of systemic toxicity. These findings underscore the potential of conjugating the PD-L1 aptamer with albumin as a promising strategy to enhance its in vivo functionality, suggesting that BSA-Apt holds promise for application in cancer immunotherapy.
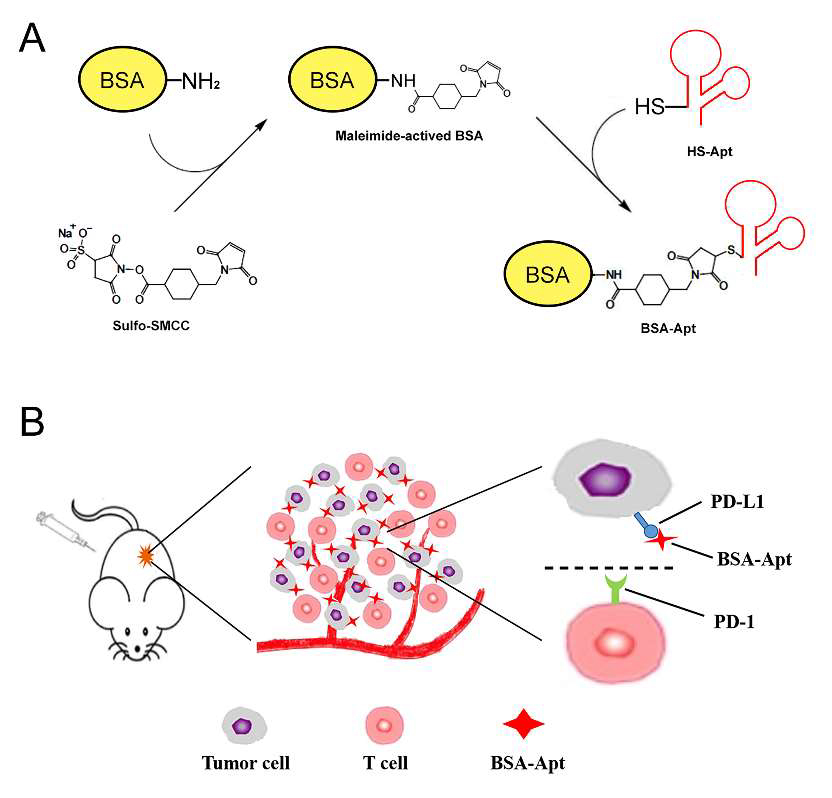 Fig. 7.
Fig. 7.
A schematic illustration of the bovine serum albumin (BSA)-Apt designed for colon cancer therapy is presented. (A) The two-step reaction scheme involves conjugating BSA with the PD-L1 aptamer using a crosslinker (sulfo-SMCC). First, sulfo-SMCC reacts with BSA to produce maleimide-activated protein. After removing the non-reacted crosslinker, the maleimide-activated BSA reacts with the thiol-modified PD-L1 aptamers to form BSA-Apt. (B) The PD-1/PD-L1 blockade scheme shows BSA-Apt binding to PD-L1 expressed on the surface of tumor cells, thereby blocking the PD-1/PD-L1 interaction [90].
In a study conducted by Jiang et al. [91], a multivalence aptamer-based nanotherapeutic is devised to augment the efficacy of PD-1 and PD-L1 aptamers in combating cancer. Through a novel approach, the researchers coalesce the PD-1 and PD-L1 aptamers onto an albumin nanoparticle (NP), forming a bispecific multivalence nanostructure known as PD1-NP-PD-L1. This innovative construct presents several distinct advantages. For example, owing to the modified NPs, there’s a propensity for enhanced accumulation in tumor tissues facilitated by the enhanced permeability and retention effect, capitalizing on the heightened permeability of tumor blood vessels. With multiple aptamers tethered to each nanoparticle, there’s the potential for multivalent binding, thereby elevating affinity compared to individual aptamers. In addition, unlike free aptamers prone to rapid renal filtration due to their small size, the designed nanotherapeutics exceed the renal filtration threshold, extending their circulation time for heightened efficacy. Lastly, PD1-NP-PD-L1, as a bispecific nanostructure, engages both PD-1-expressing T cells and PD-L1-expressing tumor cells, fostering immunological synapse formation to bolster immune responses. This innovative nanotherapeutic paradigm promises to revolutionize cancer immunotherapy and merits further exploration for clinical translation.
In another work, Zhang et al. [92] introduced a novel bispecific peptide-polymer conjugate termed octaPEG-PD1-PD-L1. This conjugate was synthesized by concurrently attaching PD1-binding and PD-L1-binding peptides onto an 8-arm-PEG structure. octaPEG-PD1-PD-L1 serves as a bridge between T cells and cancer cells, consequently augmenting T cell-mediated cytotoxicity against cancer cells. Additionally, this tumor-targeting conjugate enhances the infiltration of cytotoxic T lymphocytes into tumors while mitigating their exhaustion. As a result, it effectively activates the tumor immune microenvironment, leading to a robust antitumor response in CT26 tumor models with an impressive tumor inhibition rate of 88.9%. This study introduces a approach to bolster tumor immunotherapy by conjugating bispecific peptides onto a hyperbranched polymer, thereby efficiently engaging target-effector cells.
Lu et al. [93] have reported the development of a novel peptide-polymer conjugate, PEG-MP9-aPD-L1, which incorporates four functional motifs including a D-peptidomimetic inhibitor of PD-L1, a matrix metalloproteinase-2 (MMP-2) cleavable spacer, and MP9, conjugated with 4-arm PEG. This innovative conjugate was identified as a highly promising systemic delivery vehicle due to its specificity for PD-L1 targeting and favorable pharmacokinetic properties. Their study demonstrated that PEG-MP9-aPD-L1-induced oncolysis leads to the generation of immunogenic cell death (ICD)-relevant damage-associated molecular patterns (DAMPs) both in vitro and in vivo, which are crucial for immunotherapy utilizing PD-L1 inhibitors. Additionally, PEG-MP9-aPD-L1 exhibited remarkable immunotherapeutic efficacy in a colorectal cancer (CRC) mouse model, characterized by enhanced tumor infiltration of CD8+ T cells and induction of cytotoxic lymphocytes (CTLs) in the spleen. These findings suggest that PEG-MP9-aPD-L1 serves as a comprehensive platform for oncolytic immunotherapy and immune checkpoint blockade (ICB).
As shown in Fig. 8 (Ref. [94]), Jeong et al. [94] introduced a
groundbreaking approach utilizing peptide–dendrimer conjugates (PDCs) to
stabilize the
 Fig. 8.
Fig. 8.
A schematic illustration of the development process of a multivalent dendrimer–peptide conjugate as a PD-1/PD-L1 antagonist. Reproduced with permission from Jeong et al. [94], Journal of the American Chemical Society; published by ACS, 2020.
In a recent study, the 5′-COOH-modified MJ5C PD-L1 aptamer was integrated into spherical nucleic acids (SNAs) featuring a shell composed of PD-L1 aptamer and indocyanine green (ICG), embedded within a core of mesoporous hafnium oxide nanoparticles (Hf@ICG-Apt). This innovative approach introduces a novel avenue for enhancing biosafety in cancer treatment by enabling the monitoring of PD-L1 expression and localization, alongside robust radiation therapy against malignant cells. The versatile nanoprobe incorporates a degradable HfO2 core, improving the stability of ICG aqueous solution, which retains almost all fluorescent intensity after 30 minutes of exposure. Notably, mesoporous HfO2 degrades at the tumor site even under low pH conditions, facilitating the release of ICG, particularly in high PD-L1 tumors targeted by the PD-L1 aptamer. Furthermore, the PD-L1 aptamer is shielded against degradation by the nanoprobe. Moreover, the nanoprobe serves as a radiosensitizer, inducing apoptosis. Overall, this nanoprobe enables the tracking of PD-L1 expression evolution in checkpoint blockade immunotherapy (CBI) response and enhances radiosensitization in cancer therapy [95].
In conclusion, the current scientific literature lacks comprehensive reviews that explore both aptamers and peptides as potential tools for targeting PD-L1. Addressing this gap is crucial to consolidate recent advancements in biotechnological approaches aimed at PD-L1 targeting. Our review aims to fill this void by synthesizing available data and discussing the potential implications of aptamer- and peptide-based strategies. Such insights would offer valuable guidance for researchers and clinicians striving to optimize PD-L1-targeted therapies, enhancing efficacy and precision in cancer treatment.
HT, SA, and XT conducted a comprehensive literature review, synthesized the findings through the creation of illustrative charts, and developed the initial conceptual framework for the manuscript. All authors contributed critically to the subsequent manuscript drafts, incorporating feedback from the review process to produce the final version. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The authors thank Bowling Green State University for Startup grant (33900025) and the Building Strength Grant Programs to XT (18700).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.