





1 School of Life and Environmental Sciences, Faculty of Science, The University of Sydney, Camperdown, NSW 2050, Australia
2 Lacombe Research and Development Centre, Agriculture and Agri-Food Canada, Lacombe, AB 403, Canada
3 School of Agriculture and Food Sciences, Faculty of Science, University of Queensland, Gatton, QLD 4343, Australia
Academic Editor: Yasuhito Shimada
Abstract
Background: A foal undergoes considerable growth and development from
birth to weaning, progressing from a milk-based diet to complete herbivory. The
symbiotic relationships between bacteria, archaea and fungi substantiate this
energy demand by colonising the hindgut and remaining flexible throughout the
diet transitions. Methods: A total of 70 faecal samples were collected
from 14 mares and their foals across five studs in NSW as they aged from 0 to 5
months old. DNA was extracted from faecal samples and underwent amplification and
sequencing of the 16S rRNA gene V4 hypervariable region of archaea and bacteria, and
the fungal internal transcribed spacer-1 (ITS1) region. The fungal and bacterial
community structure was assessed using Bray-Curtis dissimilarities, and the
effect of age at sampling and location was determined using PERMANOVA.
Results: Age at sampling had a substantial effect on the foal’s
archaeal and bacterial faecal microbiota (PERMANOVA: R
Keywords
- 16S rRNA gene sequencing
- fungal ITS sequences
- equine
- microbiome
The gastrointestinal tract (GIT) microbiome represents one of the most complex and rapidly evolving biotic networks responsible for the inner workings of an animal and its functional health. Heavily involved in immune function, gut-brain connectivity and behaviour, disturbance of the equine microbiome has been linked to metabolic diseases such as laminitis, colic, equine metabolic syndrome, equine grass sickness and colitis [1, 2, 3]. The developmental period from birth to weaning represents an interval of considerable growth for horses, where foals progress from 10% of their mature body weight at birth to 50% at weaning [4]. The energy demand to substantiate this growth is met by dietary transitions from changing milk compositions to complete herbivory, supported by the microbial colonisation of the GIT. The symbiosis between bacterial, archaeal, and fungal populations maximises hindgut feed digestion, producing by-products such as volatile fatty acids and metabolites to maintain host health [5, 6, 7, 8].
Studies of the equine GIT microbiome have identified a ‘core’ bacterial population consisting primarily of the phyla Firmicutes, Bacteroidetes, Verrucomicrobia, Actinobacteria, Proteobacteria and Spirochaetes [9, 10, 11, 12, 13]. However, the functional characteristics of many bacterial and fungal taxa and their importance to the host are yet to be proven in horses [9, 11, 13]. Rather, the bulk of the current literature addresses only ruminant fermentation [14, 15]. This is significantly misleading the industry’s current understanding of the equine microbiome, causing a lag in disease treatment strategies, perception of microbial resistance, and general appreciation of the microbiome’s role in equine health. Further, comparing the findings of previous investigations of the equine microbiome is compromised by the considerable influence of diet, climate, and management practices on the intestinal microbiota [16, 17]. This study is both a continued exploration of the equine microbiome and a novel investigation into the establishment of the mycobiome in neonate foals utilising high-throughput sequencing technology.
While bacterial populations in the foal hindgut have received considerable attention [16, 18, 19, 20], fungal colonisation remains relatively overlooked. Of the few studies reporting on the mycobiome, most only describe populations in adult horses or pathogenic overgrowth in compromised hosts [21, 22, 23, 24]. Fungi are instrumental in substantiating diet transitions by increasing the digestibility of foliage through the production of cellulolytic and hemicellulolytic enzymes [25, 26]. A better understanding of the symbiotic relationships between bacteria and fungi and the metabolites they produce is needed to more effectively manage GIT conditions and the effects that any perturbation, such as antibiotics, may have on the hosts’ internal microbiome [27, 28, 29].
The authors believe this is the first study to simultaneously characterise the colonisation of bacterial and fungal species during a principal developmental period. It is hypothesised that stages of gut maturation will be classifiable based on the analyses of the faecal microbiota of foals from birth to weaning. It is expected the bacterial colonisation of the hindgut will follow ‘core microbiome’ patterns, like those previously described, despite the effect of geographical location [16, 18, 19, 20].
A more inclusive exploration of the temporal evolution of microbial diversity in foals and the interactions between bacteria and fungi will aid in bridging the knowledge gap between hindgut fermenters and current ruminant and monogastric-based research. This study will also contribute to a better understanding of the progressive colonisation of the equine microbiome, irrespective of environmental factors [11, 30] and propose a similar notion for fungal populations that are overlooked in current literature.
Faecal samples were collected from mares and foals at five Thoroughbred breeding farms across New South Wales, Australia, throughout one foaling season from late 2017 to early 2018. Farms were located in the Central West, Hunter Valley and Southern Highlands regions. A total of 70 samples were collected. Fourteen foals were sampled consecutively from their first month of life across a period of 3–5 months. Each corresponding mare was also sampled at the final foal sampling. Samples were collected from three mares and three foals at Stud 1, Stud 2, Stud 3, and Stud 4, with samples collected from two mare and foal pairs at Stud 5. The number of samples per foal ranged from 3 to 6, sampling age ranging from 7–30 days for the 0–1 m old category (n = 15), 36–58 days old for 1–2 m (n = 14), 66–83 days old for 2–3 m (n = 18), 92–121 days old for 3–4 m (n = 14), and 123–148 days old for the 4–5 m category (n = 9).
Faeces were collected with as little disturbance to the animal as possible, consistent with procedures approved by the University of Sydney Animal Ethics Committee (Project Number 1319). Targeted individuals were observed in holding yards during morning musters or later in the day within their allocated yards or paddocks until the time of defecation. Faeces were collected from the ground immediately after excretion, with the exception of 5 samples that were collected after 1–2 hours from smaller yards only containing the one mare and foal pair. A minimum of 10 g of faeces was collected with a gloved hand or a clean plastic spoon sterilised with 70% (w/v) ethanol. The exterior faecal layer was removed to ensure the samples were collected from where the specimen was least likely to have had contact with environmental contaminants. Samples were placed in sterile polypropylene falcon tubes or containers (Sarstedt, Australia) and stored on ice temporarily (approximately 1–3 hours) before transfer to a –20 °C freezer, as commonly practised [18, 31]. Details noted for each sample included age, farm, pen or field location and the clinical state of the animal.
Samples were stored at –20 °C until placed in a freeze dryer (72 h).
Samples were then finely ground using a coffee grinder and placed back into the
freezer until extraction. Genomic DNA was extracted from faecal samples through
repetitive bead beating, as described by Yu and Morrison [32]. Briefly, the
sample (
The V4 hypervariable region of the archaeal and bacterial 16S rRNA gene and the internal transcribed spacer 1 (ITS1) (ITS1-5’-CTTGGTCATTTAGAGGAAGTAA; ITS2-5’-GCTGCGTTCTTCATCGATGC-3’) region for fungi were amplified and sequenced as previously described [35] using an Illumina MiSeq instrument and v2 Reagent Kit with 500 cycles (Illumina, Inc., San Diego, CA, USA). DADA2 v. 1.16.0 (https://www.bioconductor.org/packages/release/bioc/manuals/dada2/man/dada2.pdf) [36] in R v. 4.0.3 was used to process and quality-filter all sequences. Primer sequences were removed from all reads using cutadapt v. 2.10 [37]. The forward and reverse 16S rRNA gene sequences were trimmed to 200 and 210 bp, respectively, merged with a minimum overlap of 100 bp, and chimeras removed. Taxonomy was assigned to these remaining sequences, referred to here as operational taxonomic units (OTUs) at 100% similarity, using the RDP naïve Bayesian classifier and the SILVA SSU database release 138 [38]. OTUs classified as chloroplasts and mitochondria were removed prior to analyses. For the ITS1 sequences, reads were not trimmed, and the UNITE database release 8.2 [39] was used for assigning taxonomy.
The number of OTUs per sample, Shannon diversity index, and the inverse Simpson’s diversity index were calculated in R using vegan 2.5-7 and Phyloseq 1.34.0 [40]. The fungal and bacterial community structure was assessed using Bray-Curtis dissimilarities, which were calculated with vegan, and the effect of age at sampling and location was determined using PERMANOVA (adonis2 function). The pairwise.adonis function in the pairwiseAdonis package v. 0.4 [41] was used to compare each sampling time. To account for unequal sequencing depth, the 16S rRNA gene sequence samples were randomly subsampled to 10,500 sequences per sample and the ITS1 sequence samples to 7500, prior to calculation of the diversity measures and Bray-Curtis dissimilarities.
All 16S rRNA gene and ITS1 sequences were submitted to the Sequence Read Archive under BioProject accession PRJNA699302.
A total of 70 faecal samples collected from 14 mares and their foals across five
NSW Thoroughbred breeding farms (i.e., studs) in 2017 were analysed. The average
number of 16S rRNA gene sequences per sample were 30,579
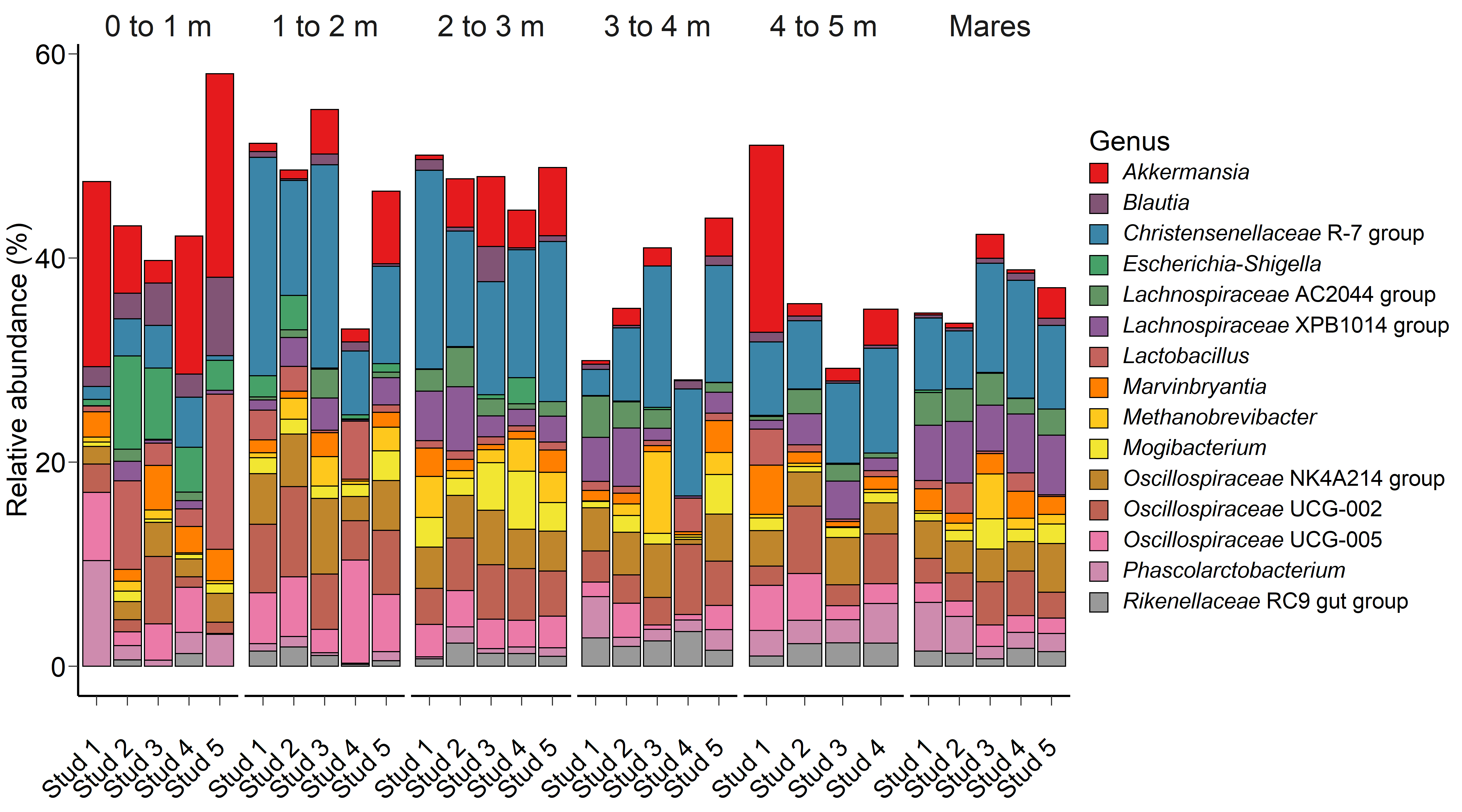 Fig. 1.
Fig. 1.The 15 most relatively abundant archaeal and bacterial genera in the equine faecal microbiota by age at sampling and geographic location.
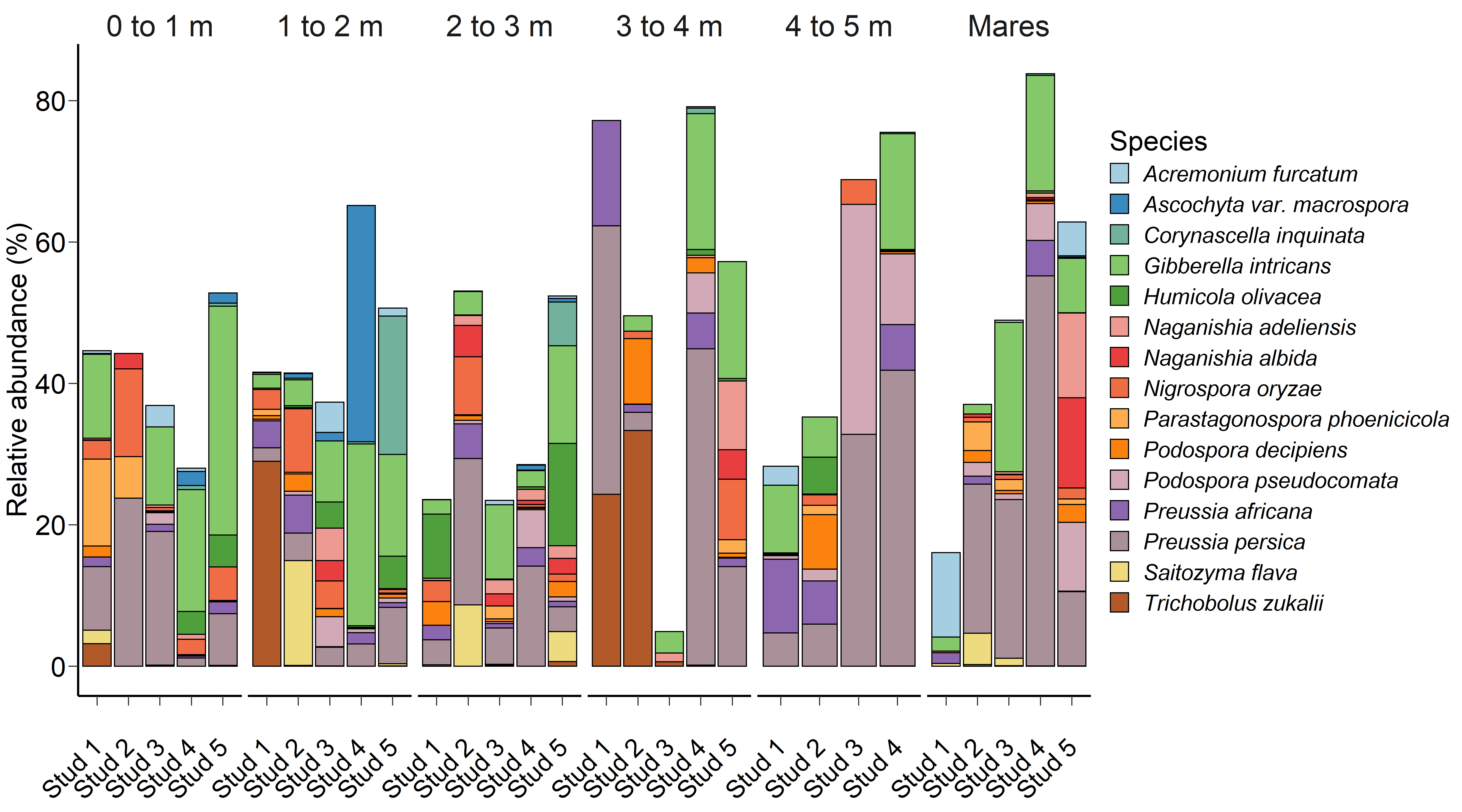 Fig. 2.
Fig. 2.The 15 most relatively abundant fungal species in the equine faecal microbiota by age at sampling and geographic location.
The archaeal and bacterial faecal microbiota was most strongly affected by age
at sampling (PERMANOVA: R
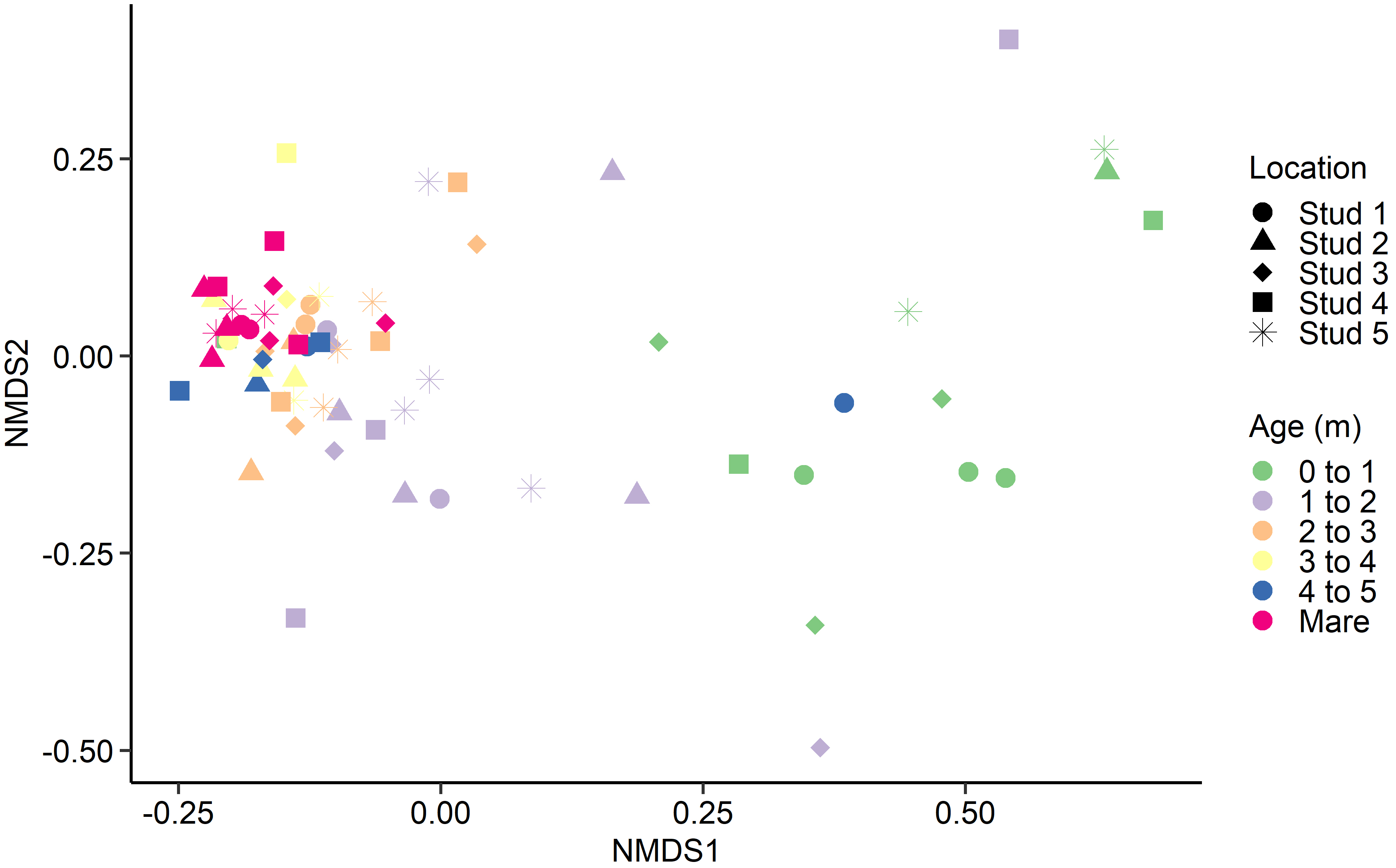 Fig. 3.
Fig. 3.Non-metric multidimensional scaling (NMDS) of the Bray-Curtis dissimilarities for the equine archaeal and bacteria faecal microbiota by age at sampling and geographic location.
| Age | p-value | |||||||||
| 0 to 1 | 1 to 2 | 2 to 3 | 3 to 4 | 4 to 5 | Mare | SEM | Location | Age | Location | |
| OTUs | 300.4c | 394.5bc | 503.5ab | 575.7a | . | 619.3a | 50.03 | 0.17 | 0.88 | |
| Shannon | 4.0c | 4.7b | 5.2ab | 5.4a | . | 5.6a | 0.23 | 0.66 | 0.99 | |
| Inverse Simpson’s | 46.3c | 44.3c | 94.1b | 123.3ab | . | 143.1a | 17.40 | 0.26 | 0.35 | |
The PRA of 15 of the most relatively abundant archaeal and bacterial genera are
presented in Fig. 1. Age had a significant effect on the PRA of the genera
Akkermansia (p = 0.04), Blautia (p
Escherichia-Shigella had a tendency (p = 0.07) to have a
greater PRA in 0 to 1 month-old foals, while the PRA of Mogibacterium
(p = 0.05) tended to be higher in foals at 2 to 3 m compared to mares
and animals at 0 to 1 and 1 to 2 m. Location was a significant factor on the PRA
of Lachnospiraceae XPB1014 group and Lachnospiraceae AC2044
group (p
Although significant, age at sampling had a smaller effect on the faecal fungal
microbiota (PERMANOVA: R
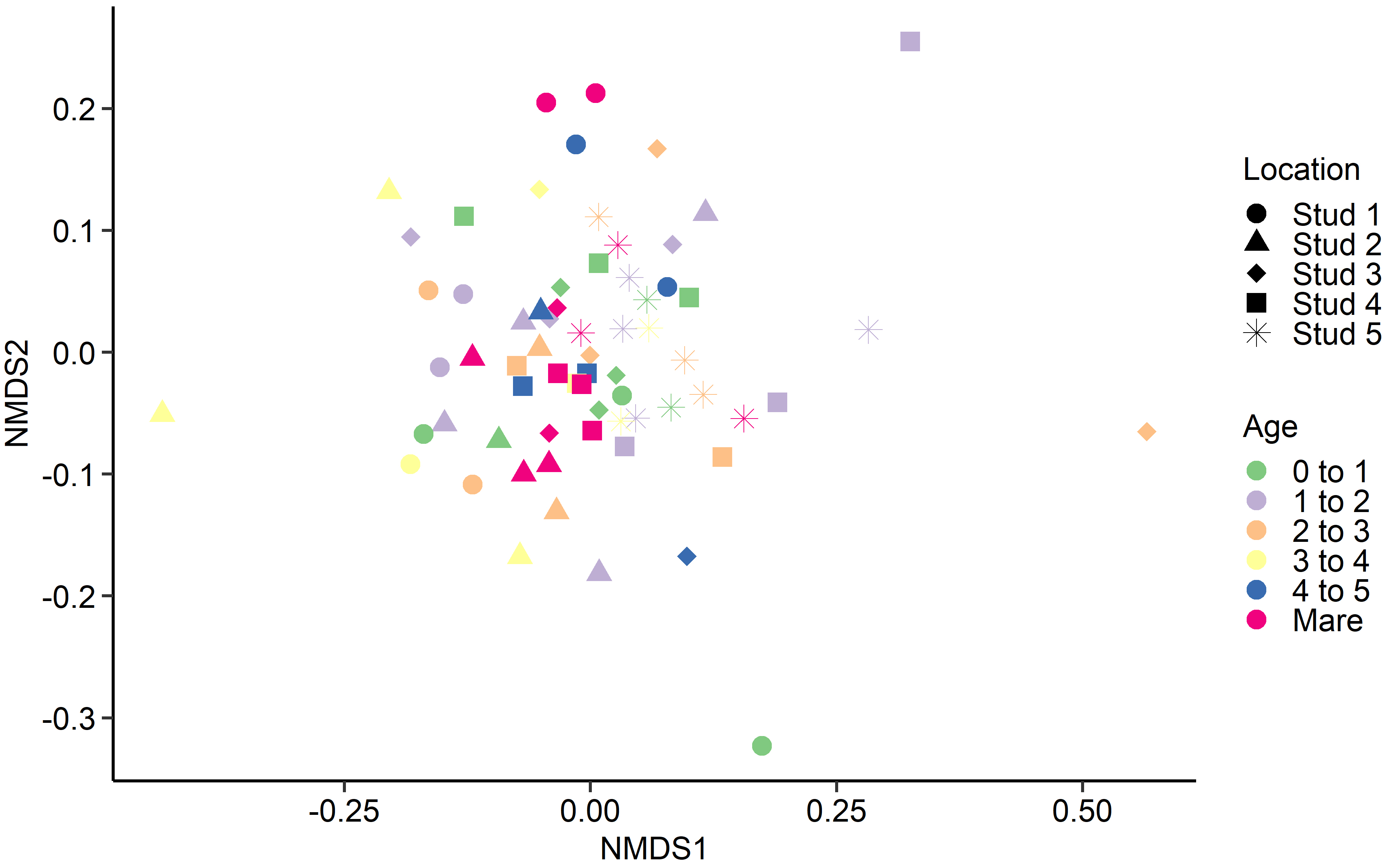 Fig. 4.
Fig. 4.Non-metric multidimensional scaling (NMDS) of the Bray-Curtis dissimilarities for the equine fungal faecal microbiota by age at sampling and geographic location.
This study has reported the identity and PRA of both fungal and bacterial microbial taxa present at each stage of foal maturation (0 to 5 months). The increase in bacterial diversity as foals aged most likely coincides with increased environmental exposure through changing diets and interaction with other animals. Costa et al. [16] reported that there are often statistically higher abundances of bacterial genera in young foals than in adult mares, with many belonging to the Firmicutes phylum, as seen in the current study. Dramatic and dynamic shifts in the bacterial microbiota have also been observed in newborn humans, puppies, and calves [42, 43, 44]. Perinatal exposure to microbial ecosystems of the meconium, amniotic fluid, and faeces aid in establishing the foal’s internal colonisation prior to birth [45]. The sudden post-partum exposure to environmental organisms, the mother’s own microbiota, and colostrum are all reflected in the increasing bacterial diversity and richness seen in young foals. Previous studies characterising the equine microbiome identified similar predominant phyla to those seen in the current investigation, including Firmicutes, Bacteroidetes, Verrucomicrobia and Proteobacteria [16, 19, 20, 46].
The identified fluctuations in the most relatively abundant bacterial and archaeal genera as the foal aged likely correlates with their metabolic function. Trends were seen in fibrolytic taxa such as Lachnospriaceae spp., where their PRA increased once foals surpassed 0 to 1 months old. Species within this family are drivers of key metabolic transformations such as reductive acetogenesis and lactate conversion. In addition, many members of the Lachnospriaceae are major butyrate-producers [47]. This would aid in substantiating the large energy demand for the growth foals experience during birth to weaning as they begin to graze and ingest carbohydrates. The Phascolarctobacterium genus also showed a positive relationship with age. Species within this genus are known to generate short-chain fatty acids such as acetate and propionate [48], likely contributing to energy utilisation and storage. In humans, the abundance of bacteria in the Christensenellaceae and Rikenellaceae families has been associated with reduced adipose tissue [49]. Their increase in PRA seen in 4 to 5 m old foals may contribute to the diversion of energy for fat and growth as foals prepare for weaning.
With few exceptions, samples from adult mares tended to cluster together, as did samples from foals in each age category, regardless of geographical location. A similar trend was identified by Costa et al. [16]. Although there was minor variation between individuals, the current study saw a general tendency for most bacterial and archaeal populations to stabilise in foals by 3 to 4 months of age. However, there are discrepancies in published observations where stabilisation timelines range from just 30 days [17] to six weeks [50] and 50 to 60 days post-partum [16, 51]. Variances in analytical methods and study design complicate the comparison of research groups. However, despite differences in findings, all studies agree that microbial reconfiguration occurs prior to weaning, corresponding with the introduction of plant fibre and solid feeds [18].
Limited studies have investigated microbial colonisation of foals from birth to weaning. Fewer still have considered fungal populations despite their critical role in digestive maturation and dietary transitions [52]. Although a study has been done to determine the effect of colostrum replacer on the foal microbiome, the small sample size and limited analysis provided little explanation [53]. Studies of the murine mycobiome have shown fungal populations display radical episodic variation over an animal’s lifetime while bacterial communities remain relatively stable [54]. Despite its superior fibrolytic activity, fungi account for only 8% of the rumen microbial biomass, which is thought to be due to their slower generation time than bacteria [55]. It is likely that a similar circumstance occurs in the hindgut of horses and may explain why such low PRAs were recorded in the current study.
Interestingly, age at sampling had a greater effect on the PRA of certain
archaeal and bacterial genera than geographical location, yet the reverse was
found for fungal species. As the 15 most relatively abundant fungal species
isolated from faecal samples were environmental microfungi, it is not surprising
that there were multiple age
The primary DNA barcode for eukaryotes like fungi is the ITS1 region between the 18S rRNA and 5.8S rRNA genes [59]. The authors chose to use ITS1 primers like those used in McGorum et al. [60], who collected post-mortem samples from the gastrointestinal tract of 54 horses with equine grass sickness and faecal samples from control horses. Results seen in McGorum et al. [60] are somewhat comparable to the current study, finding Acremonium, Preussia and Naganishia fungal species to be among the most dominant. However, it is unknown why Neocallimastigomycota species or other anaerobic fungal phyla known to colonise the alimentary tract of mammalian herbivores and hindgut fermenters were not detected in faecal samples analysed in the current study [24, 52, 61, 62, 63, 64, 65, 66]. As this is a preliminary study of the foal mycobiome, it may be that anaerobic fungi, such as Neocallimastigomycota, were present but at less abundant or undetectable levels.
Previous studies of the equine mycobiome have taken place in various geographical locations worldwide. Edwards et al. [23] collected faecal samples from mature ponies in the Netherlands fed haylage and straw, while Edwards et al. [22] sampled ponies from Concord, MA USA, across September and October that were provided predominantly pasture. Additionally, Mura et al. [15] sampled hindgut segments of a 24-year old Anglo-Arabian gelded male fed a mixed diet. Differences in pasture grass species, the area of forage being grazed, and the season during which the horses were sampled (summer vs winter pasture species) may influence the extent of a horse’s exposure to certain fungal organisms. It may be possible that the Australian Thoroughbreds sampled for this study may not have been exposed to high abundances of Neocallimastigomycota.
Multiple publications investigating the equine mycobiome have used at least one primer specific for the anaerobic phylum Neocallimastigomycota [15, 22, 23, 67] or employed anaerobic cultures [24]. This likely explains their reportedly high abundances compared to this study. It is possible that the type of samples collected in the current study did not offer the most accurate representation of the internal microbiome. However, a previous publication found cultured Neocallimastix spp. in high proportions in the left ventral colon (81%) and the rectum (75.5%), indicating that faecal samples are still viable as a qualitative reference sample [15]. Although, it is important to note that the same study used a Neocallimastigomycota specific 5.8S rRNA gene reverse primer.
Although there are discrepancies between this study’s results and previous literature, the authors are confident that the observations made here are accurate. The primary limitation of this study is the surprising abundance of aerobic species found, as the authors expected predominantly anaerobic organisms given the low-oxygen environment of the caecum. Potentially, the fungal PRA results may not truly reflect the faecal mycobiome but, in this case, may represent the highly abundant food-source species that survive the hindgut digestive processes, or environmental contamination. Similarly, a 2016 study sequenced the ITS1 region to identify over 90 fungal taxa within the human GIT and detected sequences belonging to edible fungi [68]. This suggests dietary fungal intake may be a confounding effect in mycobiota investigations [68]. Further, there is no exhaustive reference base for annotated fungal sequences as there is for bacteria [69, 70], making the establishment of definitions for ‘normal’ fungal diversity much more difficult. Future studies may need to undertake culture-based analysis to discern mycobiome phenotypes not resolved by sequencing, particularly as current databases overestimate the abundance of culturable sequences.
This study found that the microbial community within the foal hindgut stabilises by three to four months of age. As hypothesised, bacterial and archaeal genera identified in faecal samples belonged mainly to the Firmicutes phylum with several genera unique to 0–1-month-old animals. The colonisation of the equine hindgut and microbial composition corresponds to the introduction of plant fibre and solid feeds. This relates to the increase in the PRA of fibrolytic taxa, such as Lachnospriaceae, observed as foals surpassed 0 to 1 months old. Similarly there was a simultaneous steady increase in members of the Phascolarctobacterium genus, which are known for their role in feed conversion and VFA production. The age at sampling had significantly more influence over the archaeal and bacterial faecal microbiota than the effect of geographical location. However, the opposite was found for fungal species, more strongly affected by where the horse was sampled. Although, it is of note that the 15 most relatively abundant fungal species were environmental saprophytes. Therefore, it was more difficult to establish the relevance of these fungi as hindgut colonizers. Instead, authors conclude that environmental and dietary fungi can survive the digestive process and maintain relatively high abundances that may have outcompeted recognised ‘core’ anaerobic phyla such as Neocallimastigomycota.
GM and KM designed the research study. GO, DH, KM, GM, MP, SM and AC performed the research. DH and AC analysed the data. GO wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Faeces were collected with as little disturbance to the animal as possible, consistent with procedures approved by the University of Sydney Animal Ethics Committee (Project Number 1319).
The authors would like to thank the participating Thoroughbred studs located in the Central West, Hunter Valley and Southern Highlands regions for providing access to the samples required to complete this investigation.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.