






Frontiers in Bioscience-Elite (FBE) is published by IMR Press from Volume 13 Issue 2 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Biochemistry, Research and Development Centre, Bharathiar University, Coimbatore, 641046, Tamil Nadu, India
2 Department of Biochemistry, Indian Academy Degree College, Autonomous, Bengaluru- 560043, Karnataka, India
3 Department of Biochemistry, M.G.R. College, Hosur-635130, Tamil Nadu, India
Abstract
The present study was designed to evaluate the protective effect of fisetin against rotenone induced toxicity in SH-SY5Y neuroblastoma cellular modelof Parkinson's disease (PD). SH-SY5Y neuroblastoma cells were treated with fisetin (5µM) 2 hr prior to being treated with rotenone (100 nM). Following the exposure ofSH-SY5Ycells torotenone, there was marked decreased cell viability, increased oxidative stress, activation of caspase-3 and apoptosis (dual staining, expressions of pro-apoptotic and anti-apoptotic indices). However pretreatment with fisetin significantly and dose-dependently alleviated rotenone induced cytotoxicity and oxidative stress inSH-SY5Ycells. Moreover, fisetin attenuatedrotenoneinduced toxicity by down-regulatingBax, caspases-3 protein expression and up-regulating protein expression of Bcl-2, p38/JNK-MAPK and PI3K, Akt, GSK-3β pathways. Collectively, these results suggest that fisetin could prevent therotenone-induced neurotoxicity via various signaling pathways.
Parkinson’s disease (PD) is a neurodegenerative movement disorder, mainly affecting the aged population, which is characterized by decreased dopamine (DA) producing neurons in the substantia nigra (SN). The consequent deficits in striatal DA are responsible for the clinical features including tremor at rest, bradykinesia, rigidity and postural instability (1). DA itself cannot used to treat PD because it does not cross the blood brain barrier. Dopamine replenishment in the form of levodopa (L-Dopa), a precursor of dopamine that can cross the blood brain barrier (BBB), is being considered as a gold standard for treatment of PD. Chronic administration of L-DOPA is reported to have severe side effects in clinical situation. Other pharmacological therapies including the drugs that increase dopaminergic synthesis and mimics dopamine function, anticholinergic drugs, surgical procedures such as pallidotomy, cell transplantation and deep brain stimulation are reported only to alleviate the symptoms but not the disease progression.
The pathogenesis of PD is multifactorial with toxic reactions including inflammation, the glutamatergic toxicity, the dysfunction of mitochondrial activity and of the ubiquitin/ proteasome system, the activation of apoptosis pathways, the elevation of iron and nitric oxide, the alteration of the homeostasis of antioxidants/oxidation. The multifactorial etiology of this disease suggests that drugs with multiple targets such as plant extract and their phytochemicals could have therapeutical potential for these pathologies. Fisetin is a flavonoid belonging to polyphenols compounds present in fruits and vegetables (2). In cell cultures fisetin promoted neurite differentiation and cell survival following toxic oxidative insults, as well as activating extracellular signal-regulated kinase (3). Moreover it offers neuroprotective effect against various toxins through its antioxidant, anti-inflammatory and neuromodulatory role (4-5) demonstrated the protective effect of fisetin on MPTP/MPP+-induced neurotoxicity in PC12 cells by evaluating cell viability, inflammation and apoptosis.
SH-SY5Y cell line has been commonly used as a in vitro model for PD research because (i) it possess the ability to synthesize dopamine as it express tyrosine hydroxylase and dopamine-β-hydroxylases (7) and (ii) express dopamine transporter, a protein regulates DA homeostasis through specific uptake and sequestration of DA (8). Rotenone model of PD links both the involvement of pesticide exposure and electron transport chain (ETC) complex I dysfunction in PD aetiology. Emerging evidence has suggested that mitochondrial dysfunction mediated oxidative stress may contribute to the neurodegeneration observed in PD (9). Rotenone exposure inhibits mitochondrial complex I activity, enhances ROS formation (10), induces mitochondrial membrane potential (MMP) loss and releases cytochrome c (cyt-c) from mitochondria, thereby activates the caspases and finally causes cell death via regulation of JNK, p38, and ERK MAPK pathways (11-12). Moreover, rotenone treatment regulated PI3K/AKT/GSK3β signalling pathway that is also reported as a main pathway involved in the activation of neuronal survival, growth and function (13). The purpose of this study was to evaluate the neuroprotective effect of fiestin against rotenone-induced toxicity in SH-SY5Y cells by evaluating cell viability, apoptotic damage and expression of apoptotic and signaling markers.
Fisetin, rotenone, Dulbecco’s modified Eagle medium (DMEM): nutrient mix F-12 (1:1), fetal bovine serum (FBS), antibiotic/antimycotic agent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), thiobarbituric acid (TBA), phenazinemethosulphate, nitroblue tetrazolium (NBT), 5,5- dithiobis (2-nitrobenzoic acid) (DTNB), Dimethyl sulfoxide (DMSO), glutamine, penicillin–streptomycin, EDTA, trypsin, Dulbecco’s modified Eagle medium (DMEM) and acridine orange/ethidium bromide (AO/EB) were obtained from Sigma–Aldrich (St. Louis, MO, USA). Anti- caspase-3, Bax, Bcl-2, JNK, P38 MAPK, Akt, GSK3β, PI3K and β- actin antibodies were obtained from cell signaling (Beverly, MA, USA). All other chemicals used were of analytical grade.
The human neuroblastoma SH-SY5Y cell lines were obtained from the National Centre for Cell Science (NCCS), Pune, India. SH-SY5Y cells were grown and maintained in complete medium consisting of high-glucose DMEM with 10% FBS and 1% antibiotic/antimycotic reagent. They were maintained at 37°C and 5% CO2 atmosphere.
To assess the therapeutic potential of fisetin against rotenone toxicity, we evaluated the cell viability indirectly by measuring mitochondrial activity in living cells by MTT quantitative colorimetric assay (14). In the therapeutic group, cells were pretreated with different concentration of fisetin (0, 2.5., 5, 10 and 20 μM) for 2 hours, and later rotenone 100 nM was added for 24 hours (15). Then cells were incubated with MTT (5 mg/ml) at 37°C for 4 hours. After incubation, the medium was removed, and then the cells were dissolved with DMSO. The absorbance of formazan reduction product was measured at 570 nm in plate reader. Cell viability was expressed as a percentage of the value in the control.
Group I: Untreated control cells
Group II: Rotenone (100 nM)
Group III: Fisetin (5 µM) + Rotenone (100 nM)
Group IV: Fisetin (5 µM)
The SH-SY5Y cells were harvested by trypsinization and the cell pellet obtained was
suspended in PBS. The suspension was taken for biochemical analysis. The level of lipid peroxidation was determined by analyzing TBA-reactive substances (TBARS) (15). The pink chromogen formed by the reaction of 2-TBA with breakdown products of lipid peroxidation was measured. Superoxide dismutase (SOD) activity was assayed (15), based on the inhibition of the formation of reduced nicotinamide adenine dinucleotide phenazine methosulphate-NBT complex. Catalase activity was assayed (16) by quantifying the hydrogen peroxide after reacting with dichromatein acetic acid. The activity of glutathione peroxidase was assayed by a known amount of enzyme preparation and was allowed to react with hydrogen peroxide and GSH for a specified period (17). Then, the GSH content remaining after the reaction was measured. The total GSH content was measured based on the development of a yellow color when 5, 5?-dithiobis-2- nitrobenzoic acid was added to compound containing sulfhydryl groups (17).
The control and experimental cells were stained with fluorescent DNA binding dyes AO/EB to detect apoptosis. Cells were incubated with rotenone (100 nM) and fisetin (5 μM) alone and with fisetin prior to rotenone exposure. After 24 hours of incubation, the medium containing toxin and/or drug was removed. The plate was washed and dried and 200 μl of AO/EB reagent or was added to each well and incubated at 37°C for 10 minutes and observed under Nikon eclipse TS 100 inverted microscope using fluorescence filters. An excitation wavelength of 460 nm and an emission maximum of 650 nm were used for AO/EB staining. In dual staining methods, live cells were observed with normal green nuclei, early apoptotic cells with bright green nuclei and late apoptotic cells with condensed and fragmented orange colored chromatin (18).
Briefly, cells in 6-well plates were harvested and washed with PBS. Cells were lysed in 100 μl lysis buffer (20 mM Tris–HCl, pH 7.4., 150 mM NaCl, 1 mM EDTA, 30 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride) followed by centrifugation at 1000 g for 5 minutes at 4°C. The supernatants (cytosolic fractions) were saved and the pellets solubilized in the same volume of mitochondrial lysis buffer (50 mM Tris pH 7.4., 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2.% Triton X-100, 0.3.% NP-40, 100 μM PMSF, 10 μg/ml leupeptin, 2 μg/ml aprotinin), kept on ice and vortex for 20 minutes followed by pelleting at 10 000g for 10 minutes at 4°C and subjected to 12.5.% polyacrylamide gel electrophoresis. A total volume of 40 μg of protein was loaded per lane. The separated proteins were blotted onto a PVDF membrane by semi-dry transfer (Bio-rad). After being blocked for 1 hour with 5% defatted milk in Tris-buffered saline (TBS), which contained 0.0.5% Tween-20 (TBST) at 37°C, blots were incubated with primary antibody against Bcl-2, Bax at a dilution of (1:500), Caspase-3 (1:1000), p-JNK, p-P38 and p-ERK (1:1,000), pPI3K (1:1250), pAkt (1:1200), p-GSK3β (1:1250) and β-actin (1:1000) overnight at 4°C. After washing, the membrane was incubated with anti-rabbit HRP conjugated secondary antibody (1:2000), and bands were detected by treating the membranes with 3,3?-diaminobenzidine tetrahydrochloride (Western blot detection reagent, Sigma, USA) and densitometry was done using ‘Image J’analysis software (19).
All data were expressed as mean ± Standard deviation (SD) of four number of experiment. The
statistical significance was evaluated by one-way analysis of variance (ANOVA) using SPSS version 15.0. (SPSS, Cary, NC, USA) and the individual comparison were obtained by Duncan’s Multiple Range Test (DMRT). Values not sharing common superscript are significant with each other at P<0.0.5.
MTT assay was used to find out the LD50 of rotenone toxicity and optimum dose of fisetin treatment. Rotenone treatment to SH-SY5Y cells induced cytotoxicity, with approximately 50% viability at 100 nM (LD50). Pretreatment with fisetin dose-dependently (0, 0.5, 1, 2.5, 5, 10, 15, 20, 25 and 50 μM) protected against cytotoxicity induced by 100 nM rotenone (Figure 1, A, B) with 80% protection at 5 μM in fisetin. A further increase in fisetin concentration decreased cell viability as compared to 5 µM fisetin pretreatment. In addition treatment of 5 µM fisetin and 100 nM rotenone for 24 h incubation used as further in vitro studies in SH-SY5Y cells.
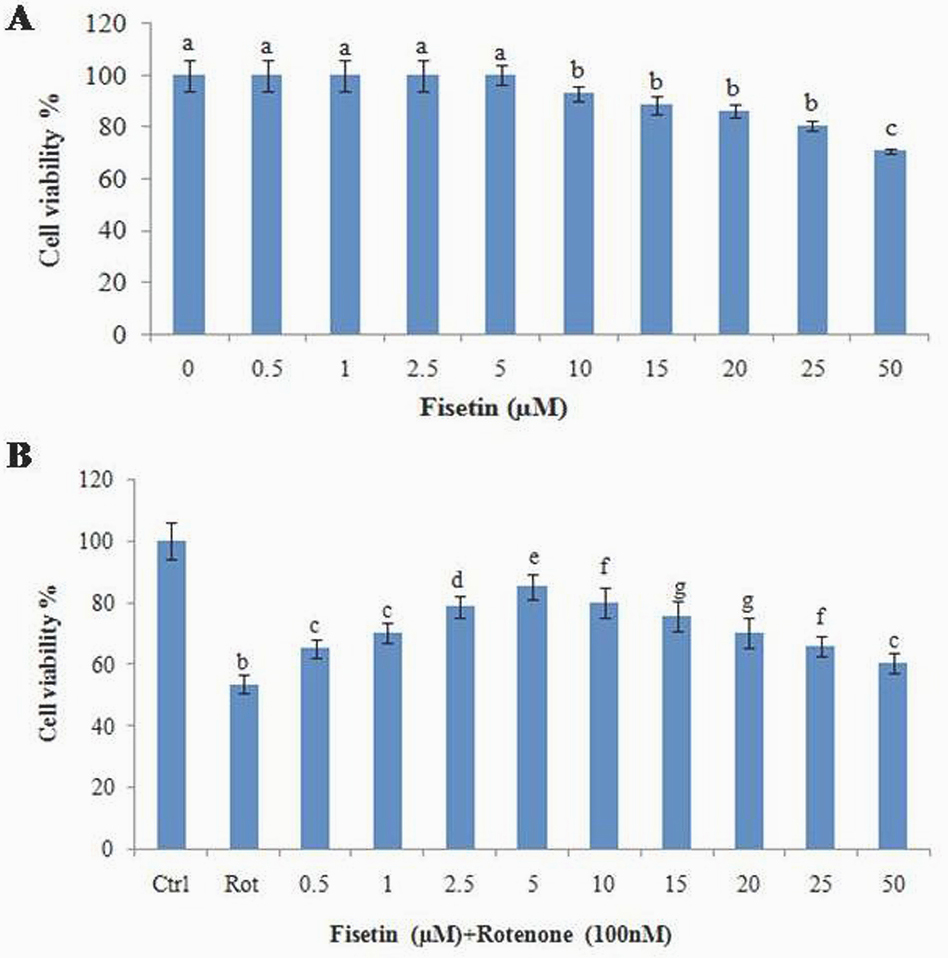 Figure 1
Figure 1Effect of fisetin on rotenone induced cytotoxicity (MTT assay) in SH-SY5Y neuroblastoma cells. (A) shows the dose-dependent effect of fisetin at various concentrations (0, 1, 2.5., 5, 10, 15, 20, 25, and 50 µM) did not induce any toxicity after 24 h treatment. (B) Shows the protective effect of fisetin against rotenone induced cell death. Values are expressed as the percentage of the untreated control and represented as mean ± SD of four independent experiments in each group.
The levels of TBARS and GSH in rotenone treated SH-SY5Y cells incubated with or without fisetin. Compared with untreated cells, rotenone treatment (100 nM) increased the levels of TBARS and decreased the levels of GSH significantly in SH-SY5Y cells. Pre-treatment with fisetin 5 µM to rotenone decreased levels of TBARS and enhanced GSH significantly, compared to rotenone alone treated group (p<0.05). The activities of SOD, catalase and Gpx in rotenone treated SH-SY5Y cells incubated with or without fisetin. Compared with untreated cells, rotenone (100 nM) treatment increased SOD, catalase and Gpx activities in SH-SY5Y cells. Pre-treatment with fisetin to rotenone decreased the activities of SOD, catalase and Gpx significantly, compared to rotenone alone treated group (Figure 2A-E) (p<0.0.5).
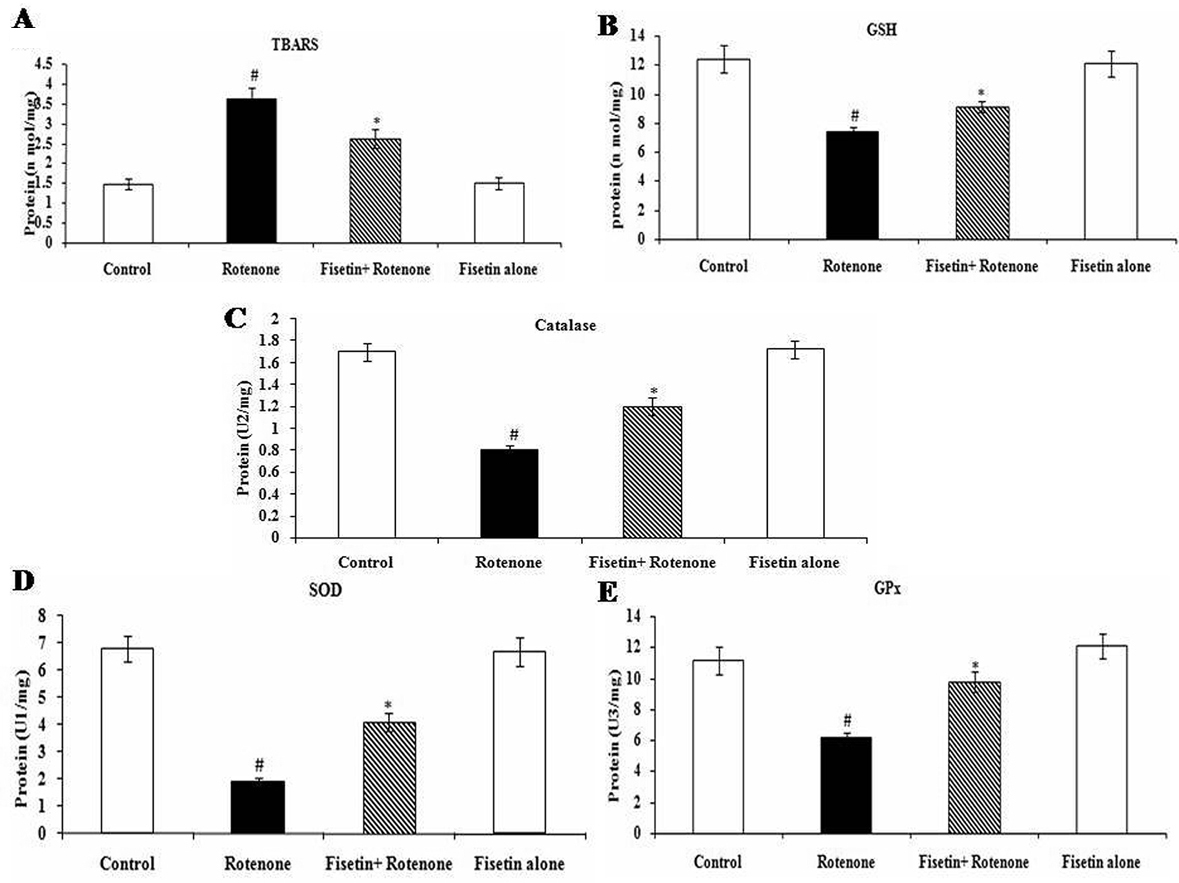 Figure 2
Figure 2Effect of fisetin on rotenone inducedoxidative stress. Rotenone (100 nM) treatment significantly increased the levels of TBARS and diminished the levels of GSH as compared with control cells, while fisetin (5μM) pre-treatment significantly attenuated rotenone-induced oxidative stress (SOD, Catalase, GPx). Values are given as mean ± SD of four independent experiments in each group. *P < 0.0.5. compared to control and #P < 0.0.5. compared to the rotenone group (DMRT). A Enzyme concentration required for 50% inhibition of NBT reduction in 1 minute. B Micromoles of hydrogen peroxide consumed per minute. C Micrograms of glutathione consumed per minute.
AO/EB staining reveals distinctive characteristic of apoptotic morphology in SH-SY5Y cells. This method discriminates viable cells with uniform bright green nuclei and non-viable cells with orange to red nuclei. The results obtained from AO/EB staining are presented in (Figure 3) Control cells fluoresced brightly with green nuclei and showed normal morphology. In contrast, at 100 nM rotenone exposure cells revealed orange luminescent apoptotic body formation. Distinctly, treatment with fisetin increased cell viability and decreased apoptotic cell death when compared to cells exposed solely to rotenone (p<0.05).
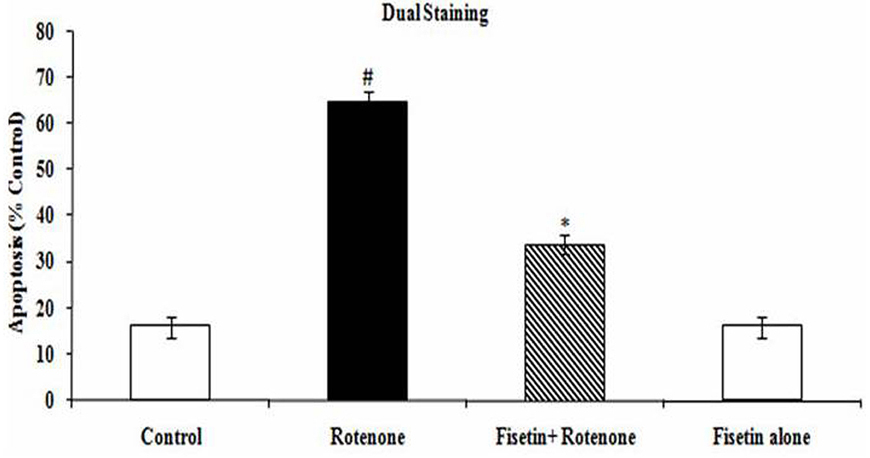 Figure 3
Figure 3Effect of fisetin on rotenone induced apoptosis. (A) Photomicrograph showing the antiapoptotic effect of fisetin (5 µM) against rotenone at a concentration of 100 nM effective dose. Rotenone treatment induced cell apoptosis compared to control cells; pretreatment with fisetin (5 µM) suppresses these apoptotic features. Values are given as mean ± SD of four independent experiments in each group. p< 0.0.5 compared to control and p< 0.0.5 compared to rotenone group (DMRT).
Rotenone treatment (100 nM) decreased the protein expression levels of antiapoptotic B-cell CLL/lymphoma 2 (Bcl-2) and increased the protein expression levels of proapoptotic Bcl-2-associated X protein (Bax). Fisetin pretreatment 5 µM attenuated the rotenone-induced reduction in Bcl-2 expression and increased expression of Bax (p<0.05). We also examined the effect of Fisetin 5 µM on the protein expression of caspases-3. Treatment with rotenone (100 nM) for 24 h increased the expression of caspases-3 at the protein level. Fisetin pretreatment significantly ameliorated the rotenone-mediated increase caspases-3 protein expression levels (p<0.05) (Figure 4A-B).
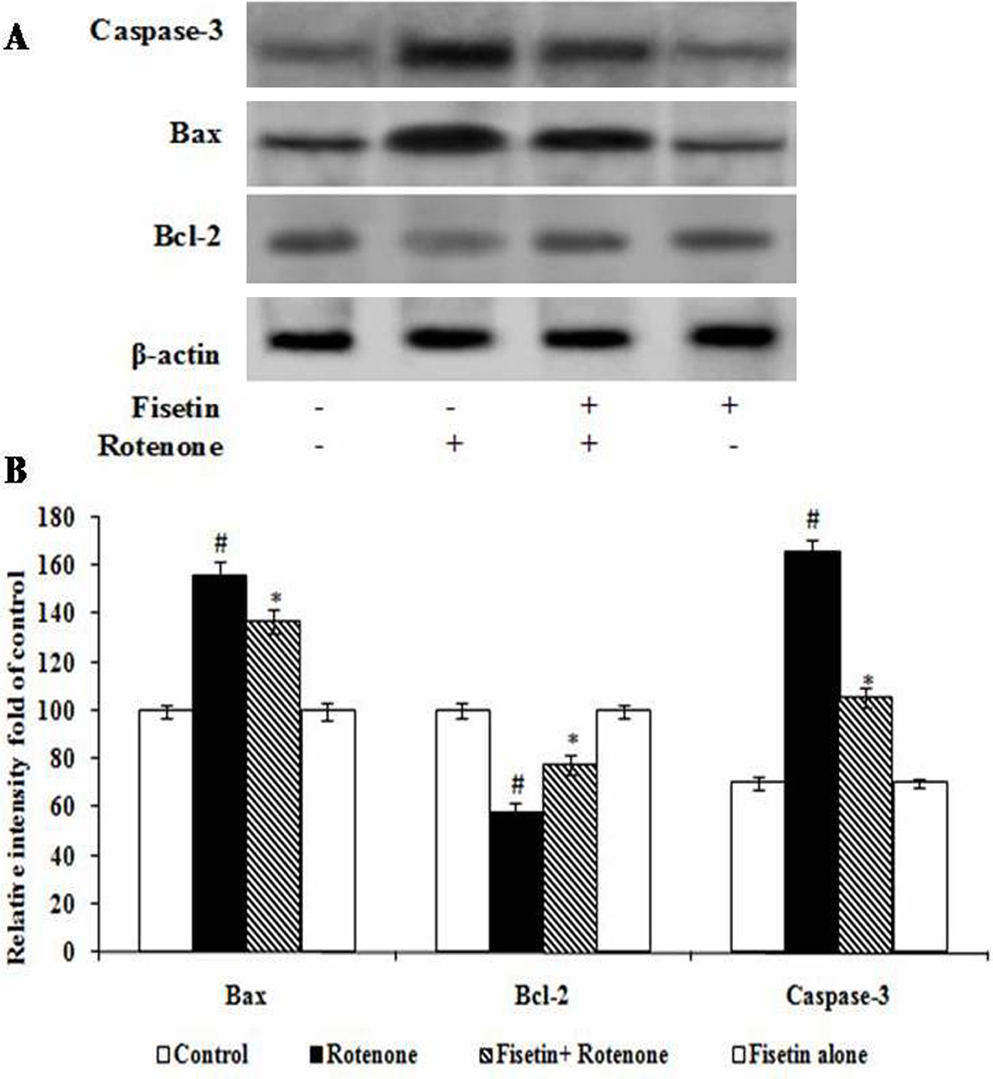 Figure 4
Figure 4Effect of fisetin on the expressions of apoptotic markers. show the expressions of Bax; caspase-3 were increased while the expression of Bcl-2 were significantly decreased by the rotenone treated group as compared with control. Pretreatment with fisetin gradually restored the imbalanced expression profile of these proteins. Values are given as mean ± SD in each group. *p < 0.0.5 compared to control and #p< 0.0.5 compared to rotenone group (DMRT).
Western blot analysis demonstrated that rotenone treatment significantly increased the expressions of p-P38, p-ERK and p-JNK as compared with the control group (p<0.05). Treatment with fisetin induced a significant decrease in p-P38 MAPK, p-ERK and p-JNK, as compared with the rotenone group (p<0.05). These results suggest that the neuroprotective effects of fisetin on rotenone induced neurotoxicity were mediated through the inhibition of JNK, ERK and P38 MAPK-mediated signalling pathways (Figure 5A, B).
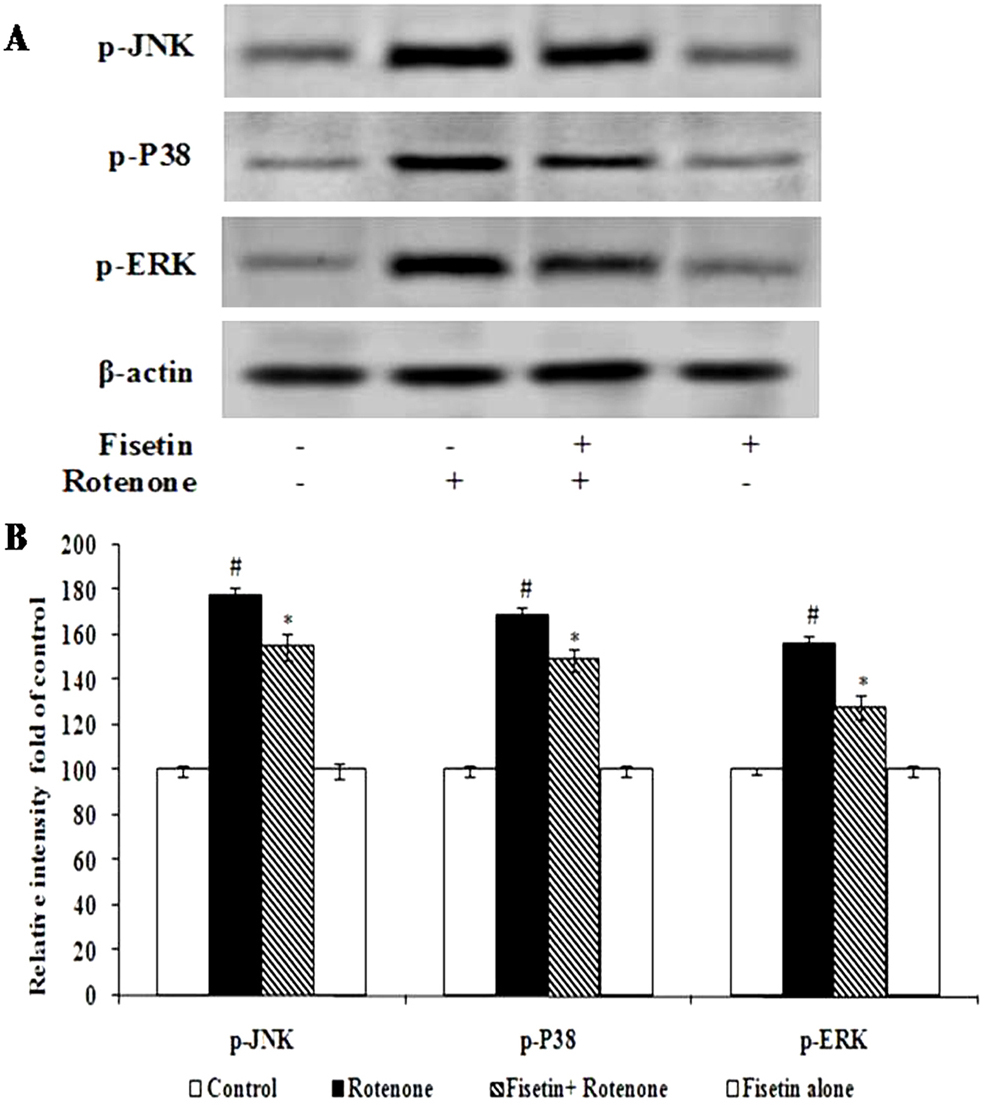 Figure 5
Figure 5Effect of fisetin on the expressions of signaling markers. Rotenone treatment stimulates the expressions of p-JNK, p-P38, and p-ERK as compared with control. Pretreatment with fisetin decreases the expressions of p-JNK, p-P38, and p-ERK significantly. Immunoblots are representative of at least four independent experiments. Values are given as mean ± SD in each group. p< 0.0.5 compared to control and p< 0.0.5 compared to rotenone group (DMRT).
Compared with the controls, the levels of p-PI3K and p-Akt (Ser473) were markedly decreased in rotenone treated cells (p <0.05). The anti-apoptotic function of Akt was induced allowing the phosphorylation of its downstream substrate: GSK3β. The phosphorylated forms at serine residue 9 for GSK3β were measured in these groups against their unphosphorylated form. rotenone administration induced a large reduction in the levels of phosphorylated-GSK3β compared with the control group (p<0.05). Pre-treatment with fisetin prevented the reduction (Figure 6A-B).
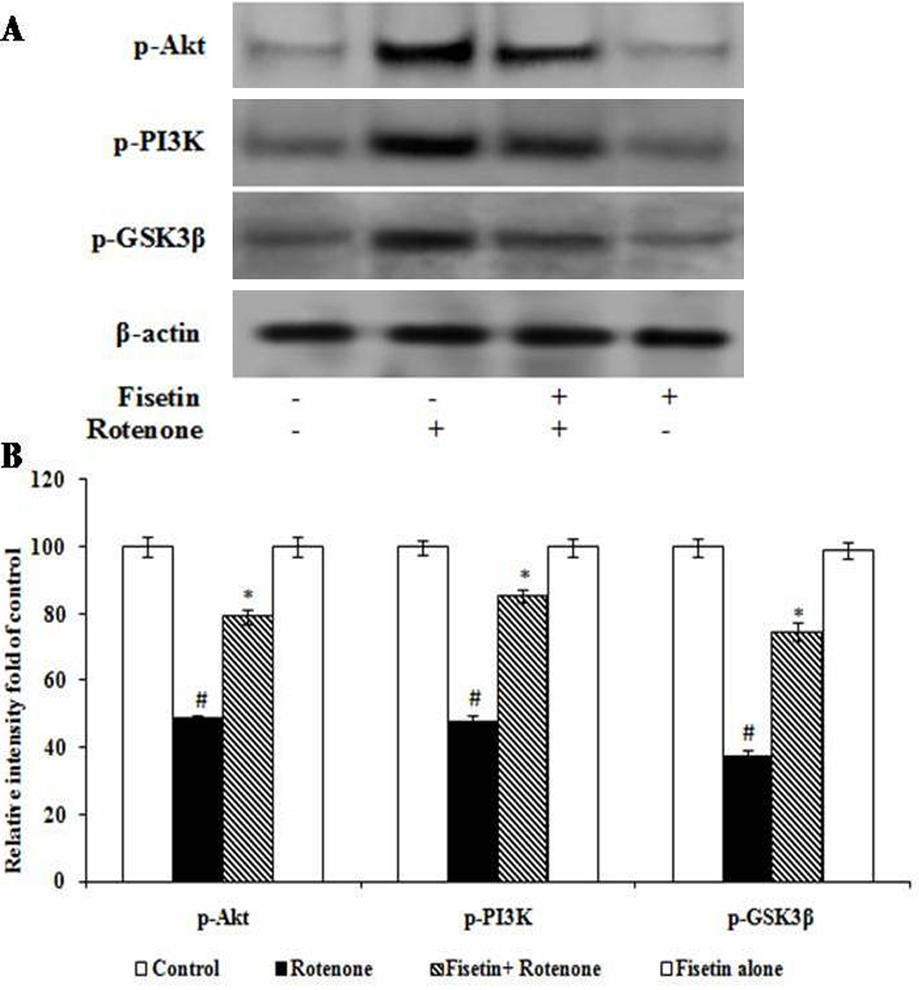 Figure 6
Figure 6Fisetin alleviated rotenone-induced P13K/AKT/GSK-3β pathway. Immunoblots of p-P13K, p-AKT, p-GSK-3 expression and relative optical density normalized to β-actin in each group as indicated. Values are expressed as arbitrary units and given as mean ± SD. *p< 0.0.5. compared to the control group, #p<0.0.5. compared to the rotenone treated cell.
Although 1-Methyl-4-phenyl pyridinium (MPP+), rotenone, and paraquat are used to induce PD in in vitro conditions, (20-21) rotenone is commonly used because of their lipophilic nature, so that it can enter into a cell without transporter, induce greater toxicity even at a lower concentration with a very shorter exposure time and well-known mechanisms of toxicity. Rotenone treatment for 24 hours induced a dose-dependent cell death in SH-SY5Y cells and about 50% of cell viability was obtained at 100 nM rotenone concentration, which is corroborated with previous studies (15, 17). Fisetin exposure enhanced the cell survival in the H2O2-induced PC12 cell death and protected PC12 cells from the oxidative insult (22), which is corroborated with our study. The neuroprotective effect of fisetin in MTT assay is also supported by the finding of the morphological analyses obtained with dual staining.
Rotenone exposure enhanced the levels of TBARS indicating overproduction of free radicals along with glutathione depletion, which is in consistent with previous studies (16, 23). Rotenone inhibits complex I activity, leaks the electrons to oxygen leading to the formation of superoxide ion (O2-) (24). O2-, undergoes spontaneous or SOD-catalyzed dismutation to form H2O2. Catalase and Gpx catalyzes the breakown of H2O2 to H2O and O2. Increased production of ROS during neurodegenerative diseases is an indication of the oxidative stress and leads to a rapid consumption and depletion of endogenous scavenging antioxidants. Decreased activities of enzymatic antioxidants such as SOD, catalase and GPx were probably due to a response towards increased concentration of reactive oxygen species and lipid peroxidation in rotenone treated cells. Moreover, GSH depletion, the first indicator of oxidative stress during PD progression, suggests a concomitant increase in reactive oxygen species. Pre-treatment of fiestin diminished the levels of TBARS and enhanced the levels of GSH and the activities of the GPx, SOD and catalase due to its potent antioxidant function. The presence of 3', 4’-dihydroxy catechol structure in B ring, 2, 3 unsaturation together with an oxo function in C ring and coplanarity of the molecule in fisetin are responsible for its antioxidant scavenger and cell protective activities (25).
The extracellular-signal regulated kinase 1/2 (ERK1/2), c-Jun NH2-terminal kinase (JNK), and P38 MAPK are belongs to MAPK subfamilies and play a key role in the neuronal differentiation, survival, and death (26-27). Rotenone treatment in our study triggers apoptosis by inducing the phosphorylation of intracellular 38/JNK MAPK proteins. Their activation enhances the recruitment of cytoplasmic Bax to mitochondria by forming permeability transition pore, causes the of cytochrome c release, which further activates apoptosis (26). Bax gets phosphorylated by JNK- and/ or p38K-signaling molecules in mitochondria, which may be a key marker for mitochondrial membrane permeability (MMP) change and apoptosis (28). Fisetin treatment attenuated the expressions of signaling and apoptotic indices may be due to its anti-apoptotic properties.
AKT regulates neuronal survival, growth and function by activating PI3K (17) through phosphorylating GSK3β. GSK3β is considered as important kinase associated with the regulation of cellular apoptosis and survival by modulating mitochondrial mediated apoptotic pathways (29), including the activation of mitochondrial complex I activity and B-cell lymphoma (Bcl-2) related family of proteins (30). Other findings indicated that the over-expression of the active form of mitochondrial GSK3β favours the apoptotic effect of rotenone (31). GSK-3β activity is regulated by the action of kinases involving phosphorylation at Ser 9 and Tyr 216 (32). Phosphorylation at Ser 9 inhibits its kinase activity, whereas phosphorylation at Tyr 216 enhances its activity. Previous studies also indicated that the active form of GSK-3β (p-GSK-3 Tyr216) was enhanced in the postmortem brain of PD patients (33). Activation of PI3K/AKT signaling pathway induces the phosphorylation of GSK-3β at Ser9, thereby inhibits its activity and hence cell death (34). Our results indicated that the rotenone treatment down-regulated the expression of Ser 9 p-GSK-3β and pAKT thereby inducing apoptosis. Fisetin pre-treatment offered antiapoptic effect by preventing the decline in the expression of p-AKT, resulting in increased expression of pSer9-GSK-3β clearly confirming its protective effects.
Beyond its wide usage and commercial values, our findings suggested that fisetin could counteract apoptosis induced by rotenone by preserving oxidative stress and antioxidant action. Though rotenone induced in vitro model of PD is considered as a good model, which resembles various pathological features of clinical studies, further extensive research is warranted to find out the effect of fisetin in animal and clinical models of PD.
Key Words: Fisetin, Oxidative stress, Mitochondria Dysfunction, Rotenone, Apoptosis, Signaling pathway