





 , Chuan Yao 1, Luqi Zhang 1, Wenbo Liu 1, Yang Liu 1, Tianqi Gao 1, Ziyu Zhang 1, Jiayi Dang 1, Chenguang Zhao 1
, Chuan Yao 1, Luqi Zhang 1, Wenbo Liu 1, Yang Liu 1, Tianqi Gao 1, Ziyu Zhang 1, Jiayi Dang 1, Chenguang Zhao 11 Key Laboratory of Micro-Nano Materials for Energy Storage and Conversion of Henan Province, Institute of Surface Micro and Nano Materials, College of Chemical and Materials Engineering, Xuchang University, 461000 Xuchang, Henan, China
Abstract
A novel electrochemical sensor for cadmium ions (Cd2+) was fabricated by electropolymerizing L-phenylalanine on a glassy carbon electrode (PPhe/GCE). The resulting polyphenylalanine (PPhe) film combines a conductive polymer backbone with abundant amino/carboxyl groups for coordination and aromatic rings for π-cation interactions, offering synergistic pre-concentration for Cd2+. Under optimized conditions, the sensor exhibited a wide linear range from 0.50 to 500 μM, a detection limit of 0.115 μM, high selectivity, and good reproducibility. Determination of Cd2+ in spiked lake water samples with recoveries of 96.2%–102.2% demonstrates its potential for environmental water monitoring.
Keywords
- L-phenylalanine
- electropolymerization
- electrochemical sensor
- anodic stripping voltammetry
- cadmium detection
The contamination of aquatic systems by heavy metals, stemming from rapid industrialization and agricultural practices, poses a severe and persistent threat to global ecosystems and human health [1, 2]. Distinguished from degradable organic pollutants, heavy metals represent persistent environmental contaminants due to their non-biodegradable and bioaccumulative nature, enabling the manifestation of toxic effects at minute concentrations [3, 4]. Cadmium ion (Cd2+) is among the most perilous heavy metals due to its high toxicity, extensive mobility in the environment, and long biological half-life [5, 6]. Cd2+ has been implicated in a spectrum of severe health disorders upon chronic exposure, notably renal dysfunction, bone demineralization, and an elevated risk of various cancers [7, 8]. It is therefore imperative to devise rapid, sensitive, and field-deployable methods for assessing Cd2+ levels in water, which is vital for safeguarding environmental integrity and ensuring public health safety.
The conventional techniques for heavy metal analysis, such as atomic absorption spectrometry (AAS) [9], inductively coupled plasma mass spectrometry (ICP-MS) [10] and surface-enhanced Raman spectroscopy (SERS) [11], are recognized as standard methods for their high accuracy and sensitivity. However, their widespread deployment for routine monitoring is often constrained by significant drawbacks, including the high cost of instrumentation and maintenance, complex and time-consuming sample preparation procedures, the necessity for highly skilled operators, and their inherent lack of portability for field analysis [12, 13]. Electrochemical methods, particularly anodic stripping voltammetry (ASV), have emerged as powerful and viable alternatives, offering a compelling combination of high sensitivity, excellent selectivity, portability, low cost, and suitability for real-time and in-situ analysis [14, 15]. The performance of an ASV-based sensor is critically dependent on the working electrode. Bare traditional electrodes often suffer from poor sensitivity, fouling, and limited selectivity. A frequently employed solution is to engineer the electrode interface with functional materials, thereby enhancing the electroactive area, facilitating electron transfer, and offering tailored recognition sites for the target analyte [16, 17]. Accordingly, considerable exploration has been dedicated to a broad spectrum of advanced materials for this purpose, including carbon nanomaterials (e.g., graphene, carbon nanotubes) [18, 19], metal/metal oxide nanoparticles [20, 21], metal-organic frameworks (MOFs) [22, 23], and conducting polymers [24, 25], serving as effective modifiers in electrochemical detection platforms.
Among these, conducting polymers are particularly attractive due to their unique
electrical conductivity, environmental stability, facile synthesis, and the ease
with which their properties can be tuned through molecular design [26].
Polyaniline, polypyrrole, and their derivatives have been widely studied. For
instance, a recently reported N-CQDs@
We present herein an electrochemical sensor for Cd2+ based on a PPhe-modified glassy carbon electrode (GCE), detailing its fabrication, thorough characterization, and performance. The key PPhe modifier was engineered through a facile, single-step electropolymerization strategy that aligns with green chemistry principles. A comprehensive investigation was conducted to optimize all critical parameters affecting the sensor’s performance. The electrochemical interface and the reaction kinetics were probed in detail. The analytical performance was evaluated, demonstrating a wide linear range, a low detection limit, excellent reproducibility, and remarkable anti-interference ability. The practical utility of the proposed PPhe/GCE sensor was convincingly demonstrated by the determination of Cd2+ in real environmental water samples with high accuracy and precision, showcasing its great potential as a reliable tool for environmental monitoring.
L-phenylalanine (L-Phe,
Electrochemical measurements were performed with a CHI 660E workstation (CH Instruments, Shanghai, China). A conventional three-electrode system was used: a bare or modified glassy carbon electrode (GCE, 3 mm diameter) as the working electrode, a platinum wire as the counter electrode, and a saturated calomel electrode (SCE, 0.241 V vs. SHE) as the reference. All potentials are reported vs. SCE. pH measurements were made with a PHSJ-3F pH meter (INESA Scientific Instrument Co., Ltd., Shanghai, China).
The pretreatment procedure involved successively polishing the bare GCE with aqueous alumina slurries of decreasing particle size (1.0, 0.3, and 0.05 µm) on a micro-cloth pad until a mirror finish was attained. It was then subjected to ultrasonic cleaning in ethanol and ultra-pure water for 2 minutes each to remove any adsorbed alumina particles. Electrochemical activation was performed in 0.5 M H2SO4 by cycling from –1.0 to +1.0 V until a stable baseline was obtained. Electropolymerization of L-Phe was conducted from a 2 mM solution (1 mM HCl + 0.1 M KCl) by applying 8 cyclic voltammetry cycles between –1.0 and +1.0 V at 100 mV s-1. The formation of a light-yellow film indicated polymerization. The modified PPhe/GCE was then thoroughly rinsed with water, air-dried, and subsequently employed for electrochemical measurements.
The electropolymerization of L-Phe proceeds via electrochemical oxidation of the amine group, generating radical intermediates that couple to form a polymeric network, similar to mechanisms reported for other aromatic amines [31]. HCl provides a protonic medium to facilitate polymerization, while KCl acts as the supporting electrolyte for ionic conductivity.
For electrochemical characterization, the modified electrodes were subjected to cyclic voltammetry (CV) analysis in a solution containing a 5.0 mM 1:1 mixture of K3[Fe(CN)6] and K4[Fe(CN)6], with 0.1 M KCl as the supporting electrolyte. For the detection of Cd2+, differential pulse anodic stripping voltammetry (DPASV) was employed. The analysis followed a standard two-step procedure: (1) Pre-concentration/deposition: the PPhe/GCE was immersed in the sample solution, and a deposition potential of –1.0 V was applied for 120 s under stirred conditions. This step reduces Cd2+ ions and accumulates elemental Cd (0) onto the electrode surface. (2) Stripping scan: after a 10 s quiet time without stirring, the stripping step was carried out by applying a differential pulse voltammetry (DPV) scan from –1.0 V to –0.6 V, employing a pulse amplitude of 50 mV, a pulse width of 50 ms, and a potential step of 5 mV. Quantification was based on the current of the anodic stripping peak (observed around –0.8 V), which signifies the oxidation of Cd (0) to Cd2+.
Water samples were collected from Luming Lake (Xuchang, Henan, China), filtered through a 0.45 µm membrane. Acidification with 0.01 M HCl converts Cd2+ into its free, electroactive form by dissociating complexes with natural organic matter. Dilution (four-fold) reduces ionic strength and mitigates matrix adsorption on the electrode surface, which is a common practice to ensure reliable quantification in complex environmental samples. Cd2+ was determined by the standard addition method using DPASV under optimized conditions.
The electrochemical behavior of bare GCE and PPhe/GCE was first examined using CV in 5 mM [Fe(CN)6]3⁻/4⁻ containing 0.1 M KCl. As depicted in Fig. 1a, both electrodes exhibited well-defined reversible redox peaks. The peak currents at PPhe/GCE were approximately 1.5 times higher than those at bare GCE. This marked augmentation of the peak current unequivocally indicates that the electropolymerized PPhe film effectively enhances the electrode performance. The electroactive area, calculated via the Randles–Ševčík equation, was ~1.4 times larger than that of the bare GCE (Supplementary Fig. 1).
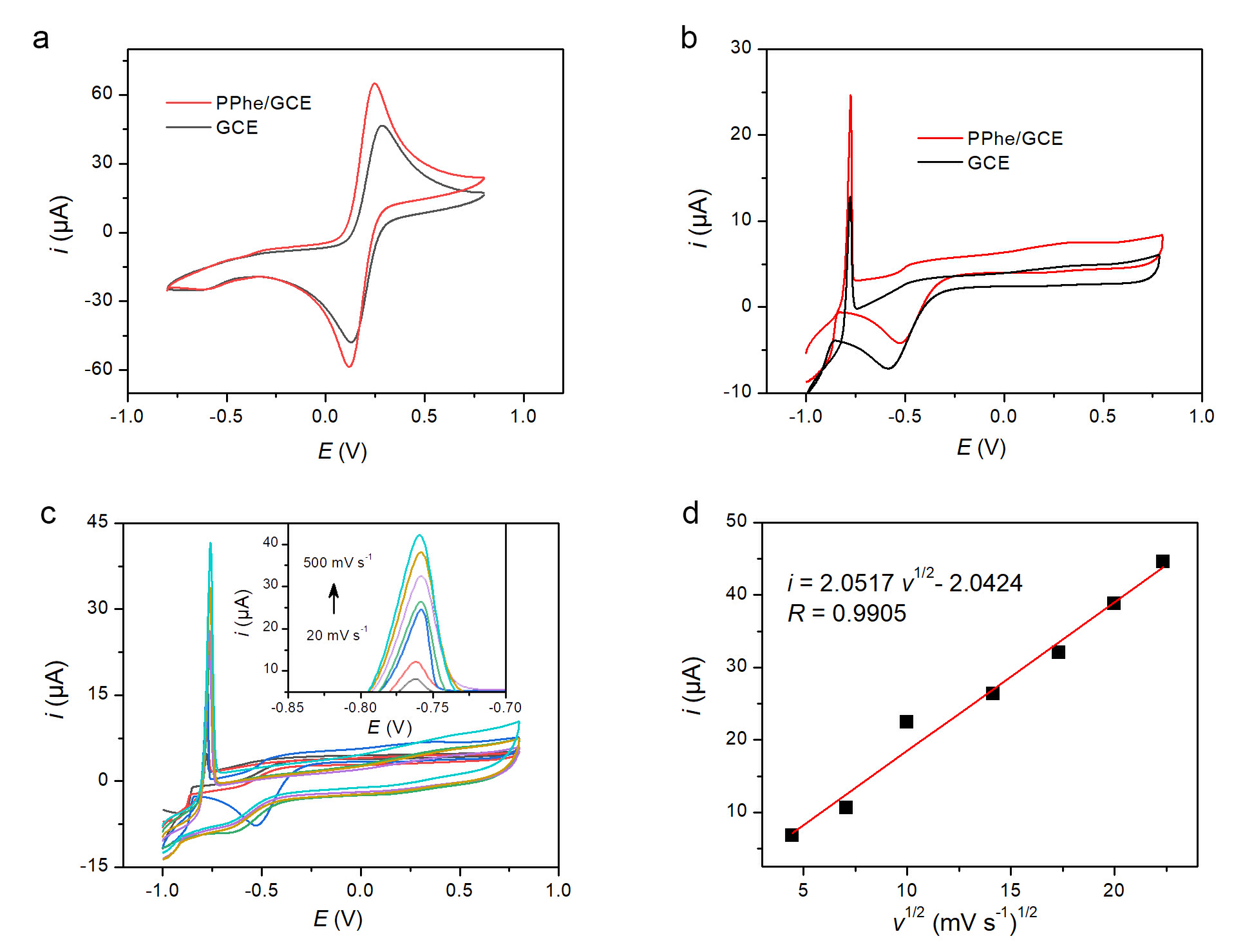 Fig. 1.
Fig. 1.
CV characterization of bare GCE and PPhe/GCE in [Fe(CN)6]3-/4- and Cd2+ solutions. (a) CVs of bare GCE and PPhe/GCE in 5 mM [Fe(CN)6]3-/4- (0.1 M KCl). (b) CVs of the same electrodes in 100 µM Cd2+ (0.1 M KCl, 0.01 M HCl). (c) CVs of PPhe/GCE in 100 µM Cd2+ at different scan rates. (d) Linear relationship between anodic peak current (i) and the square root of scan rate (v1/2). CV, cyclic voltammetry; PPhe/GCE, L-phenylalanine on a glassy carbon electrode; KCl, potassium chloride; HCl, Hydrochloric acid; Cd2+, cadmium ions.
CV responses in 100 µM Cd2+ solution (0.1 M KCl, 0.01 M HCl) are presented in Fig. 1b. A distinct anodic stripping peak appeared around –0.8 V for both electrodes. The peak current at PPhe/GCE was approximately double that at bare GCE.
The effect of scan rate on the stripping peak at PPhe/GCE was investigated (Fig. 1c). The anodic peak current increased with increasing scan rate. A linear relationship was observed between the peak current (i) and the square root of scan rate (v1/2) (Fig. 1d), yielding the regression Eqn. 1 with a correlation coefficient of 0.9905.
In-situ monitoring of the electropolymerization process (Supplementary Fig. 2) showed a characteristic current decay with increasing scan number, indicating progressive film growth. Comparison with other amino-acid-based polymers (Supplementary Fig. 3) revealed that the Cd2+ stripping peak current at PPhe/GCE was approximately 1.3 and 1.7 times higher than that at polyarginine (PArg) and polyglutamate (PGA) modified GCEs, respectively.
Key experimental parameters affecting the sensor’s performance were systematically optimized using DPASV for 100 µM Cd2+. All optimization experiments (Supplementary Fig. 4) were performed in triplicate (n = 3).
The electropolymerization potential window was evaluated (Fig. 2a). The highest oxidation peak current (74.07 µA) was obtained with the window of –1.0 to +1.0 V, compared to 37.52 µA for –0.9 to +1.0 V and 27.27 µA for –1.0 to +0.9 V.
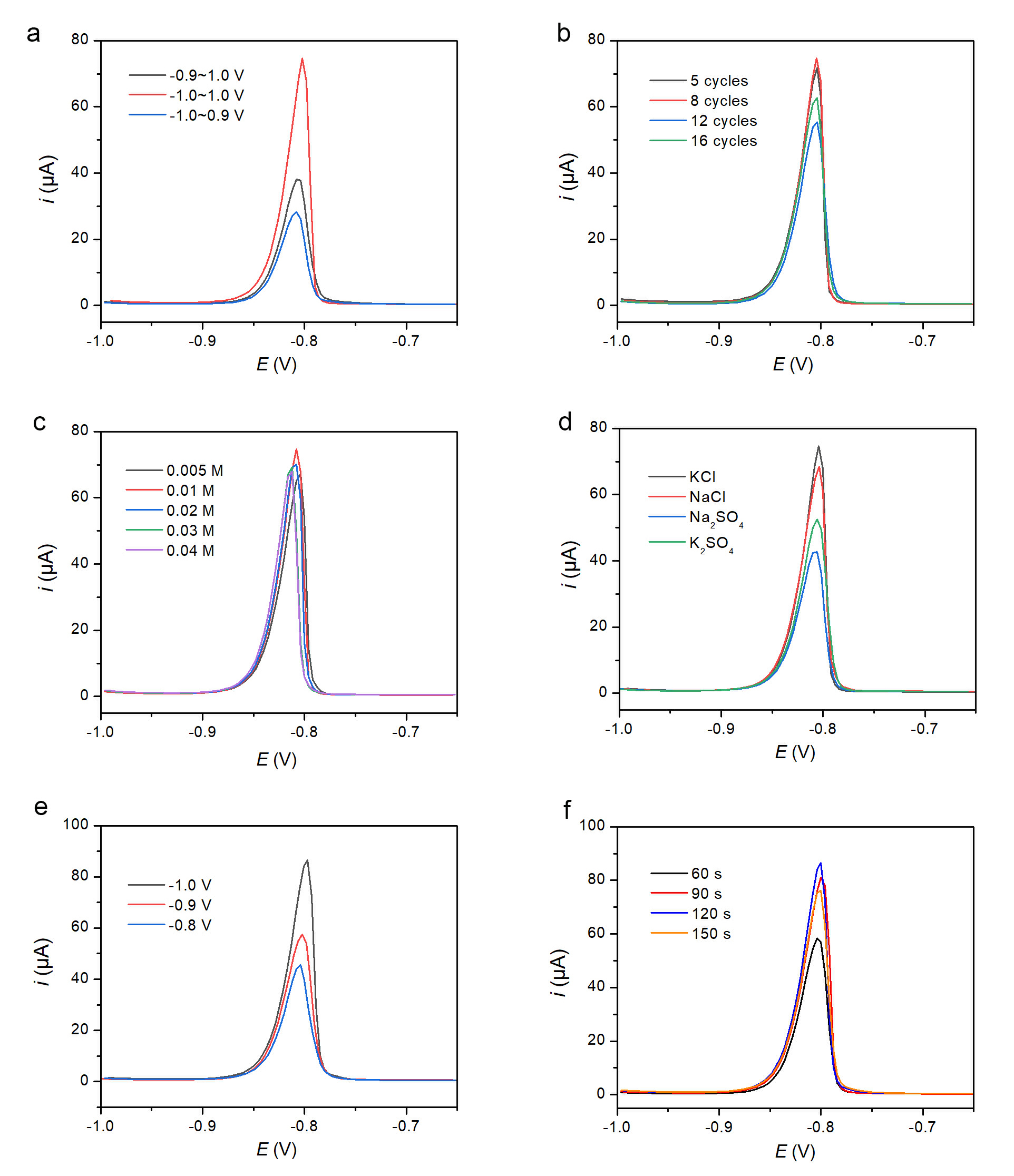 Fig. 2.
Fig. 2.
Optimization of detection parameters for 100 µM Cd2+ at PPhe/GCE. Optimization of detection conditions for 100 µM Cd2+ at PPhe/GCE: (a) Electropolymerization potential window. (b) Number of polymerization cycles. (c) HCl concentration. (d) Supporting electrolyte (0.1 M). (e) Deposition potential. (f) Deposition time.
The number of polymerization cycles was optimized (Fig. 2b). The peak current increased with cycle number up to 8 cycles (74.09 µA) and then decreased upon further cycling (12 and 16 cycles).
The effect of HCl concentration (0.005–0.04 M) is shown in Fig. 2c. The current increased from 0.005 M to 0.01 M (74.06 µA) and then declined at higher concentrations.
The Different supporting electrolytes (0.1 M) were tested in 0.01 M HCl (Fig. 2d). KCl yielded the highest oxidation peak current (74.10 µA), outperforming NaCl, K2SO4, and Na2SO4 by factors of approximately 1.2, 1.4, and 1.8, respectively.
Deposition potential was varied from –0.8 V to –1.0 V (Fig. 2e). The peak current increased with more negative potential, reaching 85.83 µA at –1.0 V, which was 1.5 times higher than at –0.9 V and 1.9 times higher than at –0.8 V.
Deposition time was investigated from 60 to 150 s (Fig. 2f). The current increased up to 120 s (85.81 µA) and then slightly decreased at 150 s.
Under the optimized conditions, DPASV responses for Cd2+ at various concentrations were recorded (Fig. 3a). Well-defined stripping peaks increased with Cd2+ concentration. The calibration plot (Fig. 3b) exhibited a linear relationship from 0.50 to 500 µM, with a regression Eqn. 2 and a correlation coefficient (R) of 0.9962.
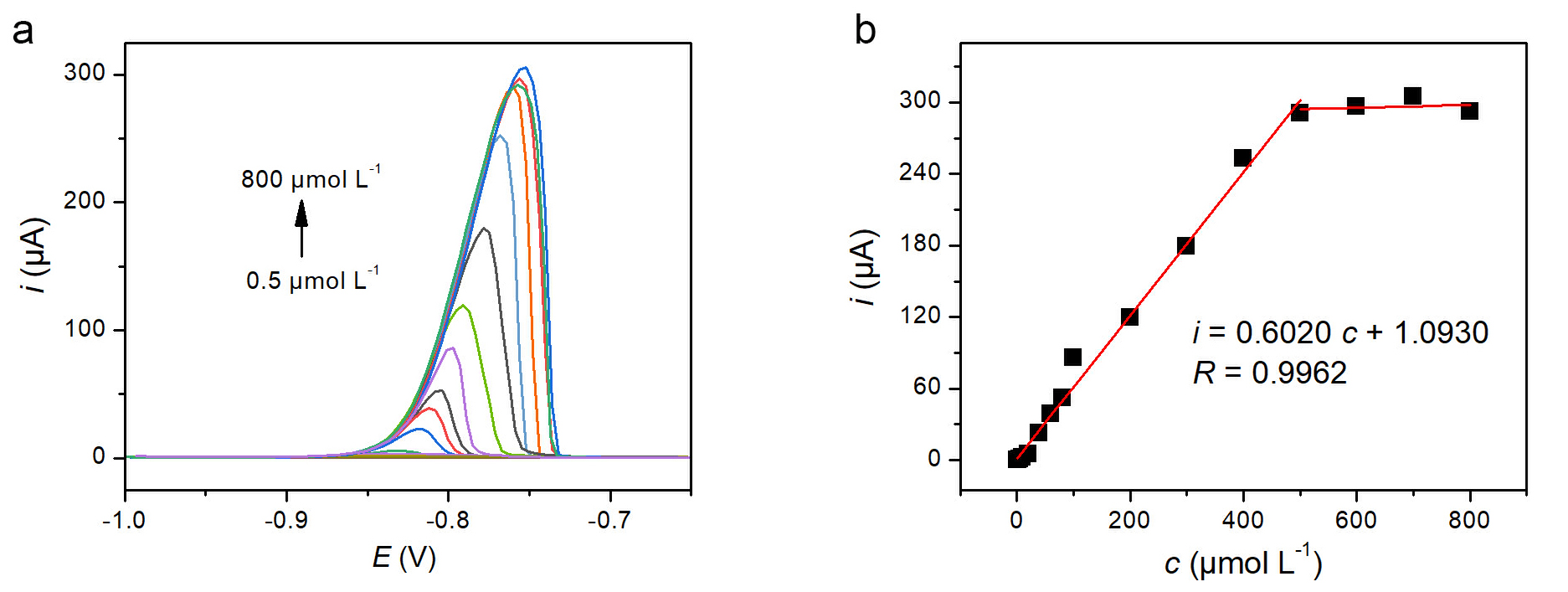 Fig. 3.
Fig. 3.
Analytical performance of PPhe/GCE for Cd2+ detection. (a) Differential pulse anodic stripping voltammetry (DPASV) curves for increasing concentrations of Cd2+ (0.50, 1, 2, 5, 10, 20, 40, 60, 80, 100, 200,300, 400, 500, 600, 700 and 800 µM) at the PPhe/GCE in 0.1 M KCl and 0.01 M HCl solution. (b) Corresponding calibration plot of peak current versus Cd2+ concentration.
The limit of detection (LOD) was calculated to be 0.115 µM based on 3N/S (where N is the standard deviation of the blank, n = 12, and S is the slope of the calibration line). A comparison of the analytical performance of PPhe/GCE with other reported electrodes for Cd2+ detection [32, 33, 34, 35, 36, 37] is summarized in Table 1.
| Electrode modification | Linear range (µM) | Detection limit (µM) | Reference |
| TFT sensor | 0~150 | 0.500 | [32] |
| Au-(Cys)PW | 0.01~0.20 | 0.009 | [33] |
| nZVI-BPC/GCE | 2.0~50.0 | 0.192 | [34] |
| G/HNFQ-CPE | 0.47~93.8 | 0.210 | [35] |
| ACOP/MWCNT-CPE | 9.09~72.9 | 0.910 | [36] |
| ZnSe/GO/EDE | 100~700 | 45.540 | [37] |
| PPhe/GCE | 0.50~800 | 0.115 | This work |
TFT, thick-film technology; BPC/GCE, biomass-derived porous carbon composite modified glassy carbon electrode; ACOP/MWCNT, composed of biomass-based orange peel activated carbon (ACOP) and multiwalled carbon nanotubes (MWCNTs); CPE, carbon paste electrode; GO, graphene oxide; EDE, electrodeposited electrode; (Cys)PW, L-cysteine tungstophosphate.
The selectivity of PPhe/GCE was evaluated by adding a five-fold excess (500
µM) of interfering ions (Ni2+, Mn2+, Mg2+, Fe3+) to 100
µM Cd2+. As shown in Fig. 4a,b, the signal changes were within
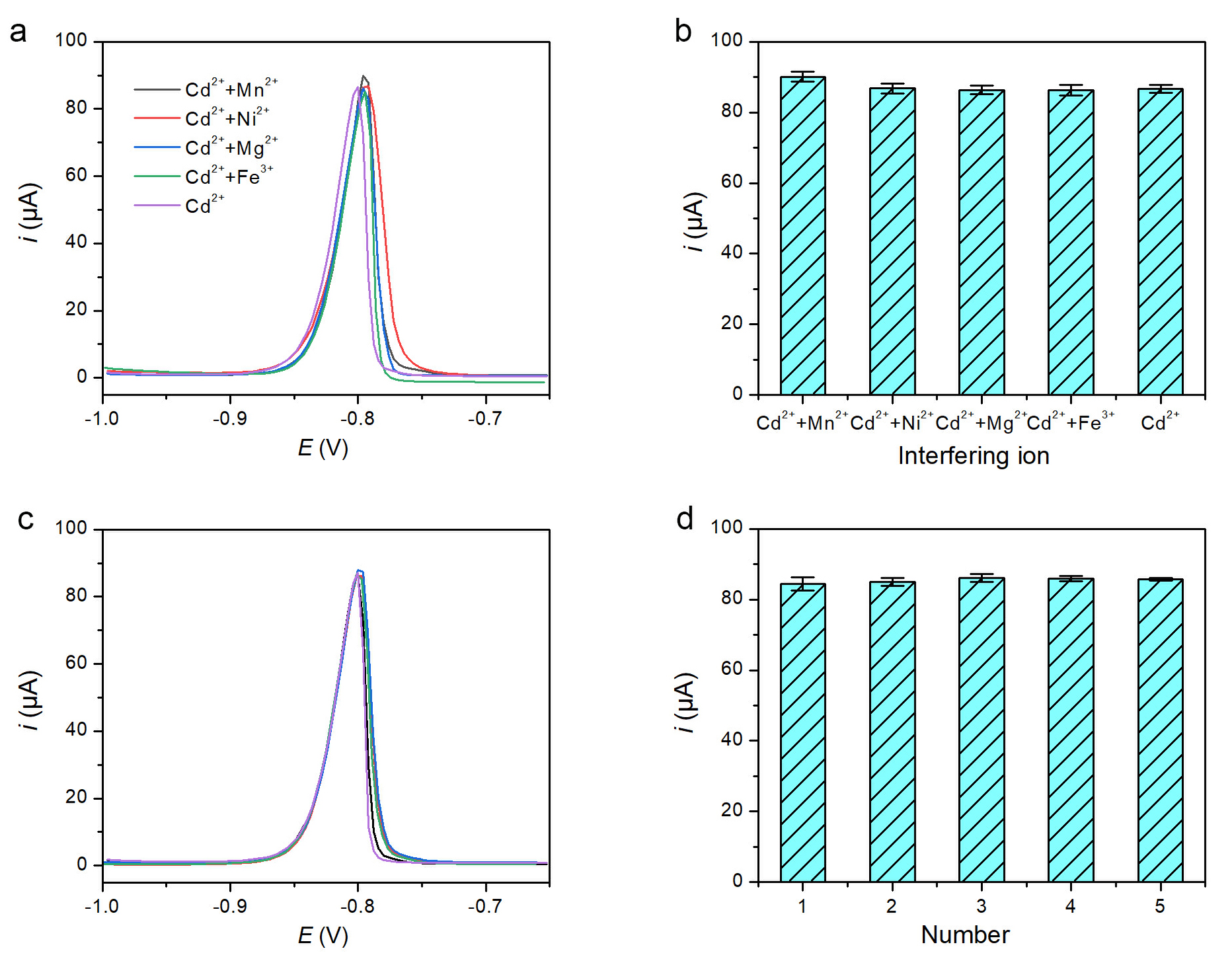 Fig. 4.
Fig. 4.
Selectivity and reproducibility of PPhe/GCE. (a) DPASV curves and (b) current histogram for 100 µM Cd2+ with a 5-fold excess of interfering ions. (c) DPASV responses and (d) current histogram for 100 µM Cd2+ obtained with five independently prepared PPhe/GCEs.
Reproducibility was assessed using five independently prepared PPhe/GCEs. The
DPASV responses and corresponding current histogram are shown in Fig. 4c,d,
yielding a relative standard deviation (RSD) of 2.4% (n = 5).
Repeatability tested with a single electrode over 20 consecutive scans gave an
RSD
The practical applicability of PPhe/GCE was tested by analyzing Cd2+ in lake water samples. No Cd2+ peak was detected in the original unspiked sample. Recovery tests were performed by spiking the samples with 5, 50, and 200 µM Cd2+. As presented in Table 2, the recoveries ranged from 96.2% to 102.2%, with RSD values below 1.5% (n = 3).
| No. | Spiked (µM) | Total (µM) | Recovery (%) | RSD (%, n = 3) |
| 1 | 0 | Not Detected | / | / |
| 2 | 5 | 5.0 | 100.4 | 1.5 |
| 3 | 50 | 51.1 | 102.2 | 1.3 |
| 4 | 200 | 192.4 | 96.2 | 1.3 |
RSD, relative standard deviation.
The results demonstrate that the PPhe-modified electrode significantly enhances the electrochemical response toward Cd2+. The increased peak currents in both [Fe(CN)6]3⁻/4⁻ and Cd2+ solutions (Fig. 1a,b) indicate that the electropolymerized PPhe film effectively enlarges the electroactive surface area and facilitates electron transfer. The ~1.4–fold increase in electroactive area and the ~2–fold enhancement in Cd2+ stripping current suggest that the film provides abundant active sites for analyte accumulation.
The superior performance of PPhe/GCE can be attributed to the synergistic
combination of multiple functional groups within the polymer film. The amino
(–NH2) and carboxyl (–COOH) groups along the polymer backbone serve as
excellent ligands for Cd2+ coordination [28], enabling effective
preconcentration of the analyte onto the electrode surface. Additionally, the
aromatic benzene rings may contribute through
The linear relationship between peak current and the square root of scan rate (Fig. 1d) confirms that the electrode process is diffusion–controlled [38], a typical characteristic of stripping analysis. This finding indicates that the PPhe film, while offering strong preconcentration capability, does not impede the mass transport of Cd2+ from the bulk solution to the electrode interface.
The optimization results (Fig. 2) provide insight into the factors governing sensor performance. The optimal electropolymerization window of –1.0 to +1.0 V (Fig. 2a) ensures complete polymerization and the formation of a conductive film with a high density of accessible binding sites. Restricted windows may yield incomplete polymerization or less favorable film morphologies, compromising electron transfer and analyte accumulation.
The dependence on polymerization cycles (Fig. 2b) reflects a trade-off between film thickness and accessibility. With too few cycles (5 cycles), surface coverage is insufficient, limiting the number of active sites. Conversely, excessive cycling (12 or 16 cycles) produces an overly thick film that may hinder charge transfer and Cd2⁺ diffusion to the electrode surface. 8 cycles provide an optimal balance, maximizing available binding sites while maintaining efficient electron transfer.
The presence of acid in the electrolyte may influence the doping state and conductivity of the polyphenylalanine film (Fig. 2c), as acidic media are known to affect the electrochemical activity of conducting polymers. At the optimal concentration of 0.01 M HCl, the film likely exhibits enhanced conductivity, facilitating electron transfer during the stripping step. Higher acidities may alter the film structure or lead to competing reactions that diminish the signal.
Among supporting electrolytes, KCl yielded the highest response (Fig. 2d). This is consistent with the high mobility of K+ ions and the ability of Cl– to form soluble chlorocomplexes (e.g., CdCl2) during the stripping step, which sharpens the voltammetric peaks. Sulfate electrolytes may form less soluble cadmium species or alter the double-layer structure, impairing stripping efficiency.
The increase in stripping current with more negative deposition potential (Fig. 2e) reflects enhanced reduction of Cd2+. The optimal potential of –1.0 V provides sufficient overpotential for efficient deposition while avoiding hydrogen evolution, which could destabilize the electrode surface. The deposition time of 120 s (Fig. 2f) allows adequate preconcentration without reaching surface saturation, which would lead to a signal plateau or decline due to the formation of a resistive cadmium layer.
The PPhe/GCE sensor exhibits a wide linear range (0.50–500 µM) and a low detection limit (0.115 µM) for Cd2+ (Fig. 3). As shown in Table 1, these analytical figures are competitive with or superior to many previously reported Cd2+ sensors [32, 33, 34, 35, 36, 37]. Notably, the detection limit is lower than that of sensors based on carbon composites and comparable to more complex systems.
In addition, the fabrication process of PPhe/GCE is simpler and more environmentally friendly than many alternative approaches. Unlike sensors requiring multi-step synthesis of nanomaterials, the one-step electropolymerization of L-phenylalanine is both time-efficient and green. This combination of high performance and simple fabrication underscores the promise of amino-acid-derived polymers as advanced sensing materials.
The excellent selectivity of PPhe/GCE against common interfering ions (signal
changes
The successful determination of Cd2+ in spiked lake water samples with recoveries of 96.2%–102.2% and low RSDs (Table 2) demonstrates that the sensor performs reliably even in complex environmental matrices. These results validate its potential for real-world environmental monitoring applications.
The present work primarily focuses on demonstrating the analytical utility and performance of the PPhe-based electrochemical platform for cadmium detection. While the sensor exhibits promising characteristics, a few aspects may be considered in subsequent studies to further support its development. For instance, future work could explore the fine structural features of the polymer film through advanced characterization techniques.
In this work, we developed a high-performance electrochemical sensor for cadmium ions based on an electropolymerized L-phenylalanine-modified GCE (PPhe/GCE). The simple, one-step fabrication aligns with green chemistry principles. The designed PPhe modifier creates a synergistic interface that significantly amplifies the anodic stripping signal for Cd2+. The optimized sensor exhibits a wide linear range (0.50 to 800 µM), a low detection limit (0.115 µM), excellent reproducibility, and high selectivity. Its practical reliability was confirmed through accurate analysis of spiked environmental water samples. This study provides a robust and economical sensing platform for cadmium detection and underscores the efficacy of amino acid-derived polymers in developing advanced electrochemical sensors.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
HS: methodology, formal analysis, data curation, writing-review and editing, supervision. CY: methodology, validation, formal analysis, data curation, writing-original draft. LZ: investigation, validation, review, and editing. WL: investigation, review and editing. YL: investigation, review and editing. TG: investigation, review and editing. ZZ: investigation, review and editing. JD: validation, review and editing. CZ: resources, review and editing. All authors reviewed and approved the final manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was financially supported by the Key Scientific Research Project of Henan Provincial Institution of Higher Learning (No. 202510480019, S202510480026).
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGPT-3.5 to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/DJNB52167.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.