

















 , Jian Ding 1
, Jian Ding 11 Fourth Institute of Geological and Mineral Exploration of Gansu Provincial Bureau of Geology and Mineral Resources, 735000 Jiuquan, Gansu, China
Abstract
A magnetic biochar–nanoscale zero-valent iron (MBC-nZVI) composite was synthesized from pine bark via an in-situ liquid-phase reduction method to immobilize arsenic in mining-impacted soils. The optimized MBC-40 exhibited uniformly dispersed Fe(0)@Fe3O4 nanoparticles (50–80 nm) anchored on a graphitic biochar scaffold, combining a high surface area (180.9 m2 g-1) with strong magnetization (32.5 emu g-1). Batch experiments revealed outstanding performance, achieving 98.5% As(V) and 96.5% As(III) removal under neutral conditions, with equilibrium reached within 120 min. Adsorption kinetics were well described by the pseudo-second-order model (R2 >0.998) and equilibrium data were consistent with the Langmuir model, yielding maximum capacities of 65.4 mg g-1 for As(V) and 82.1 mg g-1 for As(III). Based on the observed pH dependence, zeta-potential/point of zero charge (PZC) context, and competitive anion effects, the arsenic immobilization behaviour is consistent with contributions from electrostatic interactions (particularly for anionic As(V) under pH < PZC) and specific interactions with iron (hydr)oxide surfaces. Potential redox involvement of Fe(0) is discussed as mechanistically plausible for nZVI-based systems, but arsenic/iron speciation was not directly measured in this study. The composite maintained >81% efficiency after five regeneration cycles and displayed excellent selectivity in the presence of competing anions, with minimal interference from sulfate and chloride but moderate competition from phosphate. When tested in simulated mining wastewater containing 5.0 mg L-1 total arsenic and high levels of Pb2+, Cd2+, and SO42-, the MBC-40 achieved 97.0% arsenic removal, >99.9% Pb2+ elimination, and 95.6% Cd2+ reduction. The rapid magnetic separation from treated water within 30 s enables simple recovery of the sorbent using an external permanent (neodymium) magnet, without requiring electrically powered separation equipment. These findings demonstrate that the MBC-nZVI composite mitigates key practical limitations of bare nZVI (aggregation/passivation and post-treatment recovery) by integrating a biochar scaffold with a magnetically recoverable Fe(0)@Fe3O4 architecture. Consistent with prior reports of nZVI/biochar (BC)-type systems, the present work contributes an application-focused validation primarily: performance is quantified across Fe-loading levels and confirmed in a mining-relevant mixed-ion/multi-metal matrix (high sulfate and co-occurring Pb2+/Cd2+), together with reusability supported by permanent-magnet separation.
Keywords
- redox transformation
- adsorption kinetics
- magnetic separation
- heavy-metal remediation
- surface complexation
Arsenic (As) contamination associated with mining activities represents a pervasive and critical environmental health challenge, with particularly acute risks when arsenic is mobilized into aqueous pathways such as acid mine drainage, tailings seepage, and impacted surface/ground waters. Recognized as a Group 1 carcinogen, chronic exposure to inorganic arsenic is linked to cancers, cardiovascular disorders, and developmental effects [1]. From a practical treatment perspective, arsenic concentrations in drinking-water sources and mining-impacted waters are subject to stringent targets (e.g., a 10 µg L-1 guideline value is widely used as a benchmark), reinforcing the need for high-efficiency removal technologies. Mining and ore-processing activities are major drivers of arsenic mobilization, including via the weathering of sulfidic tailings and waste rock, often under acid-generating conditions [2]. In such systems, arsenic predominantly occurs as arsenite As(III) and arsenate As(V), with redox state and speciation governed by aqueous geochemistry and redox conditions relevant to both groundwater and mining-impacted waters [3].
To address arsenic risks in mining-impacted environments, a wide range of remediation strategies has been deployed, spanning source control and containment, soil-focused interventions, and water-treatment approaches such as pump-and-treat and reactive media processes [4]. However, conventional options can be constrained by cost, operational complexity, incomplete removal under variable geochemistry, and secondary waste generation [5]. These practical constraints motivate the development of sustainable, regenerable sorbents/reactive media that can achieve rapid arsenic uptake in aqueous matrices and enable straightforward post-treatment separation and recovery-features that are particularly attractive for decentralized or site-based treatment of mining-impacted waters. This study is positioned within that water-treatment need, using controlled batch experiments and a mining-relevant mixed-ion aqueous matrix to quantify performance [6].
In the search for such materials, nanoscale zero-valent iron (nZVI) has emerged as a particularly potent remediation agent. Possessing a high specific surface area and a strong reducing potential (E0 = –0.44 V), nZVI is highly effective in the reduction, precipitation, and immobilization of a wide array of contaminants, including chlorinated solvents, heavy metals, and metalloids like arsenic [7]. Despite its high intrinsic reactivity, the practical application of bare nZVI is severely hampered by three critical challenges: (1) rapid agglomeration into micron-sized clusters due to strong magnetic van der Waals forces, which drastically reduces reactive surface area; (2) rapid surface oxidation and passivation in aerobic environments, consuming its reductive capacity; and (3) difficulty in separation and recovery from environmental matrices, posing potential ecotoxicological risks [8]. Concurrently, biochar (BC), a carbon-rich solid derived from the pyrolysis of waste biomass, has gained prominence as a sustainable sorbent. Its porosity and abundant surface functional groups (e.g., carboxyl, hydroxyl) can make it an effective adsorbent; however, Brunauer-Emmett-Teller (BET) surface area is not inherently high and can vary widely with feedstock and pyrolysis conditions (e.g., ~2 m2 g-1 reported for 300 °C biochar). Various feedstocks, including sewage sludge, pomegranate peel, and sugarcane bagasse, have been explored, though biochar’s mechanism is often limited to physical adsorption, which can be non-selective and possesses a finite capacity for inorganic anions like arsenate [9].
A synergistic opportunity arises from integrating nZVI with biochar into a composite architecture that is effective in aqueous arsenic removal while mitigating key limitations of bare nZVI (aggregation/passivation and difficult post-treatment recovery) [10, 11]. Biochar can act as a dispersive scaffold and mass-transfer framework, improving accessibility of reactive iron sites and stabilizing iron phases relevant to arsenic uptake in water [10, 11]. In aqueous systems, nZVI is well known to remove both As(V) and As(III) through coupled adsorption-redox-(co)precipitation processes whose extent depends on solution chemistry and competing oxyanions [6]. In parallel, iron-biochar and iron oxide-biochar composites have been demonstrated as effective sorbents for aqueous arsenic, supporting the premise that iron-bearing carbon matrices can achieve rapid uptake with practical handling advantages [12]. The biochar matrix serves as a physical support, preventing nZVI aggregation and maximizing the available reactive sites. Its hierarchical porosity facilitates the mass transfer of arsenic species to the nZVI particles [13]. Furthermore, the aromatic, sp2-rich carbon domains formed during pyrolysis (often described as turbostratic or graphitic-like microcrystallites within an overall disordered matrix) can potentially facilitate electron transfer and thereby contribute to redox reactivity in nZVI-containing composites [14]. In turn, the nZVI provides potent reductive capabilities to transform As(V) to As(III), while its in-situ oxidized shell (e.g., magnetite, Fe3O4) not only provides magnetic separability but also acts as an exceptionally strong sorbent for both As(V) and As(III), creating stable inner-sphere complexes and serving as precipitation centers [15].
While magnetic nZVI-biochar composites have been investigated extensively, the present work does not claim conceptual novelty in the core material class [6]. Instead, it addresses a practical translation need: systematically linking Fe-loading-dependent structure/property metrics (dispersion, core-shell formation, and magnetization) with performance for both As(III) and As(V), and validating removal in a deliberately complex mining-impacted matrix (high sulfate and co-occurring metals) where simplified single-solute trends may not transfer quantitatively. This research presents a comprehensive synthesis and analysis of a magnetic biochar-nZVI (MBC-nZVI) composite derived from pine bark [16]. The objectives of this study are: (1) to synthesize and comprehensively characterize MBC-nZVI composites with varying nZVI loading ratios; (2) to comparatively evaluate their immobilization performance for both As(III) and As(V) against pristine BC and bare nZVI; (3) to interpret plausible immobilization mechanisms primarily from adsorption behaviour (pH dependence, competitive anions, and kinetic/isotherm modelling) together with the established reactivity of nZVI/iron oxide surfaces reported in the literature; and (4) to validate the composite’s practical utility via magnetic separation and performance testing in simulated mining-impacted water [12].
The collected pine bark was first washed thoroughly with deionized (DI) water to
remove surface debris, dried in an oven at 80 °C for 24 h, and then
ground using a laboratory mill. The resulting powder was sieved to obtain a
uniform particle size fraction (
The magnetic composites were synthesized via an in-situ liquid-phase
reduction method. In a typical synthesis, 2.0 g of the prepared BC powder was
dispersed in a 300 mL solution of ethanol (99.7%; Aladdin Biochemical Technology
Co., Ltd., Shanghai, China) and de-aerated deionized water (1:1 v/v) and
sonicated for 30 min to ensure a uniform suspension. A specific mass of
FeCl3
The appearance of a black precipitate and vigorous hydrogen bubbling indicated the formation of nZVI. The suspension was aged under stirring for an additional 1 h to ensure complete reaction. Finally, the black composite material was separated from the solution, washed repeatedly with de-aerated ethanol and water to remove residual ions, and dried in a vacuum oven at 60 °C for 12 h.
To effectively evaluate the synergistic effects within the composite, three
control materials were prepared. (1) Pristine BC: This is the pyrolyzed pine bark
as described in Section 2.2. (2) Bare nZVI: This material was synthesized using
the exact liquid-phase reduction method described in Section 2.3, but without the
addition of the biochar support. (3) Two different composite loading ratios were
prepared and designated: MBC-10, synthesized using a 1:10 mass ratio of Fe (from
the FeCl3
Batch experiments were conducted to evaluate the arsenic immobilization
performance of the materials under controlled aqueous conditions. Kinetic studies
were performed by adding 20 mg of sorbent (e.g., MBC-40) to 50 mL of As(III) or
As(V) solution (initial concentration C0 = 10 mg/L) in 100 mL conical
flasks. The flasks were sealed and agitated on a rotary shaker at 150 rpm and 298
K, with the solution pH maintained at 7.0
Adsorption isotherm experiments were conducted by adding 20 mg of each sorbent
(BC, bare nZVI, MBC-10, MBC-40) to a series of 50 mL solutions with varying
initial As(III) and As(V) concentrations (C0 = 5, 10, 25, 50, 100, 150, 200
mg L-1). The suspensions were agitated for 24 h at 298 K to ensure
equilibrium was reached. The equilibrium sorption capacity (qe, mg
g-1) was calculated, and the data were fitted to the non-linear Langmuir
and the Freundlich isotherm models. Unless otherwise stated, all batch sorption
experiments (kinetics, isotherms, pH, competing anions, and reusability tests)
were conducted in triplicate (n = 3), and results are reported as mean
The effect of initial solution pH on arsenic removal was investigated over a range of 2.0 to 12.0. The pH was adjusted using 0.1 M HCl or 0.1 M NaOH. The influence of common co-existing anions found in mining effluents (Cl–, SO42-, CO32-, and HPO42-) was tested by adding 100 mg L-1 of their respective sodium salts to the 10 mg L-1 As(V) solution. Reusability of the MBC-40 composite was assessed over five consecutive adsorption-desorption cycles. After each sorption cycle, the spent composite was separated using an external magnet, washed, and then desorbed by agitation in 50 mL of 0.1 M NaOH solution for 2 h. The regenerated material was then washed with DI water until neutral and reused for the next cycle.
To assess the composite’s practical applicability in a mining-relevant inorganic matrix, a SMW was prepared based on typical effluent compositions [19]. The SMW formulation represents a deliberately complex mixed-ion/multi-metal challenge (high sulfate and co-occurring Pb2+/Cd2+/Zn2+), but it does not include DOM/NOM and it does not simulate time-dependent geochemical ageing/passivation processes; these factors may influence long-term performance in natural systems and are noted as limitations and priorities for future field-oriented evaluation. The SMW contained a multi-pollutant mixture: 3.0 mg L-1 As(V), 2.0 mg L-1 As(III), 10.0 mg L-1 Pb2+, 5.0 mg L-1 Cd2+, 8.0 mg L-1 Zn2+, and 1000 mg L-1 SO42-, at a pH of 5.5. The removal efficiency of MBC-40 (dose = 0.4 g L-1) was tested in this complex matrix. Final concentrations of all contaminants were measured using an atomic fluorescence spectrometer (AFS) (Model AFS-9700, Beijing Haiguang Instrument Co., Ltd., Beijing, China) for arsenic and an Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) (Model Avio 200, PerkinElmer Inc., Waltham, MA, USA) for heavy metals.
The morphology of the BC and the synthesized composites was first investigated using scanning electron microscopy (SEM), as shown in Fig. 1. Fig. 1A reveals that the pristine BC, derived from pine bark, retained the natural, fibrous, and channel-like structure of its biomass precursor, exhibiting a porous and rough surface [20]. This inherent structure is beneficial for providing mass transfer pathways. In sharp contrast, Fig. 1B shows the morphology of bare nZVI particles synthesized without the biochar support. The particles exhibit severe, grape-like agglomeration, forming large micron-sized clusters. This aggregation, driven by strong magnetic interactions, drastically reduces the available reactive surface area and is a primary limitation of bare nZVI. Fig. 1C (MBC-10) shows that at a low loading ratio, some nanoparticles are visible decorating the biochar pores. Fig. 1D (MBC-40), however, demonstrates a dramatic change: the biochar surface is densely and uniformly coated with a layer of spherical nanoparticles [21], which still appear to maintain their discrete nature, confirming that the biochar scaffold effectively acts as a dispersant, preventing the severe agglomeration observed in Fig. 1B.
 Fig. 1.
Fig. 1.
Morphological evolution of biochar and magnetic biochar composites revealed by SEM. SEM micrographs of (A) BC showing its fibrous and porous morphology derived from pine bark; (B) bare nZVI exhibiting severe agglomeration into micron-sized clusters; (C) MBC-10 composite with sparsely distributed nZVI nanoparticles on the biochar surface; and (D) MBC-40 composite displaying a dense, uniform coating of spherical nZVI particles. BC, biochar; nZVI, nanoscale zero-valent iron; MBC, magnetic biochar; SEM, scanning electron microscopy. Scale bar = 2 µm.
Transmission electron microscopy (TEM) analysis provided further insight into
the nanoscale structure. Fig. 2A again confirms the severe aggregation
of bare nZVI. Fig. 2B, a high-magnification image of MBC-40, clearly shows that
the biochar matrix (the lighter, amorphous region) is decorated with discrete,
near-spherical nZVI nanoparticles with an average diameter of 50–80 nm. This
high dispersion is the key to maximizing reactive sites. Most critically, the
High-Resolution TEM (HR-TEM) image in Fig. 2C reveals a distinct core-shell
structure for a single nanoparticle on the MBC-40 composite. Analysis of the
lattice fringes in Fig. 2D provides definitive identification: the core exhibits
a lattice spacing of 0.202 nm, which corresponds perfectly to the (110)
crystallographic plane of body-centered cubic
 Fig. 2.
Fig. 2.
TEM and HR-TEM characterization of nZVI and MBC-40
composite. (A) Bare nZVI showing extensive aggregation. (B) MBC-40 displaying
well-dispersed, near-spherical nanoparticles (50–80 nm) anchored on the
amorphous biochar matrix. (C) HR-TEM image revealing a clear
Fe(0)@Fe3O4 core-shell structure. (D) Lattice fringe analysis showing
spacings of 0.202 nm and 0.253 nm corresponding to the (110) plane of
The intimate association and uniform distribution of the iron component were further confirmed by SEM–energy dispersive X-ray spectroscopy (EDX) elemental mapping (Fig. 3). The maps for Carbon (C), Oxygen (O), and Iron (Fe) on the MBC-40 composite show a near-perfect spatial overlap. The Fe signal is distributed evenly across the entire C-O matrix, with no evidence of large, isolated Fe-only regions. This confirms that the in-situ synthesis method was highly successful in anchoring the iron nanoparticles directly onto the biochar support.
 Fig. 3.
Fig. 3.
SEM-EDX elemental mapping of the MBC-40 composite showing uniform spatial distributions of Carbon (C), Oxygen (O), and Iron (Fe), confirming the homogeneous dispersion and strong interfacial association of iron nanoparticles within the biochar matrix. EDX, energy dispersive X-ray spectroscopy. Scale bar = 10 μm.
The crystalline phases of the prepared materials were analyzed by X-ray
diffraction (XRD) (Fig. 4A). The pristine BC pattern exhibits a broad diffraction
feature centered at 2
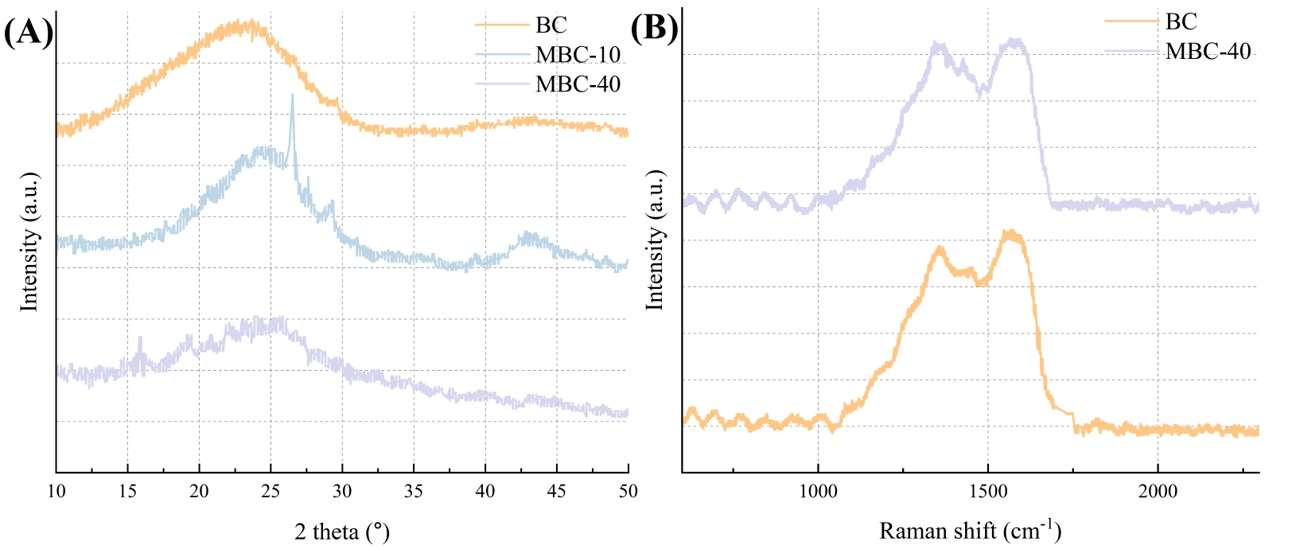 Fig. 4.
Fig. 4.
Crystalline structure and carbon framework characteristics of biochar-based composites. (A) XRD patterns of BC, bare nZVI, and the MBC composites. (B) Raman spectra of BC and MBC-40 composite showing characteristic D-band (~1350 cm-1) and G-band (~1590 cm-1). XRD, X-ray diffraction.
Raman spectroscopy was employed to investigate the structural nature of the biochar support (Fig. 4B). The spectra for both pristine BC and the MBC-40 composite exhibit two prominent bands: the D-band at approximately 1350 cm-1, which is associated with disordered carbon structures and defects, and the G-band at approximately 1590 cm-1, which corresponds to the in-plane vibrations of sp2-hybridized graphitic carbon. The intensity ratio of these bands I(D)/I(G) is a common metric for the degree of graphitization [24]. The spectra show D (~1350 cm-1) and G (~1590 cm-1) bands typical of disordered sp2 carbon. The I(D)/I(G) ratio (0.92) is consistent with a condensed aromatic carbon structure containing defects and finite crystallite sizes; however, I(D)/I(G) in biochar is not a definitive measure of ‘graphitization’ to true graphite and should be interpreted cautiously as reflecting disorder/ordering trends within amorphous/turbostratic carbon [25, 26]. This graphitic nature is hypothesized to be beneficial, potentially enabling the carbon matrix to act as an electron shuttle, which could facilitate the redox reactions between the Fe(0) core and adsorbed arsenic species [27].
The specific surface area (SSA) and porous properties of the materials, crucial for adsorption processes, were determined by N2 adsorption-desorption analysis (Table 1). The pristine BC exhibited a high SSA of 285.4 m2 g-1 and a total pore volume of 0.198 cm3 g-1, confirming its potential as an excellent porous support. Conversely, the bare nZVI had a very low SSA of only 22.1 m2 g-1, a direct consequence of the severe agglomeration observed in SEM/TEM. Upon loading of nZVI, the SSA of the composites decreased to 210.3 m2 g-1 for MBC-10 and 180.9 m2 g-1 for MBC-40. This reduction is expected, as the dense iron nanoparticles (which have a low intrinsic surface area) occupy or block some of the micropores of the biochar [28]. However, it is crucial to understand that while the total surface area decreased, the reactive surface area was vastly increased. The biochar support ensures that the 180.9 m2 g-1 of the MBC-40 composite is populated with highly dispersed, accessible nZVI reactive sites, in contrast to the inaccessible, agglomerated 22.1 m2 g-1 of bare nZVI [29].
| Material | BET surface area (m2 g-1) | Total pore volume (cm3 g-1) | Avg. pore diameter (nm) | Saturation magnetization (emu g-1) |
| Pristine BC | 285.4 | 0.198 | 3.5 | 0.2 |
| Bare nZVI | 22.1 | 0.045 | 8.1 | 85.6 |
| MBC-10 | 210.3 | 0.165 | 4.0 | 12.7 |
| MBC-40 | 180.9 | 0.142 | 3.8 | 32.5 |
BET, Brunauer-Emmett-Teller.
The surface charge characteristics of the MBC-40 composite were investigated via
zeta potential analysis (Fig. 5). The curve shows that the point of zero charge
(PZC), the pH at which the net surface charge is zero, was 6.8. This magnitude is
consistent with reported pH-PZC values for iron-bearing carbonaceous composites,
while also falling within the broad variability expected for Fe-modified biochars
due to differences in biomass feedstock, thermal history, iron phase composition,
and surface oxygen functionality. For example, a heat-treated biochar impregnated
with nanoscale zero-valent iron (HBC/nZVI) exhibited a pH-PZC of 5.1, reflecting a
more acidic surface-charge transition for that specific nZVI–biochar system
(likely linked to its surface functional group distribution and iron speciation)
[30]. In an iron oxide–hydrochar composite prepared from pomegranate peel waste,
pH-PZC values of 5.3–5.6 were reported for iron oxide–carbon composites, again
indicating that Fe–carbon materials can display moderately acidic PZC values
depending on synthesis conditions and oxide/hydroxide surface chemistry [31].
Conversely, Fe-oxide-modified biochars can also present higher pH-PZC values; for
rice husk biochar systems modified with Fe3O4 or FeS2, pH-PZC
values of 9.23 and 7.78, respectively, were reported, highlighting that mineral
phase identity and post-processing can shift the surface-charge neutrality point
to more alkaline values. Related magnetic biochars incorporating iron oxides
likewise show pH-PZC spanning approximately 4.5–9.9, depending on the
modification route and co-present metal oxides, and Fe3O4 itself is
commonly reported to have a pH-PZC near ~7.0, supporting the
reasonableness of the PZC
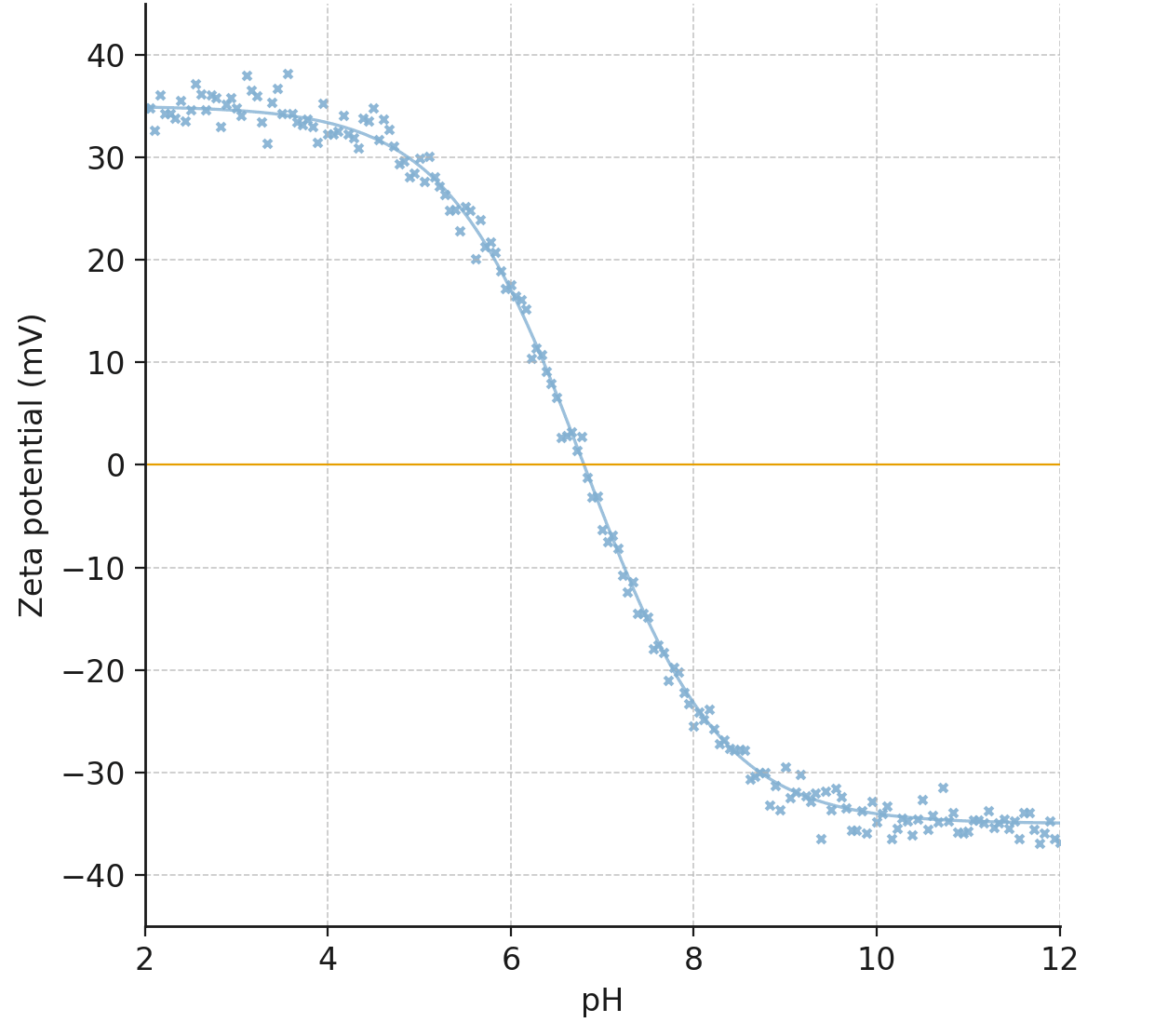 Fig. 5.
Fig. 5.
Zeta potential of the MBC-40 composite as a function of pH, showing a PZC of 6.8. PZC, point of zero charge.
This value is critical for understanding the electrostatic interactions during
sorption. At solution pH
Fourier transform infrared spectroscopy (FTIR) spectroscopy was used to identify the surface functional groups on the materials (Fig. 6). The pristine BC spectrum shows a broad peak at ~3430 cm-1 (O-H stretching of hydroxyl groups), a small peak at ~2850 cm-1 (C-H stretching), a prominent peak at ~1660 cm-1 (C=C stretching in aromatic rings and C=O stretching of carboxyl/carbonyl groups), and a band around 1050 cm-1 (C-O stretching). These groups are all characteristic of lignocellulosic biochar. In the spectrum for MBC-40, these biochar-related peaks are still present but are attenuated [35], suggesting they are partially shielded by the nZVI coating. More importantly, a new, strong and sharp absorption peak appears at ~580 cm-1. This peak is the characteristic Fe-O stretching vibration associated with the magnetite (Fe3O4) lattice. Its presence is definitive confirmation of the successful formation of the iron oxide (magnetite) shell on the biochar support, corroborating the TEM and XRD data [36].
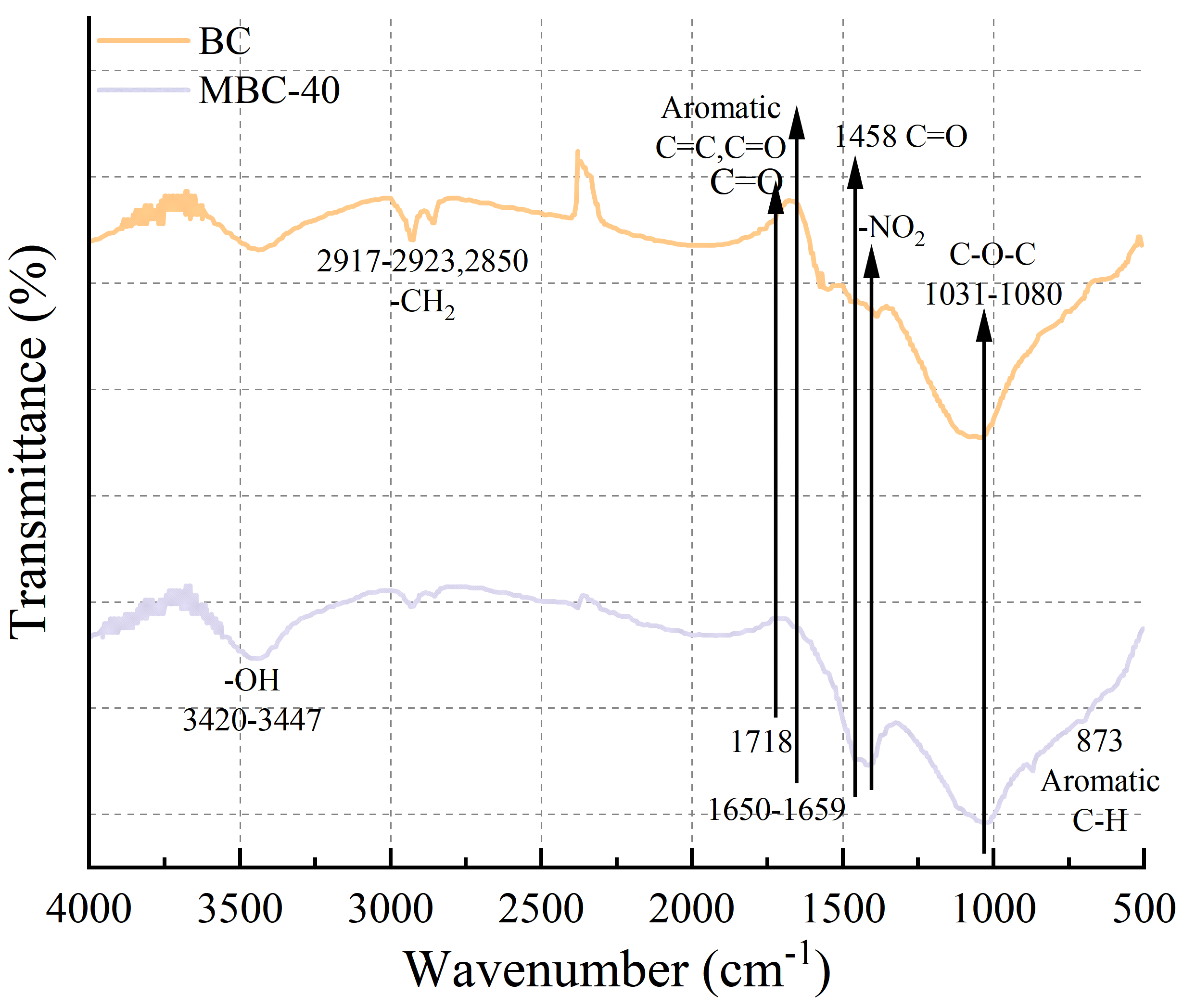 Fig. 6.
Fig. 6.
FTIR spectra of BC and MBC-40 composite. FTIR, Fourier transform infrared spectroscopy.
Thermogravimetric analysis (TGA) was conducted in an air atmosphere to determine the thermal stability and quantify the actual iron loading in the composites (Fig. 7). Pristine BC shows a major weight loss of over 60% between 200–500 °C, corresponding to the combustion of the carbon matrix. In contrast, the nZVI-loaded samples show significantly enhanced thermal stability and a much larger residual mass. At 800 °C, after all carbon has been burned off and all iron has been oxidized to its most stable form (Fe2O3), the final residual mass was 3.1% for BC (ash content), 14.5% for MBC-10, and 41.2% for MBC-40. By back-calculating from the mass of Fe2O3, these residuals confirm that the actual iron loadings in the composites are highly consistent with the theoretical 10% and 40% (by Fe) targets of the synthesis [37].
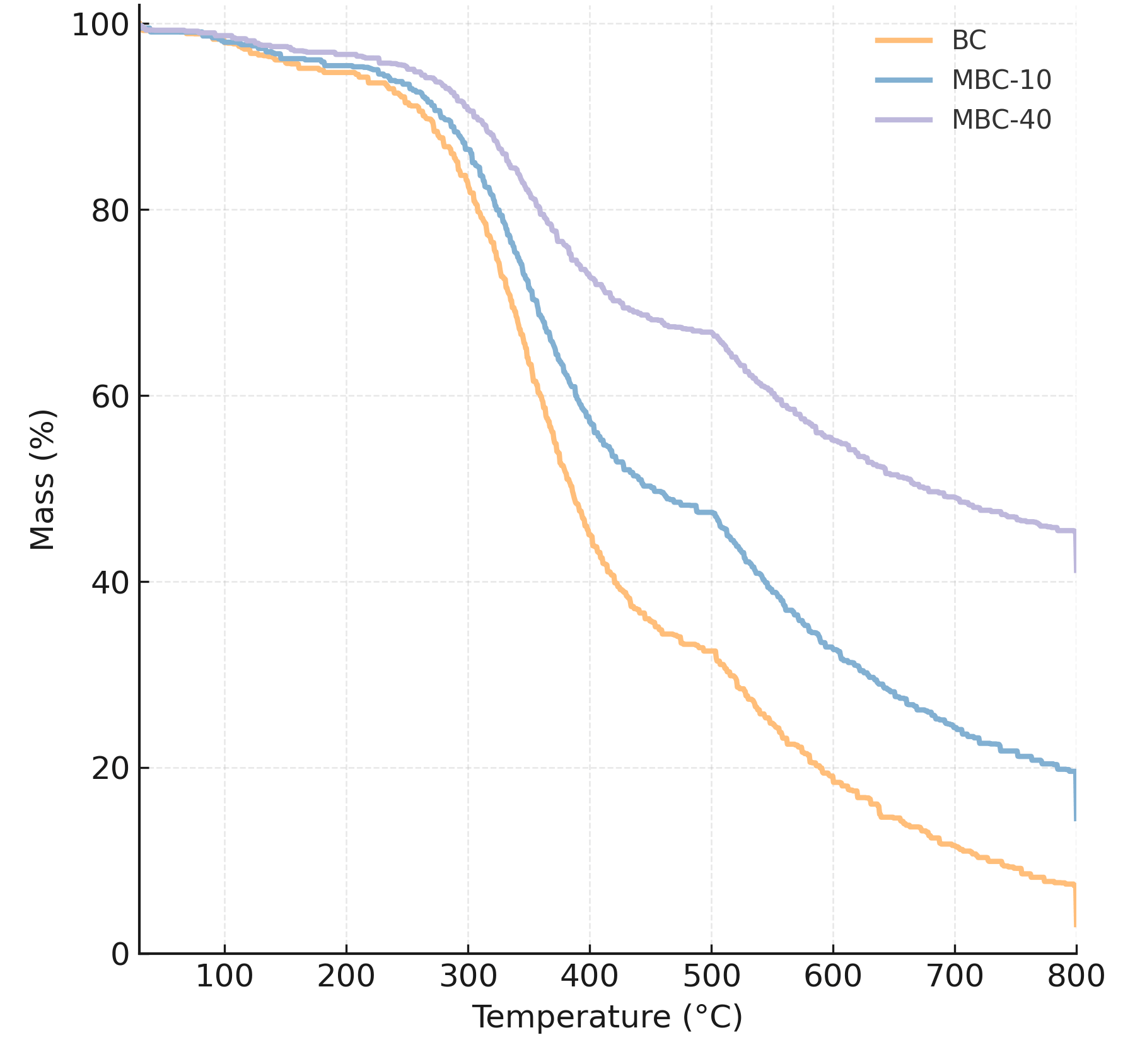 Fig. 7.
Fig. 7.
TGA curves of BC, MBC-10, and MBC-40 composites recorded in
air. Pristine BC exhibits a major weight loss of
The magnetic properties of the materials, essential for the “magnetically assisted separation” component of this study, were measured by vibrating sample magnetometry (VSM) (Fig. 8). As expected, pristine BC exhibits no significant magnetic response. MBC-40, however, displays a strong “S”-shaped magnetic hysteresis loop, which is characteristic of superparamagnetic or soft ferromagnetic materials [38]. The saturation magnetization (Ms) was determined to be 32.5 emu g-1 (Table 1). This value is substantially higher than the ~16 emu g-1 commonly referenced as a practical threshold for rapid magnetic solid–liquid separation and sorbent recovery from aqueous suspensions.
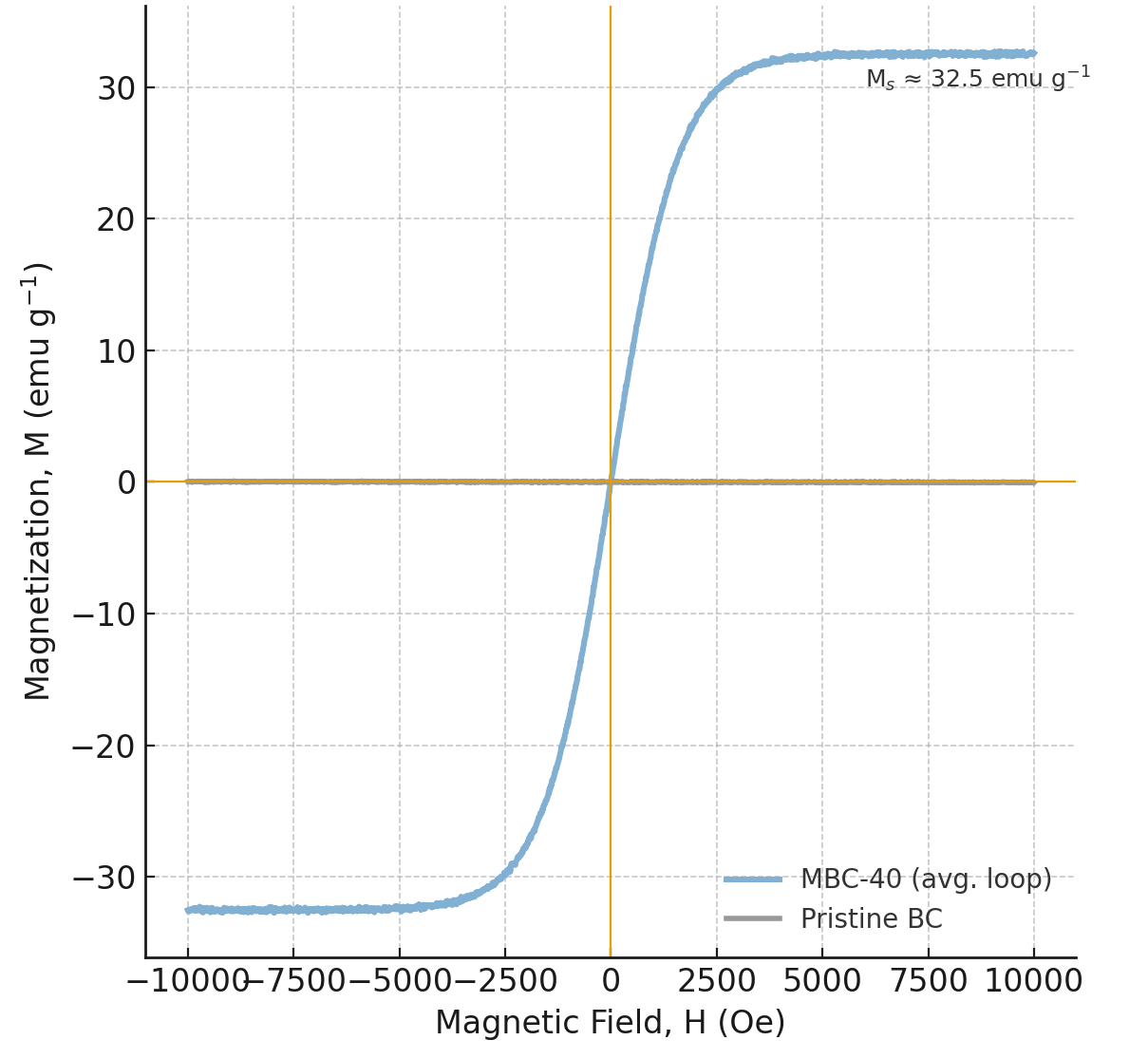 Fig. 8.
Fig. 8.
VSM of pristine BC and MBC-40 composite. VSM, vibrating sample magnetometry.
The practical implication of this strong, superparamagnetic behavior is demonstrated visually in Fig. 9. A suspension of MBC-40 in water (Fig. 9A) is turbid and black. Upon the application of an external neodymium magnet to the vial wall, the black MBC-40 particles are rapidly pulled from the solution [39]. Within 30 s (Fig. 9B), the supernatant is rendered completely clear and transparent, with all the composite material aggregated at the magnet’s location. This demonstration validates the potential for easy, rapid, and non-energetic recovery of the sorbent from treated aqueous matrices, which is a practical advantage over conventional solid–liquid separation steps such as filtration or centrifugation.
 Fig. 9.
Fig. 9.
Photographic demonstration of the magnetic separation capability of MBC-40 in water. (A) A turbid black suspension of MBC-40 before magnetic application. (B) After exposure to an external neodymium magnet for 30 s, the composite is rapidly attracted to the vial wall, leaving a clear and transparent supernatant.
The synergistic effect of the composite structure was evaluated by comparing the
arsenic removal performance of the four prepared materials under identical
conditions (Fig. 10). For As(V) removal, the pristine BC showed negligible
efficacy, removing only 12.2%. Bare nZVI, despite its high intrinsic reactivity,
performed moderately, removing 45.7%; its effectiveness was clearly limited by
the agglomeration and passivation observed in the characterization. The
composites, however, showed dramatically improved performance. MBC-10 removed
62.5%, while the high-load MBC-40 composite achieved an outstanding 98.5%
removal of As(V). The trend of MBC-40
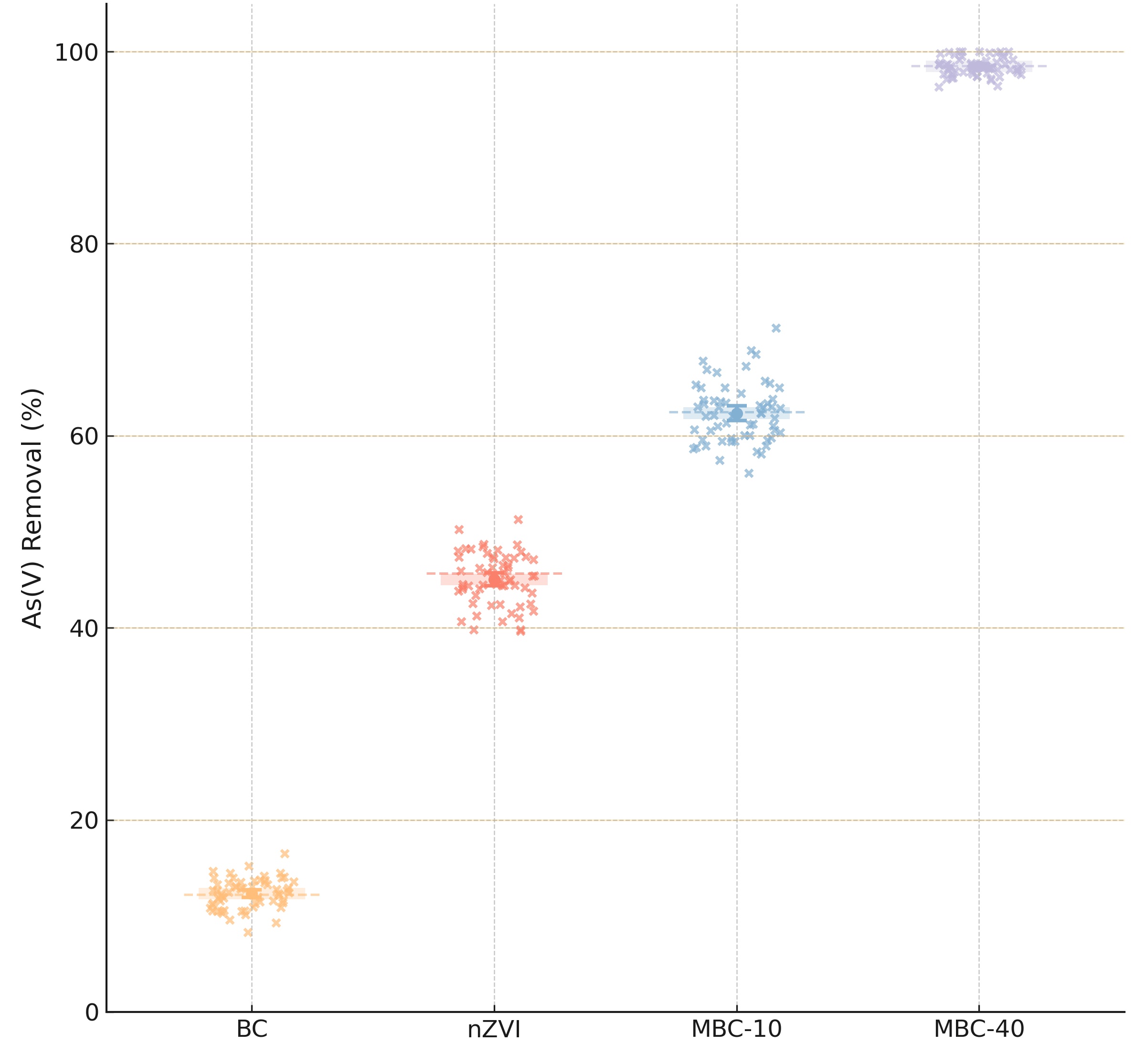 Fig. 10.
Fig. 10.
Comparative As(V) removal performance of BC, bare nZVI, and magnetic biochar composites (MBC-10 and MBC-40) under identical experimental conditions.
The solution pH is a master variable in arsenic chemistry and sorption
processes. Fig. 11 shows the effect of initial pH (2.0–12.0) on the removal of As(V)
and As(III) by MBC-40. The removal of As(V) was highly efficient (
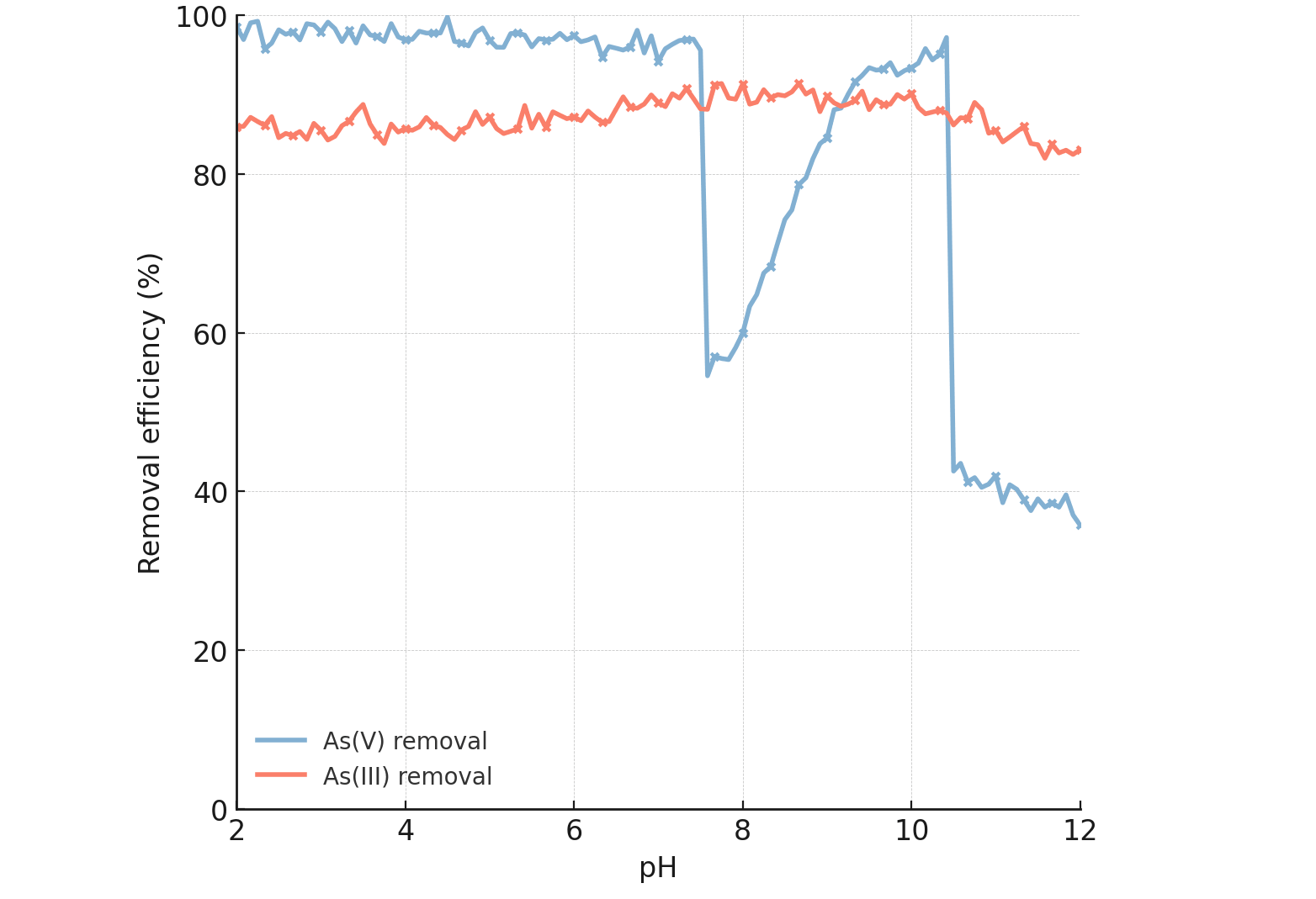 Fig. 11.
Fig. 11.
Effect of initial solution pH (2.0–12.0) on the removal efficiencies of As(V) and As(III) by MBC-40 composite (C0 = 10 mg L-1, 298 K, 150 rpm, 24 h).
The rate of arsenic uptake by MBC-40 was investigated (Fig. 12). The removal process was extremely rapid for both arsenic species. More than 80% of the total arsenic uptake occurred within the first 30 minutes, with the reaction reaching equilibrium by approximately 120 minutes. This rapid kinetics is highly desirable for practical water treatment applications, as it implies shorter contact times and smaller reactor volumes [42]. The data were fitted to PFO and PSO kinetic models (Fig. 13), with the calculated parameters summarized in Table 2.
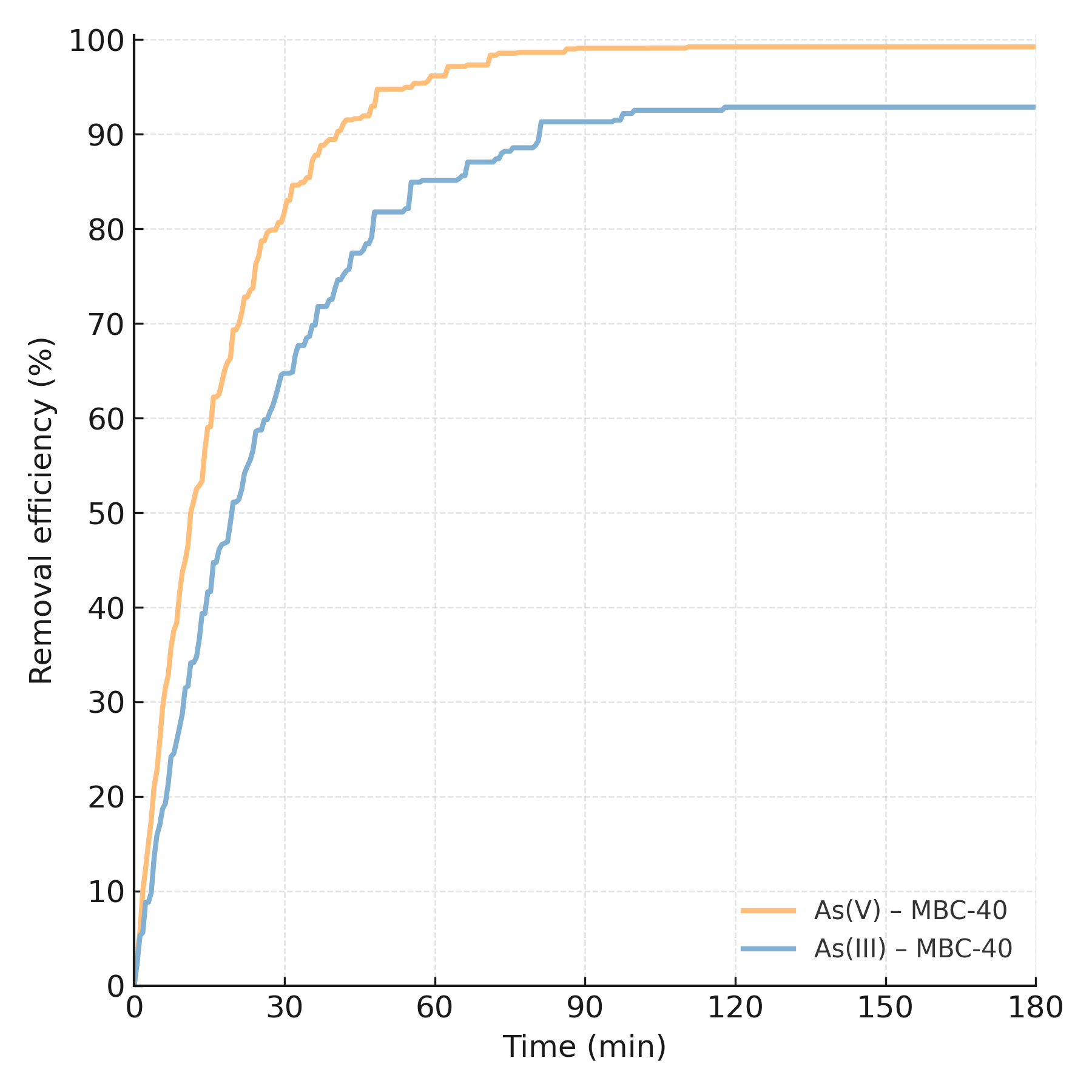 Fig. 12.
Fig. 12.
Time-dependent adsorption profiles of As(V) and As(III) onto
MBC-40 at C0 = 10 mg L-1, pH = 7.0
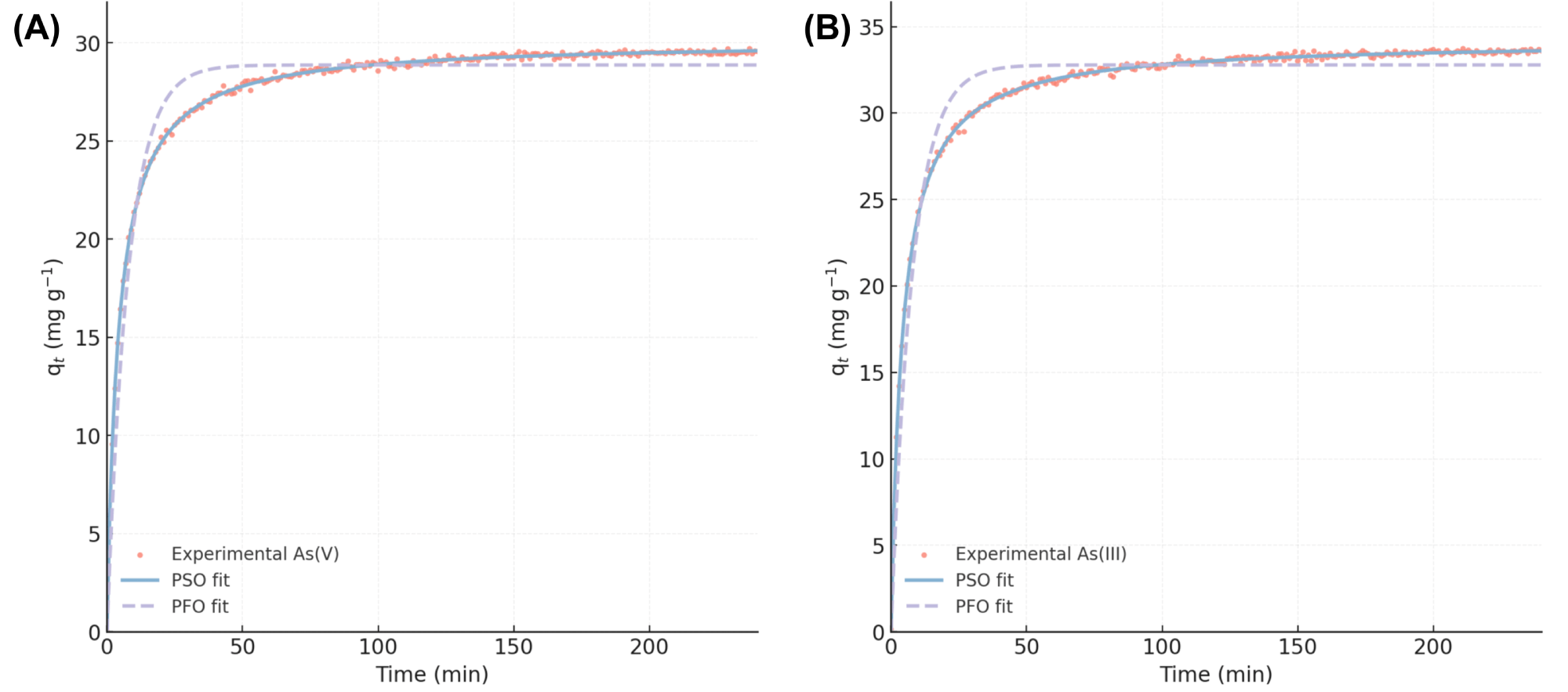 Fig. 13.
Fig. 13.
Adsorption kinetic behavior of arsenic species on MBC-40. Kinetic model fitting for (A) As(V) and (B) As(III) adsorption onto MBC-40 using PFO and PSO models. PFO, pseudo-first-order; PSO, pseudo-second-order.
| Species | Model | qe,calc (mg g–1) | k1 (min–1) or k2 (g/mg·min) | R2 |
| As(V) | PFO | 22.4 | 0.045 | 0.913 |
| PSO | 30.1 | 0.008 | 0.998 | |
| As(III) | PFO | 25.6 | 0.051 | 0.899 |
| PSO | 34.2 | 0.007 | 0.999 |
qe,calc, equilibrium capacity.
As shown in Table 2, the PSO model provided a far superior
fit to the experimental data for both As(V) and As(III). The correlation
coefficients (R2) for the PSO model were
Adsorption isotherms (Fig. 14) were studied to determine the maximum immobilization capacity of the materials. The equilibrium data were fitted to the Langmuir and Freundlich models, and the resulting parameters are presented in Table 3. The Langmuir model assumes monolayer adsorption onto a homogeneous surface, while the Freundlich model describes multilayer adsorption on a heterogeneous surface.
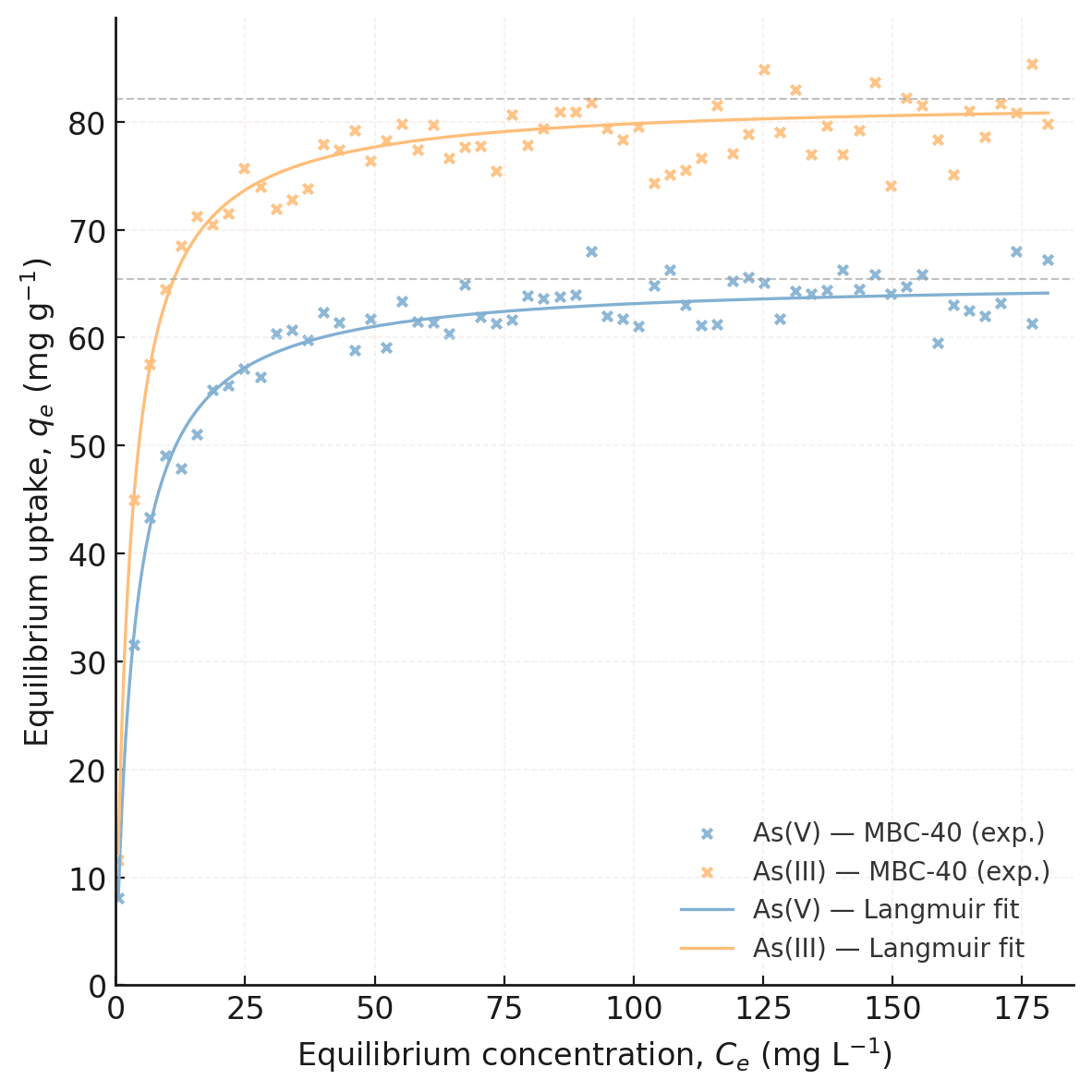 Fig. 14.
Fig. 14.
Adsorption isotherms of As(V) and As(III) onto MBC-40 at 298 K.
| Material | Species | Langmuir model | Freundlich model | ||
| Qmax (mg g–1) | KL (L mg–1) | R2 | 1/n | ||
| BC | As(V) | 8.2 | 0.03 | 0.921 | 0.65 |
| Bare nZVI | As(V) | 24.5 | 0.09 | 0.989 | 0.42 |
| MBC-40 | As(V) | 65.4 | 0.28 | 0.997 | 0.31 |
| MBC-40 | As(III) | 82.1 | 0.35 | 0.995 | 0.29 |
Qmax, maximum adsorption capacity; KL, Langmuir adsorption constant.
For the MBC-40 composite, the Langmuir model provided a superior fit for both
As(V) and As(III), with R2 values
Real-world mining effluents are not clean systems; they contain a complex
mixture of co-existing ions. Fig. 15 illustrates the impact of common anions on
As(V) removal by MBC-40. The presence of high concentrations (100 mg/L) of
Cl– and SO42- had a negligible impact, with removal efficiency
remaining
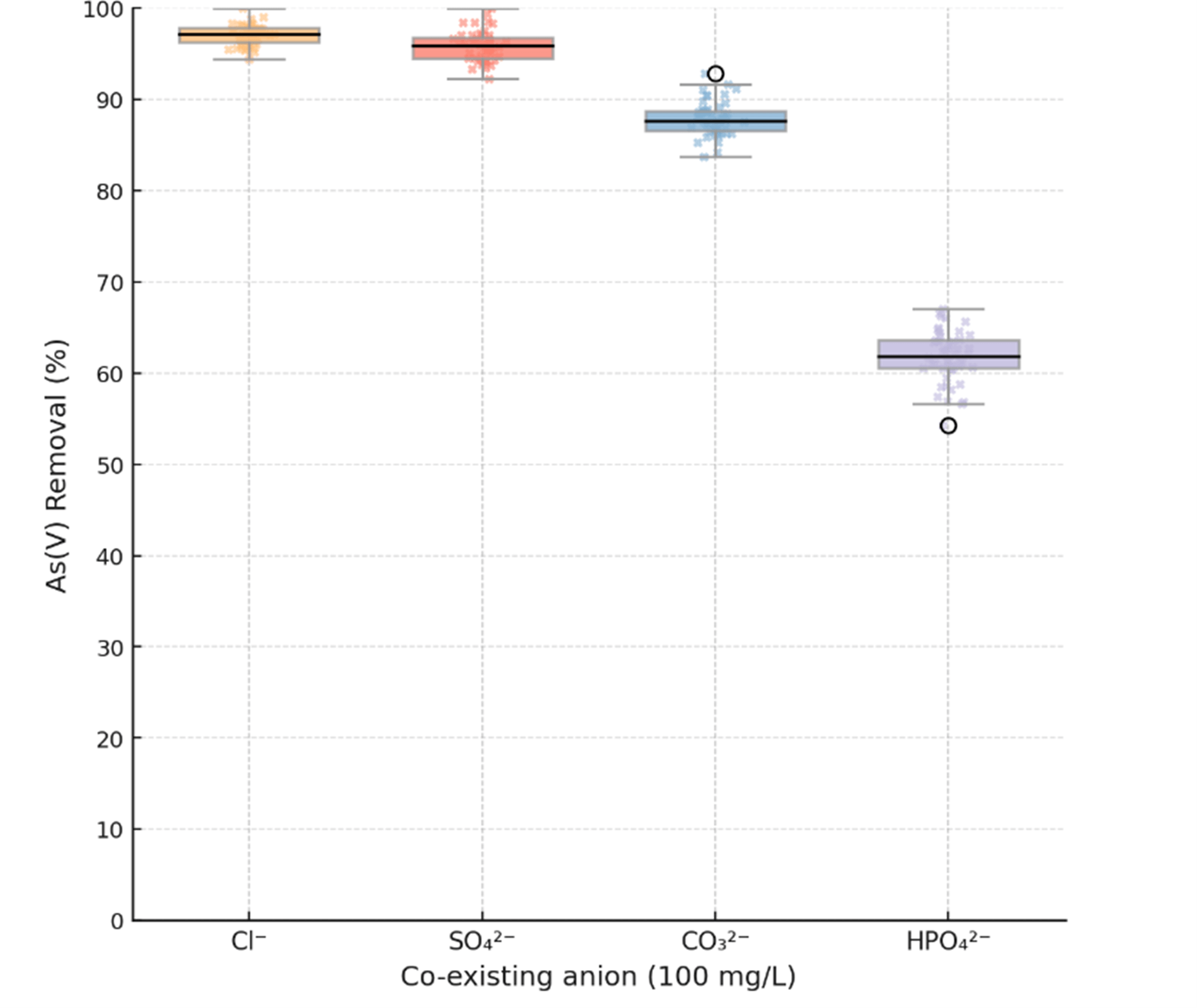 Fig. 15.
Fig. 15.
Effect of co-existing anions (100 mg L-1 each of
Cl–, SO42-,
CO32-, and HPO42-) on As(V)
removal by MBC-40 at C0 = 10 mg L-1, pH = 7.0
For a sorbent to be economically viable, it must be reusable. The MBC-40 composite was subjected to five adsorption-desorption cycles, with regeneration achieved using a 0.1 M NaOH wash (which desorbs arsenic) and recovery via an external magnet. As shown in Fig. 16, the composite demonstrated good reusability. After five cycles, the removal efficiency for As(V) remained at 81.3%, a moderate decrease from the 98.5% in the first cycle. This slight decrease in performance is likely due to two factors: (1) incomplete regeneration, as some arsenic may be irreversibly bound in inner-sphere complexes, and (2) the progressive consumption and loss of the Fe(0) reductive capacity, which is not restored by the NaOH wash. Nonetheless, an 81.3% efficiency after five uses is highly promising.
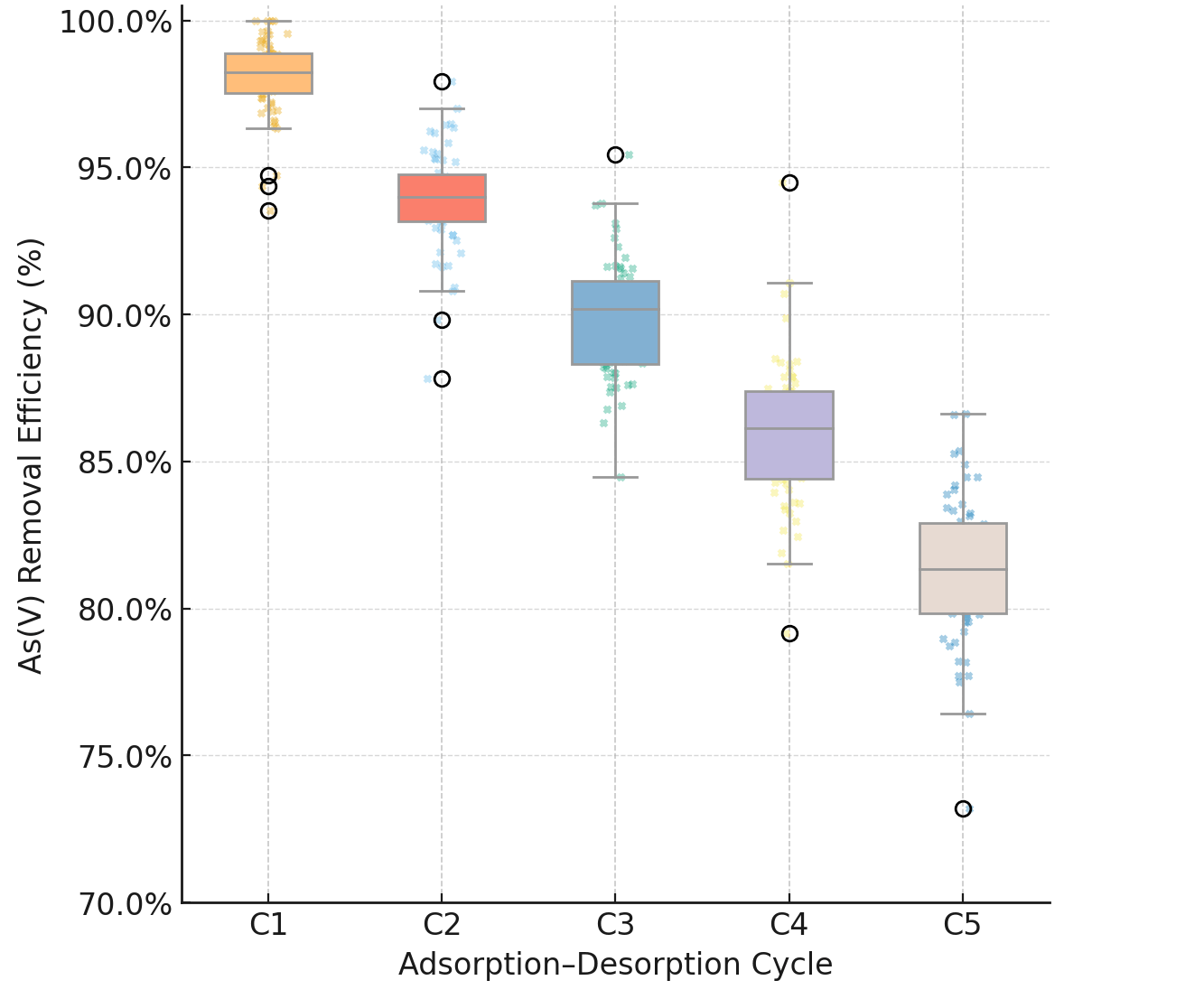 Fig. 16.
Fig. 16.
Reusability performance of the MBC-40 composite over five
adsorption–desorption cycles for As(V) removal (C0 = 10 mg L-1, pH =
7.0
The final and most critical test was the performance of MBC-40 in the complex,
multi-pollutant SMW, designed to mimic a real-world challenge. The results,
summarized in Table 4, are outstanding. Despite the extremely high background
concentration of sulfate (1000 mg L-1) and the presence of competing heavy
metals, the composite demonstrated excellent efficacy. It removed 97.0% of the
total initial arsenic (5.0 mg L-1), reducing the concentration to 0.15 mg
L-1. It was equally effective against both As(V) (97.3% removal) and
As(III) (96.5% removal). Furthermore, the composite exhibited excellent
co-removal capabilities, removing
| Parameter | SMW initial Conc. (mg L–1) | Final Conc. (mg L–1) | Removal efficiency (%) |
| As (Total) | 5.0 | 0.15 | 97.0% |
| As(V) | 3.0 | 0.08 | 97.3% |
| As(III) | 2.0 | 0.07 | 96.5% |
| Pb2+ | 10.0 | ||
| Cd2+ | 5.0 | 0.22 | 95.6% |
| SO42– (interferent) | 1000.0 | - | - |
SMW, simulated mining-impacted water.
To contextualize these outcomes, we emphasize that several macroscopic adsorption behaviors observed here (e.g., pseudo-second-order fitting, Langmuir-type isotherms, phosphate competition, and magnet-assisted recovery) have been reported previously for iron–carbon adsorbents and magnetic composites. For example, Singh and Mohan reported that adsorption behavior consistent with pseudo-second-order kinetics and Langmuir/Freundlich modeling, identified phosphate as a strong competitor, and noted that the sorbent could be recovered by a magnet [46]. The present study therefore contributes primarily at the applied systems level: (i) Fe-loading-controlled synthesis (MBC-10 vs MBC-40) is linked to quantified magnetization and arsenic uptake, (ii) the Fe(0)@Fe3O4 core–shell architecture is verified directly by HR-TEM lattice spacing assignments, and (iii) performance is validated under mining-relevant mixed contamination (5.0 mg L-1 total As with 1000 mg L-1 sulfate and co-occurring Pb2+/Cd2+/Zn2+), demonstrating robust arsenic removal alongside strong co-removal of Pb2+ and Cd2+.
The present study demonstrates that integrating nanoscale zero-valent iron with a biochar scaffold yields a structurally coherent and functionally synergistic composite capable of efficient arsenic immobilization in aqueous systems relevant to mining-impacted environments. The superior performance of MBC-40 relative to pristine biochar and bare nZVI highlights the importance of coupling structural dispersion with reactive phase stabilization. SEM, TEM, and HR-TEM analyses confirm that the biochar matrix effectively suppresses the severe aggregation typically observed for unsupported nZVI, producing a well-dispersed Fe(0)@Fe3O4 core–shell architecture. This structural configuration rationalizes the enhanced adsorption capacity and rapid kinetics observed experimentally. The Fe(0) core provides reductive potential, while the magnetite shell contributes surface hydroxyl groups favorable for arsenic binding. The measured PZC of 6.8 aligns with the strong pH dependence observed for As(V), supporting a significant electrostatic contribution under acidic to neutral conditions. Meanwhile, the comparatively weak pH sensitivity of As(III) removal suggests that specific surface interactions on iron (hydr)oxide phases play an important role. The pseudo-second-order kinetic fit and Langmuir-type isotherm behavior indicate that uptake occurs predominantly at well-defined reactive sites; however, consistent with established adsorption modeling principles, these macroscopic fits should not be interpreted as definitive mechanistic proof of chemisorption. Instead, the data collectively support a coupled adsorption–surface interaction process consistent with known behaviors of iron-based composites.
Beyond mechanistic considerations, the applied significance of this work lies in demonstrating robust performance under chemically complex conditions. The simulated mining water matrix imposed high sulfate concentrations and co-existing divalent metals, yet arsenic removal remained above 96%, and strong co-removal of Pb2+ and Cd2+ was achieved. The negligible interference from sulfate and chloride suggests selective binding preference toward arsenic oxyanions, whereas phosphate competition confirms expected ligand-exchange competition on iron oxide surfaces. Importantly, the saturation magnetization of 32.5 emu g⁻1 enabled rapid magnetic recovery within 30 s, directly addressing one of the major practical limitations of conventional nano-adsorbents—post-treatment separation. Although reusability declined moderately after five cycles, the composite retained over 80% removal efficiency, indicating structural stability and partial preservation of reactive iron phases. Nevertheless, long-term geochemical ageing, Fe(0) passivation, and dissolved organic matter interactions were not examined and may influence field-scale durability. Therefore, while the present findings validate the composite as an effective, magnetically recoverable sorbent under controlled and mining-relevant aqueous conditions, future research should prioritize speciation-resolved mechanistic verification and long-term stability assessment to fully establish environmental applicability.
While the MBC-40 composite demonstrates high efficacy for arsenic immobilization in the tested environments, several limitations of the current study should be noted:
Mechanistic uncertainty: The proposed immobilization mechanisms are interpreted as plausible based on macroscopic adsorption data rather than direct speciation measurements. This study did not utilize advanced spectroscopic tools such as X-ray absorption spectroscopy (XAS) or XPS to definitively distinguish between surface adsorption, inner-sphere complexation, and the formation of specific surface precipitates.
Aqueous matrix simplification: Experimental evaluations, including the SMW tests, were conducted in the absence of DOM or NOM. Because DOM can compete for reactive sites or alter arsenic mobility, these findings primarily reflect material performance in simplified inorganic matrices rather than complex natural or field conditions.
Geochemical ageing and passivation: The study focused on short-term performance and did not account for time-dependent geochemical ageing or the long-term passivation of the nZVI core under oxic or anoxic environmental conditions. Assessing the long-term stability and reductive lifespan of the Fe(0)@Fe3O4 architecture remains a priority for future field-oriented research.
Kinetic and structural interpretations: The application of PSO kinetic models serves as a macroscopic comparison of uptake behavior across different conditions but does not uniquely diagnose a specific rate-limiting step or provide definitive proof of chemisorption. Additionally, the I(D)/I(G) ratio obtained via Raman spectroscopy is a measure of relative disorder and should not be interpreted as a definitive quantification of true graphitization within the biochar matrix.
Loss of reductive capacity: Reusability experiments showed a moderate decline in arsenic removal efficiency after five cycles (from 98.5% to 81.3%), which is likely attributed to the progressive consumption of the Fe(0) core’s reductive capacity and the irreversible binding of arsenic to certain reactive sites.
In this work, a magnetic biochar–nZVI composite (MBC-40) was synthesized by
in-situ liquid-phase reduction using pine-bark biochar (600 °C)
as a support. The composite comprised well-dispersed Fe(0)@Fe3O4
core–shell nanoparticles (50–80 nm) anchored on the biochar matrix and showed
strong magnetic recoverability (32.5 emu g-1), enabling rapid solid–liquid
separation with an external permanent magnet. MBC-40 achieved high arsenic
immobilization performance, with Langmuir capacities of 65.4 mg g-1 for
As(V) and 82.1 mg g-1 for As(III), retained
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
RZ and JD designed the research study. RZ performed the primary research, including the synthesis of the magnetic biochar–nanoscale zero-valent iron composites and the execution of the batch arsenic immobilization experiments. JD provided help and advice on the physicochemical characterization and the testing in simulated mining wastewater. RZ and JD analyzed the experimental data and performed the kinetic and isotherm modeling. RZ drafted the manuscript. Both authors contributed to the critical revision of the manuscript for important intellectual content. Both authors have read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGPT-5.1 in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.