


1 Department of Genetics and Cell Biology, School of Basic Medicine, Qingdao University, 266071 Qingdao, Shandong, China
2 School of Life Sciences, Jining Medical University, 273500 Jining, Shandong, China
3 Department of Laboratory Medicine, Linyi Peoples’ Hospital, 276000 Linyi, Shandong, China
4 Department of Pathology, The Affiliated Hospital of Qingdao University, 266071 Qingdao, Shandong, China
5 Medical School, Shandong Xiehe University, 250000 Jinan, Shandong, China
6 Clinical Laboratory, WeiFang People’s Hospital, Shandong Second Medical University, 261000 Weifang, Shandong, China
Abstract
The application of chemotherapy using the combination of platinum and taxane agents is now considered the standard treatment option for ovarian cancer. Notably, cells respond to DNA damage by promoting DNA repair; however, when repair is insufficient, cell death is induced. The abnormality of the nuclear pore complex (NPC) also leads to nuclear envelope destruction and disrupts transmembrane transport, leading to abnormal substance exchange. Therefore, this study aimed to analyze the mechanisms through which DNA damage induced by carboplatin and paclitaxel contributes to the treatment of ovarian epithelial cancer and to explore the role of DNA damage in mediating the anti-tumor effects of these compounds.
Cell proliferation, cell cycle progression, and apoptosis were examined in four ovarian epithelial cancer cell lines following treatment with carboplatin, paclitaxel, or a combination of both compounds. A water-soluble tetrazolium-1 (WST-1) assay was employed to assess proliferation, flow cytometry was used for cell cycle analysis, and an immunofluorescence (IF) assay was utilized to detect apoptosis in the MES-OV (MOV), A2780, OVCAR-3, and OVCAR-5 cell lines. An IF assay was also used to evaluate nuclear structure and assess DNA damage.
Our study revealed that carboplatin and paclitaxel inhibited cell proliferation and induced cell cycle arrest, which was caused by DNA damage. In addition, paclitaxel induced an irreversible DNA damage, characterized by multimicronucleation, which is associated with NPC inhibition.
Carboplatin and paclitaxel inhibit the proliferation of ovarian cancer cells, induce cell cycle arrest, and promote apoptosis by inducing DNA damage. This is associated with irreversible micronucleation and may be linked to NPC suppression, as well as the disruption of the microfilament and microtubule assembly. Moreover, studying the formation and anti-tumor mechanism of irreversible micronucleation has potential implications for enhancing the efficacy of cytotoxic chemotherapy in killing tumor cells.
Keywords
- carboplatin
- paclitaxel
- ovarian cancer
- nuclear pore complex
- micronucleation
- F-actin fiber
Epithelial ovarian cancer is the fourth leading cause of cancer-related mortality among women in developed countries, accounting for over 200,000 deaths worldwide in 2022 [1, 2]. Among gynecological malignancies, ovarian cancer has the highest fatality rate and is predominantly diagnosed at advanced stages. This is primarily due to the absence of effective screening methodologies and the non-specific nature of early-stage symptoms [3, 4]. Epithelial ovarian cancer is a major component of several germline genetic mutation syndromes, as demonstrated by epidemiological and genetic studies [1]. However, the molecular mechanisms involved have yet to be fully understood, although a likely association is through defects in homologous recombination DNA repair, particularly involving the BRCA1 and BRCA2 genes [1, 5].
Ovarian cancer generally exhibits an initial response to the standard therapeutic regimen, which includes primary surgical cytoreduction followed by platinum-based chemotherapy [6]. In certain instances, the disease remains sensitive to intermittent platinum-based chemotherapy, leading to a chronic phase with a relatively symptom-free period until chemoresistance develops, limiting further treatment options [7]. A recent study has indicated that SLC7A11 expression is significantly elevated in several platinum-resistant ovarian cancer cell lines, including A2780 and SKOV3. This elevation supports intracellular glutathione synthesis, thereby reducing the production of reactive oxygen species (ROS) by platinum-based drugs [8].
Platinum-based regimens have been the global standard of care for ovarian cancer for nearly five decades [1]. However, following pivotal trial outcomes in the 1990s, combining platinum agents (either carboplatin or cisplatin) with taxane agents, such as paclitaxel, has become the established standard of care [9, 10, 11, 12]. Indeed, a pivotal study demonstrated improved survival following the concurrent use of paclitaxel and cisplatin, which led to the current protocol of administering six cycles of the less toxic carboplatin in combination with paclitaxel [6, 10]. Consequently, carboplatin and paclitaxel have remained the cornerstone of global treatment guidelines for the past two decades [7]. Additionally, this combination is recognized as an effective therapeutic strategy, with an overall response rate of 75% and a complete remission rate of 67% previously observed in a cohort of 24 patients with a measurable disease status [10].
Carboplatin, an analog of cisplatin, has lower non-hematologic toxicity and reduced gastrointestinal and metabolic toxicity than cisplatin. Moreover, carboplatin may reduce the risk of death by 16%, suggesting that the efficacy of carboplatin could be slightly superior to that of cisplatin [10]. The pharmacological effects of carboplatin are exerted through a mechanism analogous to that of cisplatin, forming 1,2-intrastrand adducts with the nitrogenous bases of DNA. This interaction results in the cross-linking of DNA strands, thereby inhibiting DNA synthesis and replication. Consequently, this leads to cell cycle arrest and the activation of DNA damage response pathways [13, 14]. DNA damage recognition proteins have been shown to transmit signals from DNA lesions to downstream proteins, including p53 and p73, thereby triggering apoptosis and other cell death mechanisms [15]. Meanwhile, in instances where DNA-damaged cells are unable to undergo apoptosis, alternative cell death pathways, such as necroptosis [16], autophagy [17], and ferroptosis [18, 19], may be activated.
Paclitaxel has been shown to induce apoptosis [20], autophagy [21], necroptosis [22], and senescence [23, 24]. Peripheral neurotoxicity is a major non-hematologic adverse effect of paclitaxel, typically emerging early in the treatment course [25]. Clinical trials have demonstrated that substituting carboplatin for cisplatin in paclitaxel combination regimens reduces neurotoxicity while maintaining comparable anti-tumor activity in the treatment of ovarian cancer [10, 11]. European studies have reported that carboplatin combined with paclitaxel induces lower neurotoxicity than cisplatin combined with paclitaxel [11, 12].
Thus, to optimize the therapeutic efficacy of paclitaxel, research has focused on enhancing the chemotherapeutic potential of paclitaxel while minimizing its associated adverse effects. Notably, apoptosis resistance has been recognized as a key factor reducing the effectiveness of the drug; meanwhile, autophagy has been linked to the development of resistance to platinum-based therapies [26, 27]. Meanwhile, senescence has been observed to have a negative impact on the progression of neoplastic growth [28]. The anti-tumor mechanisms of carboplatin and paclitaxel have been extensively studied; however, the interactions between these compounds and the cytoskeleton, including microfilaments and microtubules (MTs), remain poorly understood. Furthermore, the precise effects of paclitaxel withdrawal or its combination with carboplatin on ovarian cancer cells remain unclear. Therefore, this study aimed to elucidate the impact of combining and withdrawing paclitaxel and carboplatin chemotherapy on the cytoskeletal structure of ovarian epithelial cancer cells.
All ovarian epithelial cancer cell lines, including MES-OV, A2780, OVCAR-3, and
OVCAR-5 cells, were obtained from the Sylvester Comprehensive Cancer Center in
the University of Miami Miller School of Medicine upon conducting the research study “Anti-cancer molecular mechanism of carboplatin and
paclitaxel in ovarian epithelial cancer cells” nearly two years ago. The Sylvester Comprehensive Cancer Center
is a reputable institution known for its high-quality cell lines. Moreover, the cell line
was recently authenticated using the short tandem repeat (STR)
profiling method, which confirmed the identity and purity of the cell line.
Mycoplasma testing was also recently conducted, and the cell line was confirmed
to be free of mycoplasma contamination. The testing was performed using
microscopic examination and polymerase chain reaction (PCR).
Carboplatin and paclitaxel were purchased
from the University of Miami Hospital. Water-soluble tetrazolium-1 (WST-1) (CELLPRO,
Suzhou, Jiangsu, China) was used for the cell proliferation assay. Mouse anti-human cleaved
caspase-3 (sc-271028, 1:100 for immunofluorescence (IF)), lamin B1 (sc-56144, 1:400 for IF),
The MOV, A2780, OVCAR-3, and OVCAR-5 cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (11995, Solarbio, Wuhan, Hubei, China), supplemented with 10% fetal calf serum, and incubated at 37 °C in a humidified 5% CO2 atmosphere. Only cells in the logarithmic growth phase were used for experimental procedures.
The WST-1 assay was used to evaluate the effect of carboplatin and paclitaxel
on cellular proliferation. Cells were seeded at a density of 1
Cell fixation was performed using a 4% paraformaldehyde solution (P1110, Solarbio, Wuhan, Hubei, China). Membrane permeabilization was achieved using 0.5% Triton X-100 (T8200, Solarbio, Wuhan, Hubei, China) for 5 minutes. Non-specific binding was blocked using a solution of 5% bovine serum albumin (BSA) (SW3015, Solarbio, Wuhan, Hubei, China) in phosphate-buffered saline (PBS) (P1020, Solarbio, Wuhan, Hubei, China) with 0.1% Tween-20 (T8220, Solarbio, Wuhan, Hubei, China) for 30 minutes. Subsequently, cells were incubated with primary antibodies at 4 °C overnight, followed by incubation with secondary antibodies labeled with Alexa Fluor 488 for green fluorescence or Alexa Fluor 594 for red fluorescence (Molecular Probes/Invitrogen, Carlsbad, CA, USA). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (S2110, Solarbio, Wuhan, Hubei, China). The cells were mounted and sealed using an anti-fade reagent composed of 0.1 M n-propyl gallate (A420948, Sangon Biotech, Shanghai, China) at pH 7.4, mixed with 90% glycerol (A600232, Sangon Biotech, Shanghai, China) in PBS. Imaging was performed utilizing a Leica confocal microscope equipped with AxioVision software version 4.8 (Leica, Wetzlar, Germany).
For IF analysis, cells were fixed and resuspended in a chilled ethanol/PBS solution (70% v/v) and stored at –20 °C for subsequent use. Before flow cytometric analysis, cells were rehydrated in a solution containing propidium iodide and RNase A. The cell cycle kit (A10798, Invitrogen, Carlsbad, CA, USA) was used before conducting flow cytometric analyses on a BD LSR Fortessa instrument (Becton Dickinson, San Jose, CA, USA).
Comparisons between two independent sample groups were performed using the t-test, while comparisons among multiple groups were conducted using the analysis of variance (ANOVA) followed by Tukey’s honestly significant difference (HSD) post hoc test for multiple comparisons.
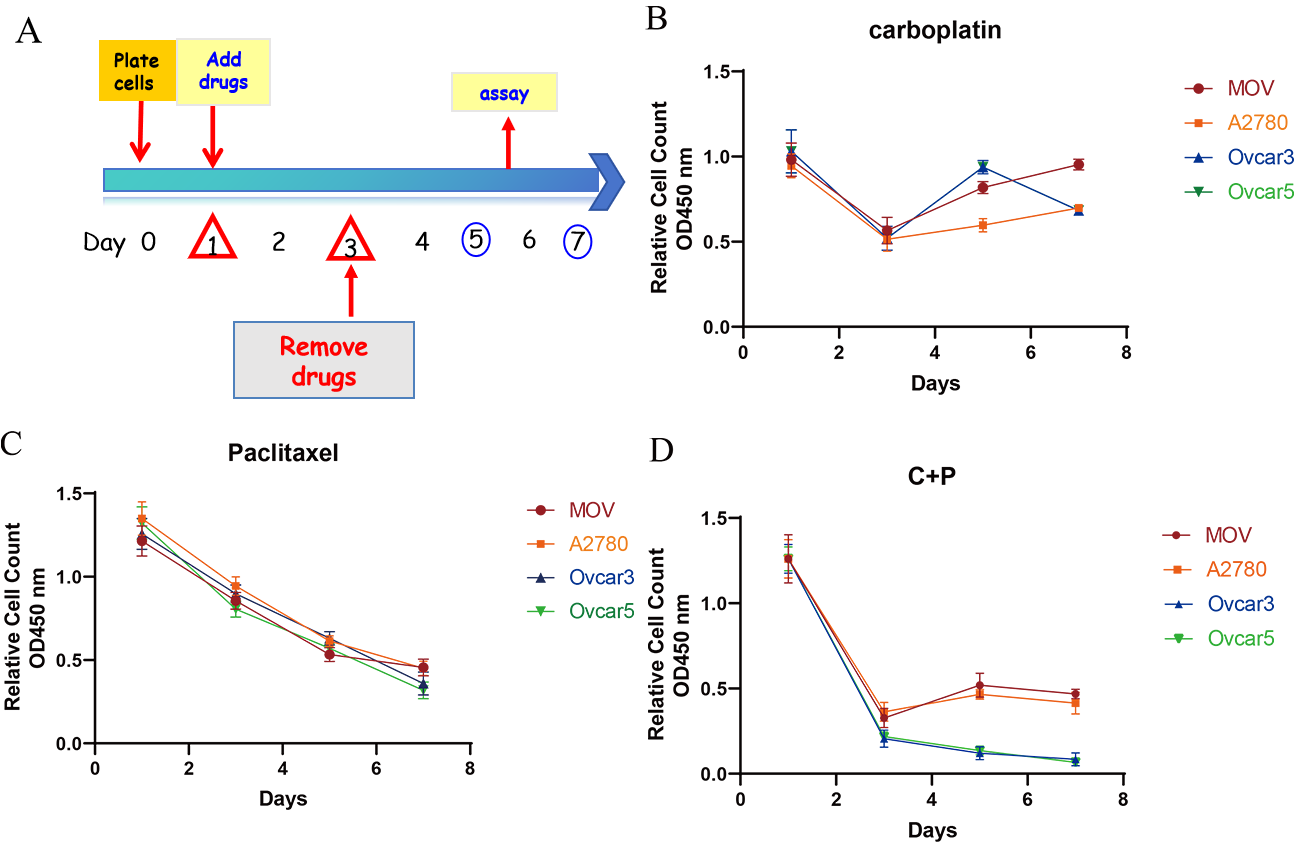
The inhibitory effects of carboplatin and paclitaxel on ovarian cancer cells were first evaluated in vitro using the WST-1 assay. As shown in Fig. 1, all cell lines (MOV, A2780, OVCAR-3, and OVCAR-5) exhibited a similar dose-response to carboplatin and paclitaxel.
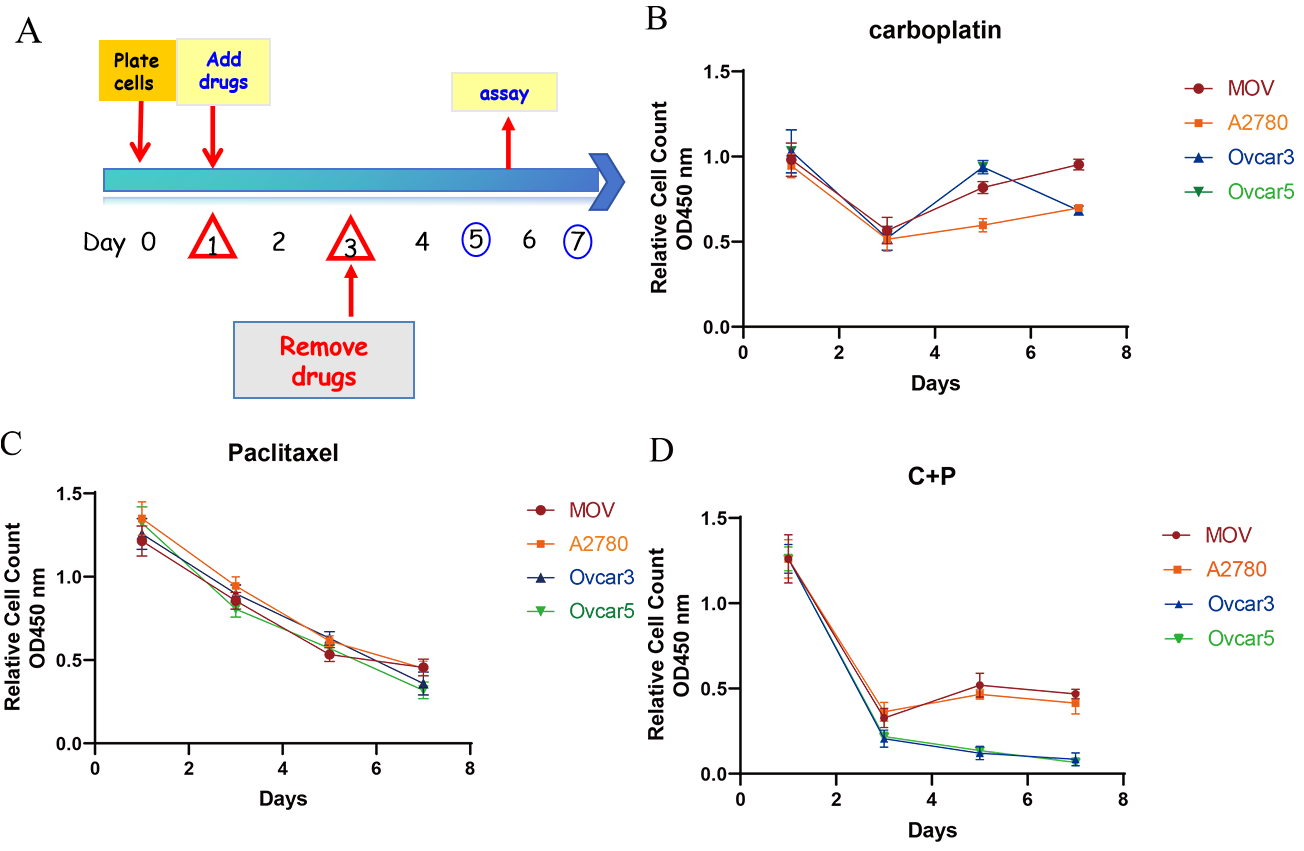 Fig. 1.
Fig. 1.
Growth inhibition effects of carboplatin and paclitaxel in vitro. (A) A protocol was established to simulate the administration
of chemotherapeutic drugs: cells were seeded and incubated for one day, followed
by a two-day drug treatment with either paclitaxel (1 nM), carboplatin (1 mM), or
a combination of both. Subsequently, the drugs were removed, and cell recovery
was assessed. (B–D) The cell viability of four ovarian cancer cell lines (MOV,
A2780, OVCAR-3, and OVCAR-5) was evaluated using the WST-1 assay after treatment with
carboplatin (B), paclitaxel (C), or a combination of both agents (D), as per
the manufacturer’s protocol. Cell viability was determined in triplicate,
and the results are presented as the mean
A protocol was established to investigate the synergistic effects of paclitaxel and carboplatin in the context of ovarian cancer treatment. The methodology involved a 48-hour exposure of ovarian cancer cells to either paclitaxel, carboplatin, or a combination of both compounds. Subsequently, the drugs were removed to allow for the recovery of the surviving cell population (Fig. 1A).
A significant reduction in cell count was observed following 48 hours of drug exposure, with values ranging from 40% to 70% of the initial count on the third day. The data presented in Fig. 1B–D correspond to paclitaxel, carboplatin, or a combination of both compounds, respectively. Notably, upon the withdrawal of carboplatin, the cells exhibited a resurgence in proliferation. Conversely, cells treated with paclitaxel failed to regain their proliferative capacity, showing a sustained decline in cell number even after a seven-day post-treatment period (Fig. 1C,D).
These findings suggest that while carboplatin-induced growth inhibition is reversible upon drug removal, paclitaxel treatment results in irreversible cell death and growth suppression. Despite the cessation of paclitaxel administration, the inability of the cells to proliferate was attributed to the transmission of DNA structural abnormalities to subsequent generations, ultimately leading to the death of progeny cells.
According to the WST-1 assay results, we speculated that carboplatin and paclitaxel could induce DNA damage or disrupt the cytoskeleton disorder in ovarian cells. Therefore, we examined the structures of the nuclear pore complex (NPC) and F-actin fibers in ovarian cells treated with carboplatin and/or paclitaxel using IF staining. These four cell lines (MOV, A2780, OVCAR3, and OVCAR5) were used in various experiments, all of which showed similar responses to carboplatin and paclitaxel, with only some representative results provided (Figs. 2,3,4).
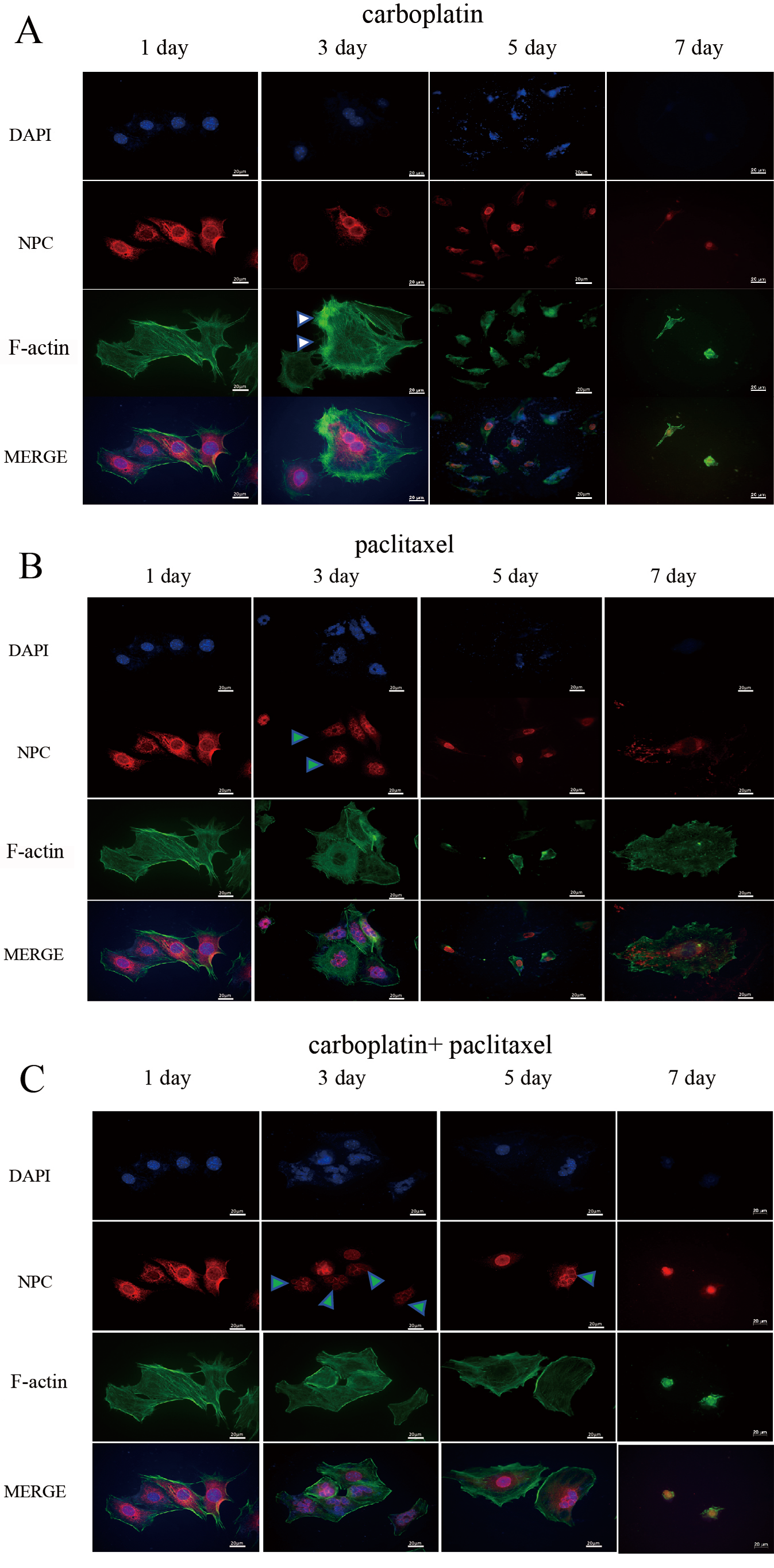
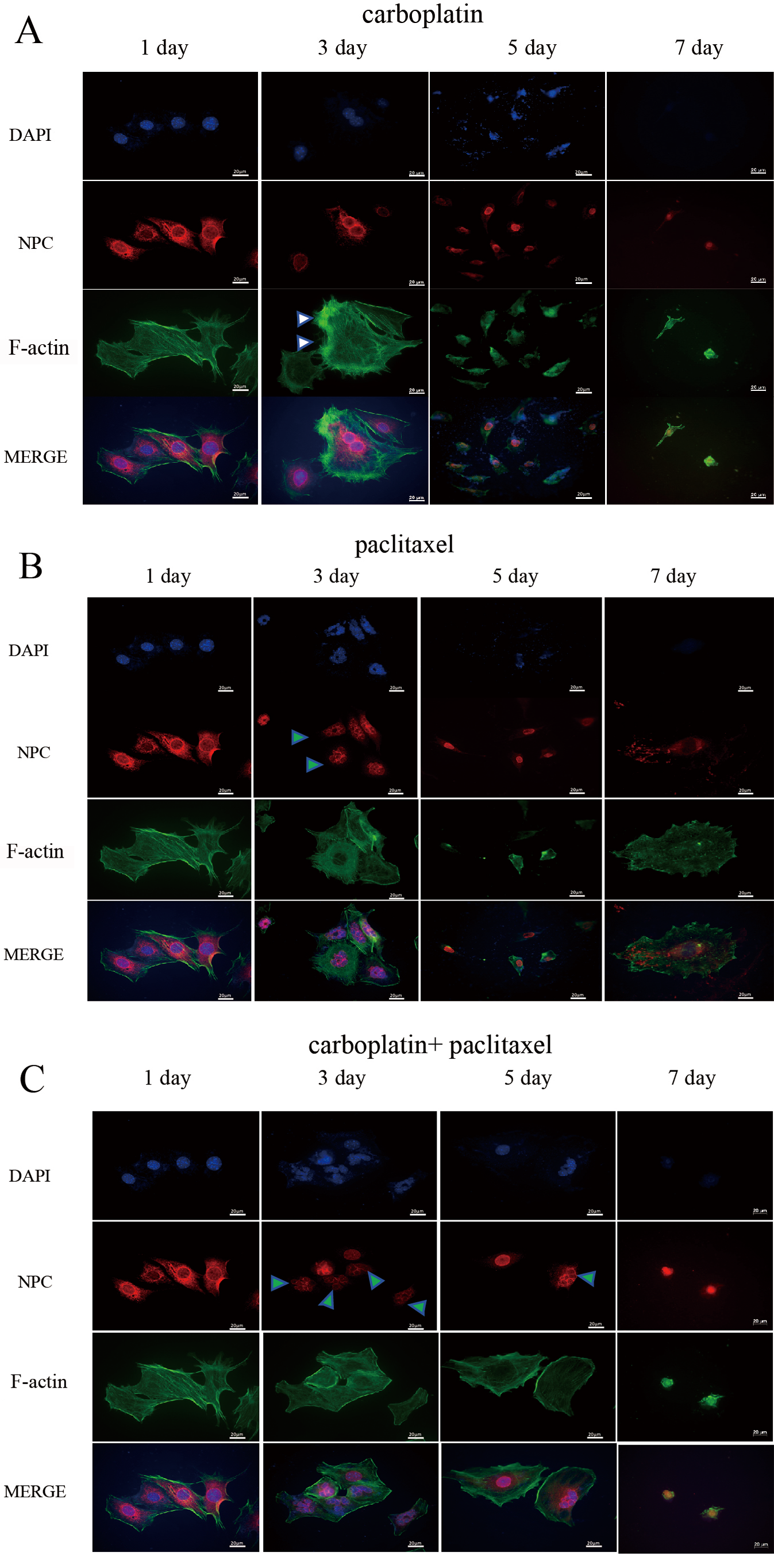 Fig. 2.
Fig. 2.
Detection of NPC and F-actin fiber structures by IF staining in MOV cells. Cell morphology and the nuclear structure of MOV cells were examined by IF staining after treatment with carboplatin (A), paclitaxel (B), or a combination of carboplatin and paclitaxel (C) for 2 days. The corresponding drugs were then removed, and cells were analyzed at 1, 3, 5 and 7 days (Fig. 1A). The white triangle indicates an increase in cell size and the formation of F-actin stress fibers in the cells. After removing carboplatin, a large number of cells underwent nuclear burst or multinucleation. (B) The nuclei of a large proportion of MOV cells became smaller and exhibited a mininucleated shape. After removing paclitaxel, a large number of cells underwent nuclear burst or multinucleation. The green triangle indicates multimicronucleation. (C) The shape and the nuclei of the MOV cells became irregular, with the nuclei of most cells enlarging, and the overall cell morphology becoming enlarged. The green triangle represents multimicronucleation. Scale bar = 20 µm. NPC, nuclear pore complex; IF, immunofluorescence; MOV, MES-OV; DAPI, 4′,6-diamidino-2-phenylindole.
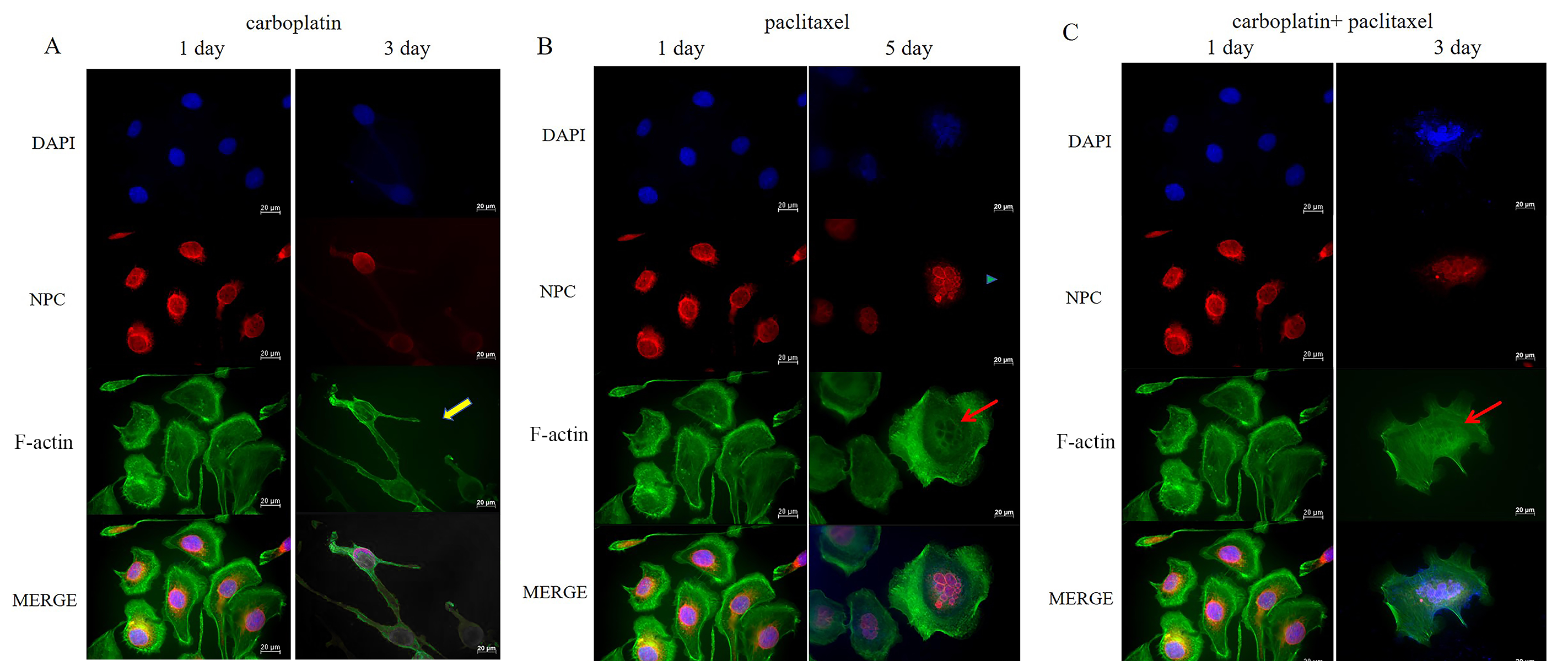
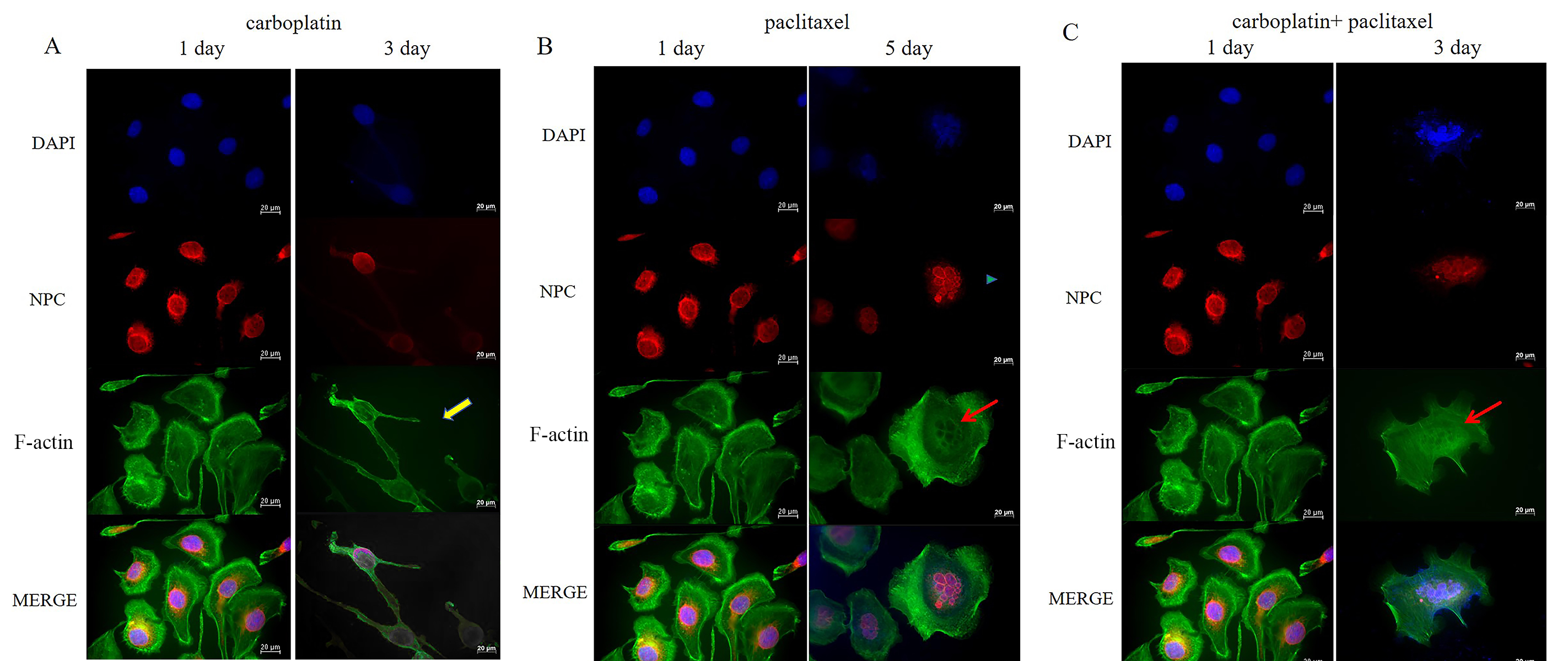 Fig. 3.
Fig. 3.
Detection of NPC and F-actin fiber structures by IF staining in OVCAR-3 cells. (A) Cell morphology and the nuclear structure of OVCAR-3 cells were examined by IF staining before and after treatment with carboplatin for 2 days. The yellow arrow represents the formation of actin spikes and multipolar morphology in OVCAR-3 cells. (B) Cell morphology and nuclear structure of OVCAR-3 cells were examined by IF staining before treatment with paclitaxel and after paclitaxel removal for 2 days. The blue triangle indicates multimicronucleation, while the red arrow represents F-actin bundles around the multimicronucleations. (C) Cell morphology and the nuclear structure of OVCAR-3 cells were examined by IF staining before and after treatment with the combination of carboplatin and paclitaxel for 2 days. The red arrow represents F-actin bundles around the multimicronucleations. Scale bar = 20 µm. NPC, nuclear pore complex; IF, immunofluorescence.
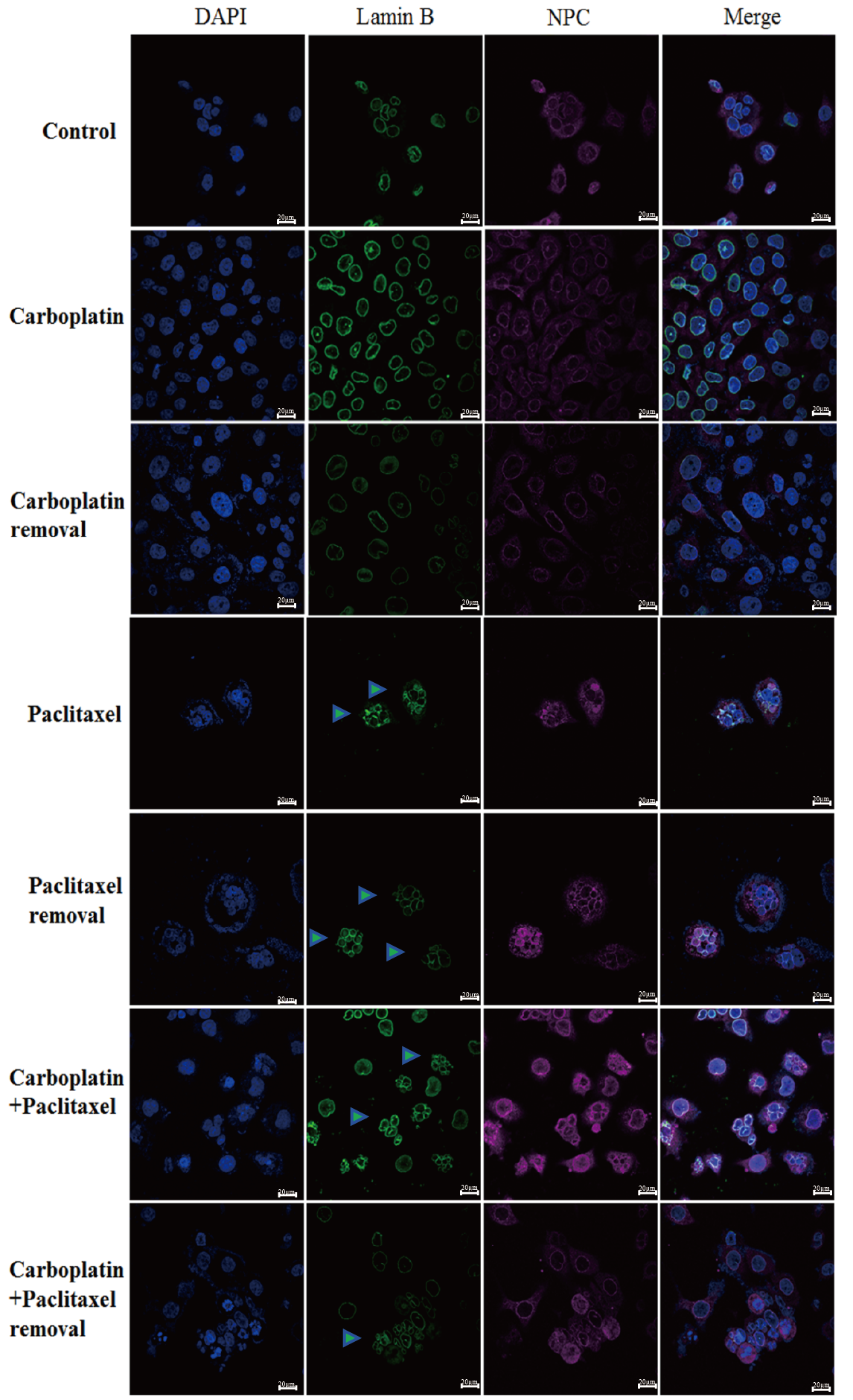
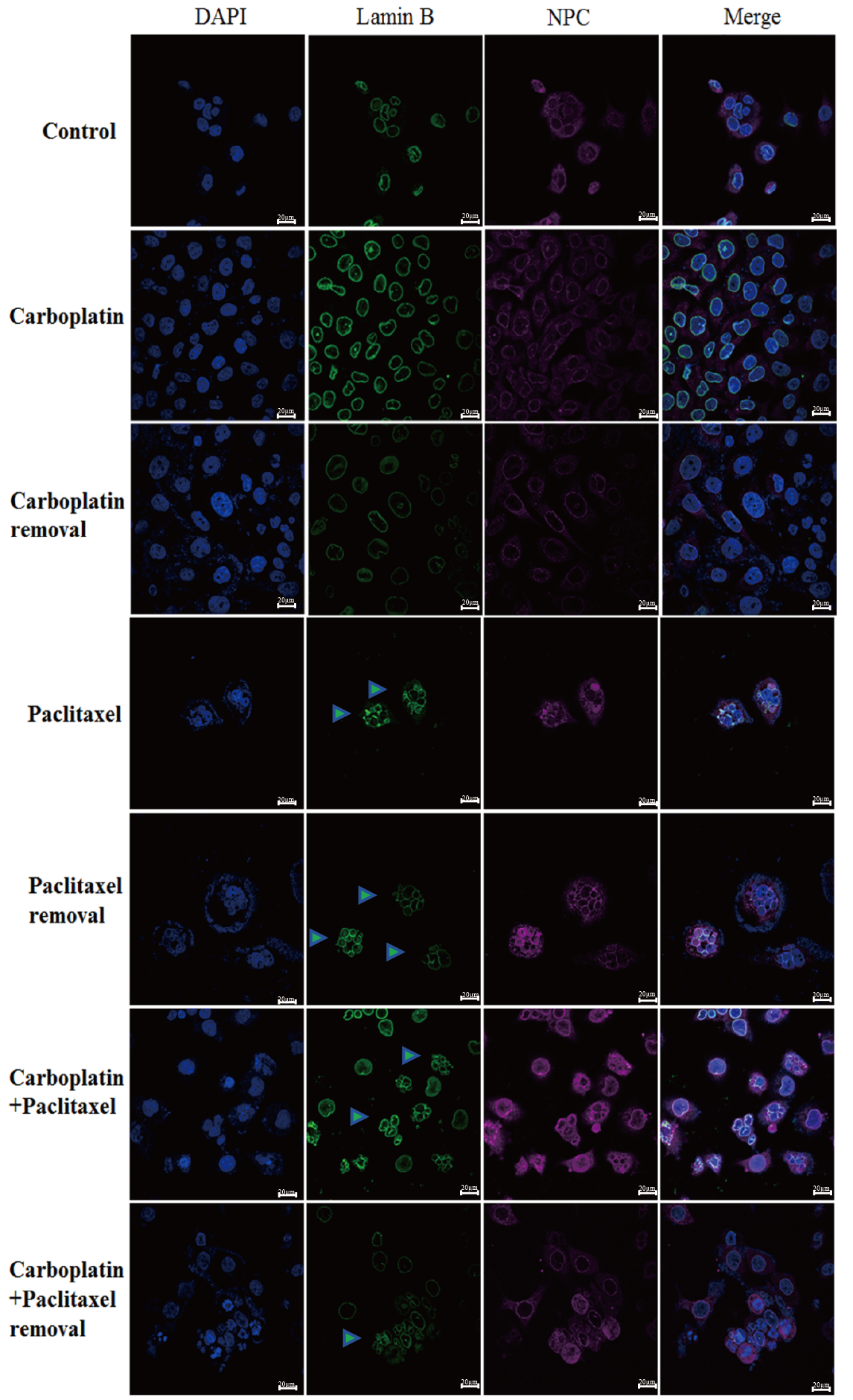 Fig. 4.
Fig. 4.
Detection of NPC and lamin B in OVCAR-5 by IF staining using a confocal microscope. Control: wild type. Carboplatin: carboplatin treatment for 2 days. Carboplatin removal: carboplatin removal for 2 days. Paclitaxel: paclitaxel treatment for 2 days. Paclitaxel removal: paclitaxel removal for 2 days. Carboplatin + paclitaxel: carboplatin + paclitaxel treatment for 2 days. Carboplatin + paclitaxel removal: carboplatin + paclitaxel removal for 2 days. The green triangle represents multimicronucleation. Scale bar = 20 µm. NPC, nuclear pore complex.
We found that the shape and the nuclei of the MOV cells became irregular after treatment with carboplatin for 48 h. Additionally, following treatment, the cells increased in size. After removing carboplatin, we observed that a large number of cells underwent nuclear bursting or exhibited multiple nucleation (Fig. 2A).
In contrast to carboplatin, paclitaxel treatment caused a large proportion of the nuclei in the MOV cells to exhibit micronucleation, with multiple smaller nuclei clustered together, rather than a single contiguous mass, as exemplified in NPC-stained specimens (Fig. 2B). The mininucleated structure has previously been reported as multimicronucleation [29]. This is potentially due to paclitaxel disrupting the assembly of MTs, preventing proper chromosome segregation during replication, which leads to the formation of multinucleated cells within a single cell. After removing paclitaxel from the MOV cells for 96 h, a significant number of cells similarly underwent nuclear burst or increased nucleation (Fig. 2B). The multimicronucleation remained irreversible even after the combination treatment of carboplatin and paclitaxel was removed for 2 days (Fig. 2C).
In addition to substantially increasing cell size, carboplatin enhanced actin stress fiber formation in MOV cells (Fig. 2A) and even induced tripolar microfilament structures in the OVCAR-3 cells after 48 h of treatment (Fig. 3A). F-actin bundles were also observed surrounding the multimicronucleation caused by paclitaxel in OVCAR-3 cells (Fig. 3B,C). After the removal of carboplatin for 96 hours, F-actin microfilaments condensed into a small spot, localized around the fragmented nuclear (Fig. 2A). In contrast, following the removal of paclitaxel for 96 h, the microfilaments expanded, becoming more diffused and weaker (Fig. 2B). No significant difference in the concentration of F-actin microfilaments was observed between the effects of paclitaxel alone and the combination of carboplatin and paclitaxel after 2 days of treatment (Fig. 2B,C).
Using IF staining and confocal microscopy, we further examined the expressions of NPC and lamin B, and found that the nuclei in most cells were significantly enlarged after treatment with carboplatin. Moreover, after removing carboplatin from the OVCAR-5 cells for 48 h, the nuclei in most cells remained significantly enlarged. In addition, the surface of the nuclear envelope remained intact. In contrast to the carboplatin treatment group, paclitaxel treatment promoted the destruction and rupture of the nuclear envelope. Even after removing paclitaxel, the nuclear membrane remained ruptured (Fig. 4).
Thus, these data suggest that carboplatin and paclitaxel induce DNA damage and disrupt F-actin microfilaments. Even after removing the combination of carboplatin and paclitaxel, the DNA damage could not be repaired autonomously. Notably, with increasing drug concentrations leading to multimicronucleations, even after eliminating paclitaxel, the cell survival rate continued to decline.
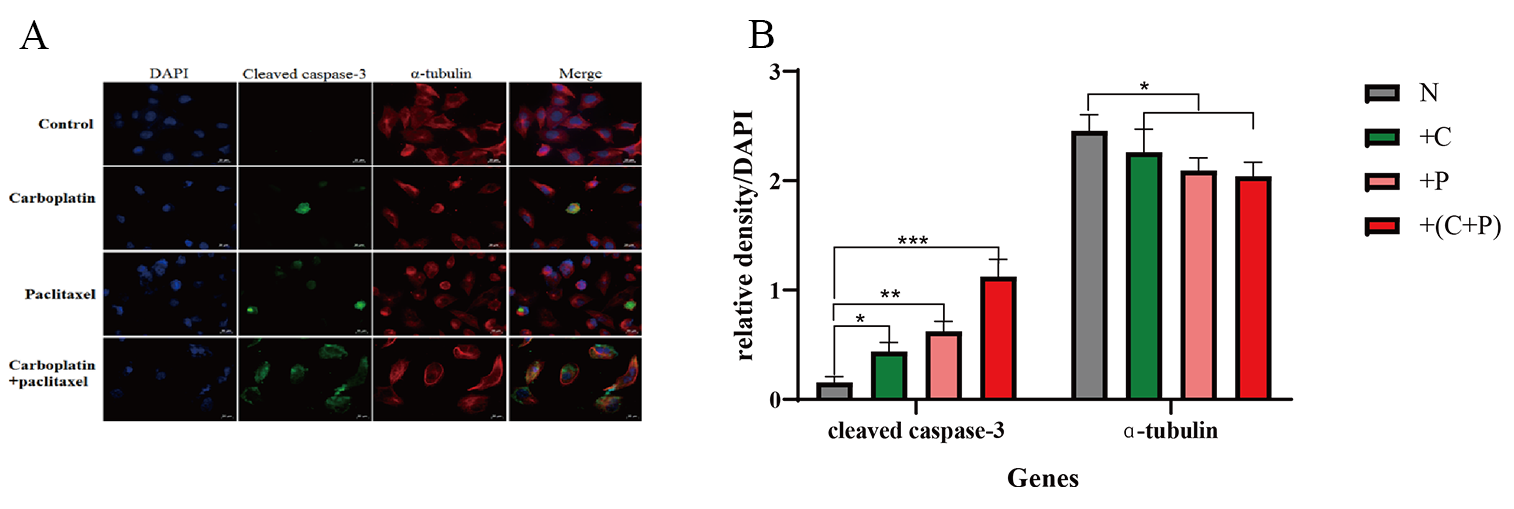
We determined the expressions of cleaved caspase-3 and
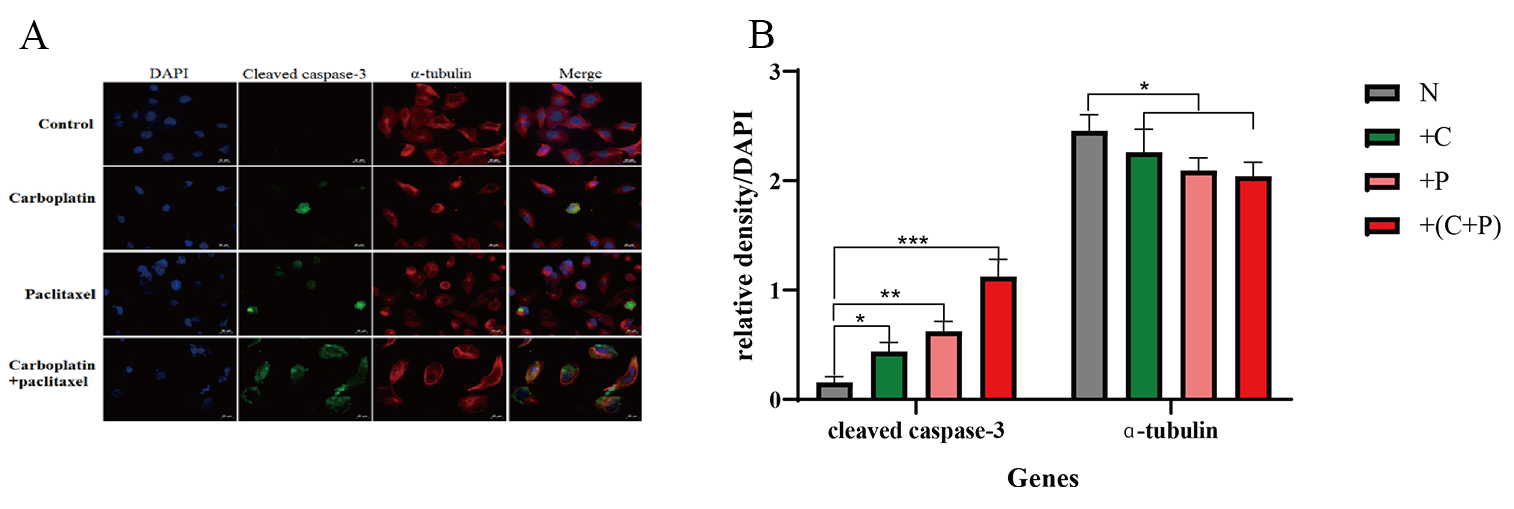 Fig. 5.
Fig. 5.
Detection of cleaved caspase-3, and
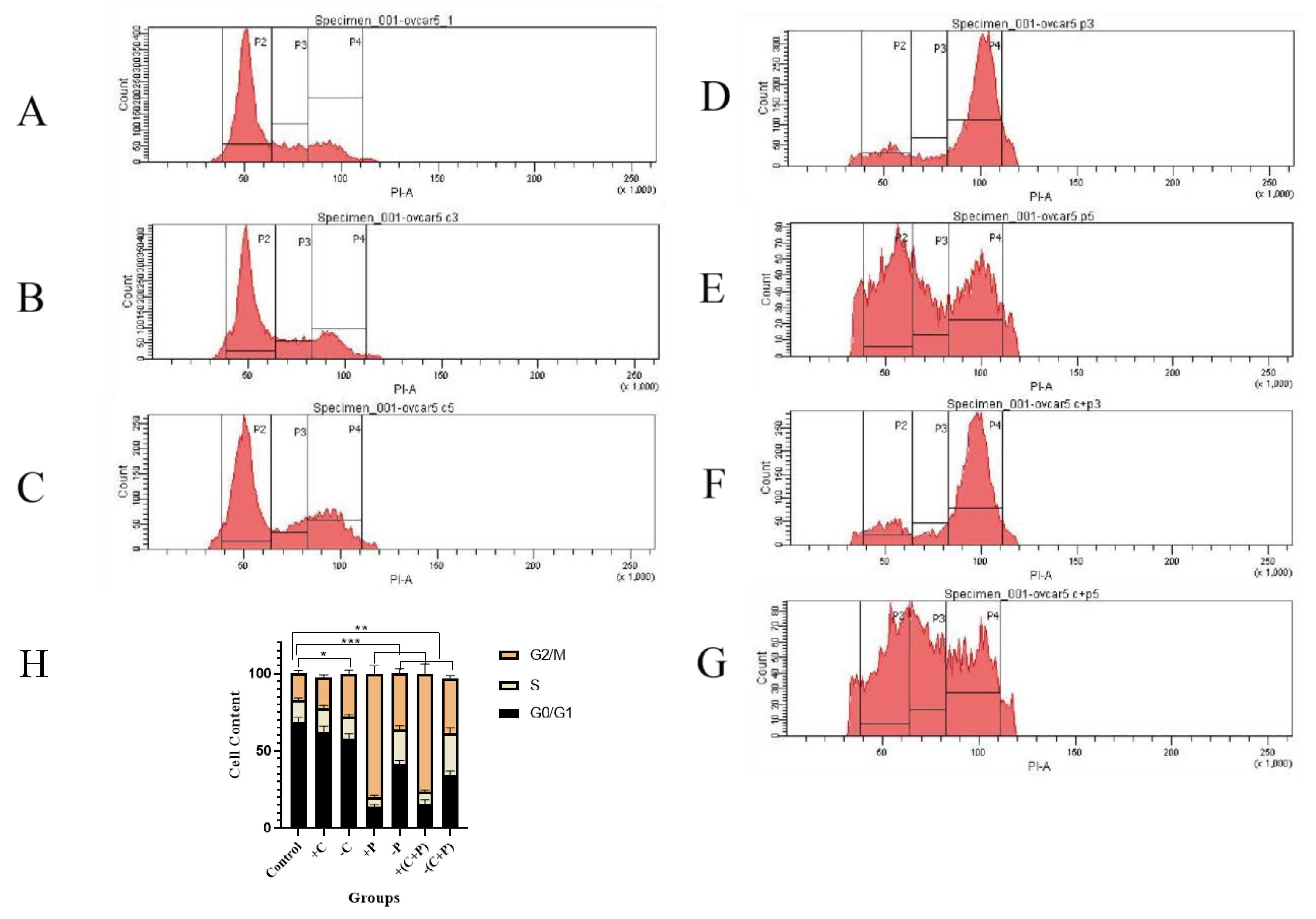
Flow cytometry showed that carboplatin had minimal effects on the cell cycle; however, cell numbers in the G1 phase decreased after carboplatin was removed, while the number of cells in the G2/M phase increased, likely due to the induction of DNA damage. Subsequently, DNA damage induced cell cycle arrest in OVCAR-5 cells and promoted cell apoptosis (Fig. 6A,B,C,H).
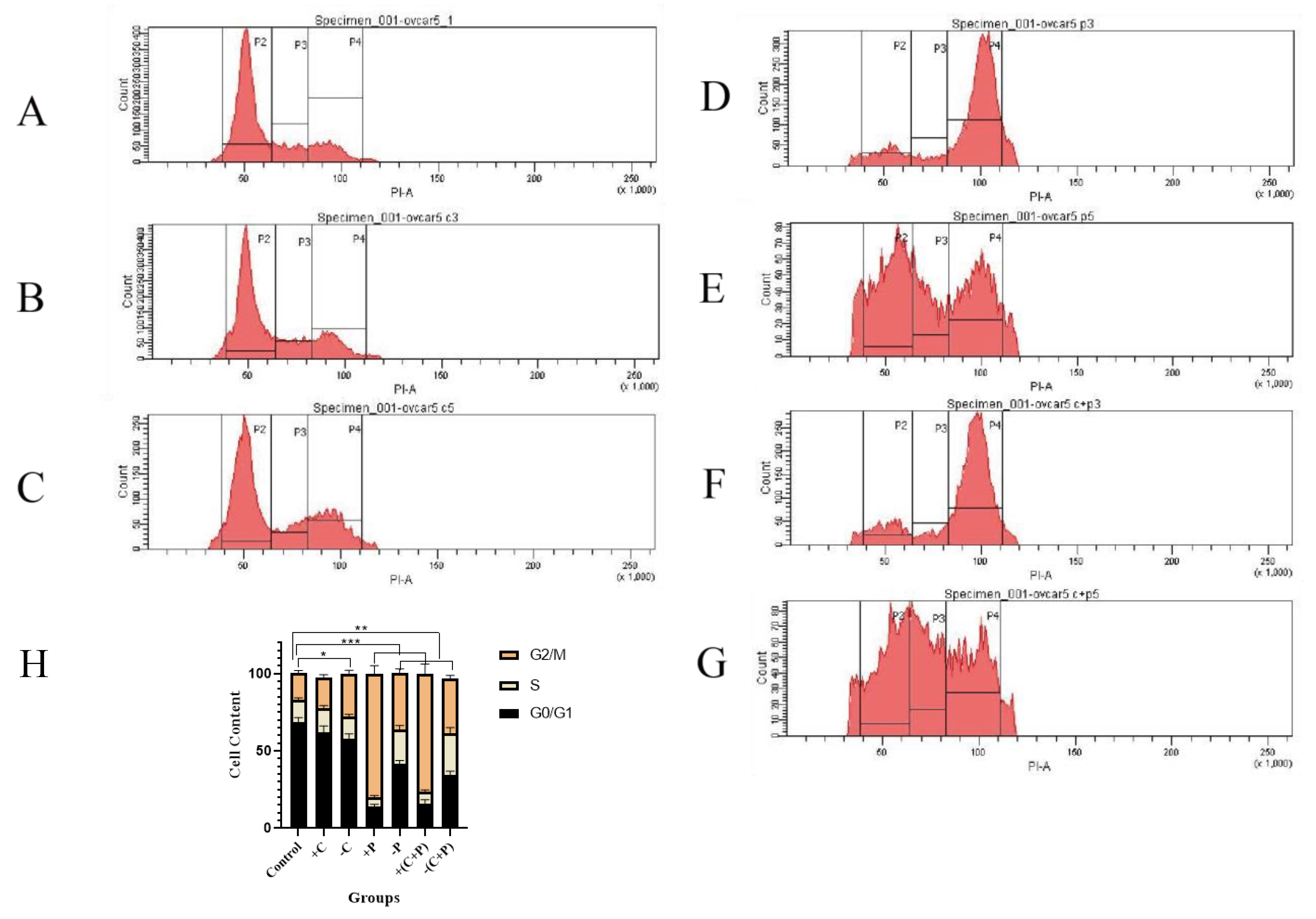 Fig. 6.
Fig. 6.
Cell cycle distribution and DNA content changes in OVCAR-5
cells assessed by flow cytometry. (A)
Control. (B) Carboplatin treatment for 2 days. (C) Carboplatin removal for 2
days. (D) Paclitaxel treatment for 2 days. (E) Paclitaxel
removal for 2 days. (F) Carboplatin + paclitaxel treatment for 2
days. (G) Carboplatin + paclitaxel removal for 2 days. (H) Cell
cycle alterations. P2: G0/G1 phase, 2n; P3: S phase,
2n~4n; P4: G2/M phase, 4n. “*” represents
p
Paclitaxel significantly increased the number of cells in G2/M phase arrest after 2 days of treatment and decreased the number of cells in the G1 phase. Even after removing of paclitaxel, the cells remained arrested in the G2/M and S phases (Fig. 6D–H).
In summary, our data suggest that carboplatin, and particularly paclitaxel, can induce G2/M phase arrest in ovarian cancer cells, even after the compounds are removed.
The six-cycle intravenous carboplatin/paclitaxel regimen represents the current standard primary chemotherapy treatment following surgical debulking in ovarian cancer patients [30, 31]. Although the majority of patients with advanced disease initially respond well to chemotherapy, most eventually develop recurrent disease that becomes refractory to further chemotherapy. Drug resistance, often due to reduced drug uptake, increased detoxification, dysregulation of apoptosis, and heightened DNA repair mechanisms, remains the predominant factor contributing to treatment failure [32]. Thus, a critical need exists for novel diagnostic and therapeutic approaches for ovarian cancer. Presently, no biomarkers with sufficient specificity have been identified to predict the onset of drug resistance. Hence, further research into the cellular and molecular mechanisms underlying drug resistance in ovarian cancer is warranted.
Our experimental data suggest that both carboplatin and paclitaxel inhibit the proliferation of ovarian cells, with paclitaxel causing a more pronounced and irreversible reduction in cell proliferation. Additionally, the synergistic effects of carboplatin and paclitaxel were more pronounced. Furthermore, cisplatin depletes intracellular glutathione, resulting in the inhibition of glutathione peroxidases (GPX) [18], which induces oxidation, F-actin depolymerization, and ultimately results in the condensation of microfilaments into spots around the fragmented nucleus. Meanwhile, paclitaxel weakens microfilaments by inducing apoptosis [20], autophagy [21], necroptosis [22], and senescence [23, 24], even after the compound is removed.
Carboplatin and paclitaxel can cause DNA damage, while the resulting DNA damage, such as multimicronucleation, cannot be repaired by the cells even after paclitaxel is removed. Since lamin B levels recover after removing the agents, lamin B is potentially not involved in multimicronucleation. Thus, we hypothesize that the irreversible multimicronucleation induced by paclitaxel is caused by the suppression of the NPC. Additionally, the small GTPase Ran regulates nucleocytoplasmic shuttling of components and drives chromatin-dependent MT assembly [33]. Furthermore, elevated Ran expression in tumors has been correlated with increased malignancy and reduced survival compared to tumors with lower expression levels [34]. Ran has also been reported to contribute to three hallmarks of cancer: proliferation, resistance to apoptosis, and invasion/metastasis [34]. In our study, the expression of tubulin was significantly reduced after treatment with the paclitaxel and carboplatin combination, and a positive correlation was observed between Ran and tubulin assembly. Furthermore, paclitaxel induced the formation of microfilament bundles around multimicronucleations in ovarian cells. Finally, we hypothesize that the suppression of the NPC, along with the disruption of microfilament and MT assembly, may contribute to the irreversible multimicronucleation.
Low levels of DNA damage activate cellular repair mechanisms and promote survival, whereas high levels of DNA damage induce cell death [35]. Carboplatin and paclitaxel are genotoxic anti-cancer drugs that target DNA and induce various DNA lesions. Multimicronucleation is a form of irreversible DNA damage that can activate pathways that promoting cell elimination, such as apoptosis and necrosis [36]. During apoptosis and cell cycle arrest, the expression level of p53 is critical in DNA damage repair (DDR) [37].
DNA damage can activate cell cycle checkpoints, allowing time for DNA repair before replication occurs. These checkpoints are pivotal for pharmacological interventions targeting the cell cycle, making the cell cycle checkpoints prime targets for anti-cancer drugs. Should the DNA repair pathway fail, the unrepaired damage disrupts replication and transcription, thereby activating the DDR and triggering downstream signaling pathways that lead to cell death [35]. Flow cytometry results showed that carboplatin induced DNA damage, preventing OVCAR-5 from entering the cell cycle. Paclitaxel significantly increased G2/M phase arrest and decreased the number of cells in the G1 phase. Carboplatin and paclitaxel induced DNA damage, leading to dysfunction in cell cycle regulation and ultimately causing the death of OVCAR-5 cells.
Current research suggests that low levels of DNA lesions trigger DNA repair mechanisms, as indicated by the upregulation of repair genes, including DDB2, XPC, XRCC1, XPF, XPG, and others [38]. However, at high levels of DNA damage, repair mechanisms become saturated, and unrepaired DNA damage triggers the death programs, including apoptosis, necrosis, and autophagy. The induction of necrosis results from the activation of poly (ADP-ribose) polymerase 1 (PARP1) during the DDR process. Activation of PARP1 depletes cellular pools of nicotinamide adenine dinucleotide (NAD+) and adenosine triphosphate (ATP), thereby triggering necrotic cell death [39]. Furthermore, following treatment with paclitaxel and carboplatin, the proportion of OVCAR-5 cells exhibiting high levels of cleaved caspase-3 protein expression was significantly higher than that in the control group. Specifically, when OVCAR-5 cells were exposed to a combination of carboplatin and paclitaxel, nearly all cells exhibited an upregulation in cleaved caspase-3 expression. Although we have investigated some of the molecular structures of multimicronucleation and the mechanisms by which it promotes apoptosis, the molecular mechanisms underlying the formation of multimicronucleation still need to be further explored.
Overall, carboplatin and paclitaxel can inhibit cell proliferation and induce cell cycle arrest by causing DNA damage. Additionally, paclitaxel induces the destruction of nuclear structure, leading to irreversible multimicronucleation, which is associated with the suppression of the NPC and the disruption of microfilament and MT assembly. The cleaved caspase-3 pathway promotes cell apoptosis in ovarian cells following treatment with carboplatin and paclitaxel treatment. However, the mechanisms underlying the suppression of the NPC and the disruption of microfilament and MT assembly induced by carboplatin and paclitaxel warrant further investigation. Therefore, elucidating the mechanisms through which irreversible multimicronucleation leads to cell death is crucial for enhancing the cytotoxic efficacy of genotoxic chemotherapy.
The data analyzed during the current study are not publicly available due to the management of Sylvester Comprehensive Cancer Center but are available from the corresponding author on reasonable request.
BL, FZ and LJ designed the research study. FZ, JL and BL performed the cell proliferation, cell cycle, western blot research. HJ and GL performed the immunofluorescence experiments. WH and HZ analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This project was supported by the Grants from the Youth Innovation and Science and Technology Plan of Colleges and Universities in Shandong Province (2019KJK016), Shandong Taishan Scholars Young Experts Program (No.tsqn202103056), Natural Science Foundation project of Shandong Province (ZR202209280042), Shandong Provincial Natural Science Foundation, China (ZR2021MF095), Shandong Provincial Natural Science Foundation (ZR2020QH189), Shandong Provincial Medical and Health Science and Technology Development Program (2019WS123).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.