


 , Jiarui Li 2, Jie Tan 1, Ping Tang 1,*
, Jiarui Li 2, Jie Tan 1, Ping Tang 1,*
1 Department of Orthopedic Surgery, Jiaxing Maternity and Children Health Care Hospital Fetal Medicine, 314009 Jiaxing, Zhejiang, China
2 Department of Orthopedic Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, 310016 Hangzhou, Zhejiang, China
Abstract
Fetal skeletal dysplasia is a group of disorders that cause abnormal bone growth and development in the fetus, resulting in severe complications and economic burdens on healthcare systems. This study aims to enhance the diagnosis and management of fetal skeletal dysplasia by examining its phenotypes, genetic causes, and the connection between genetic mutations and observed traits.
We performed a retrospective analysis of 28 prenatal cases diagnosed with fetal skeletal dysplasia using advanced genetic testing methods such as whole exome sequencing, chromosomal analysis, and single nucleotide polymorphism (SNP) array analysis.
Our findings revealed diverse phenotypic presentations, with 24 cases exhibiting limb shortening, and distinct genetic inheritance patterns: parental dominant (PAD), de novo mutations (DNMs), autosomal recessive (AR), and cases without pathogenic mutations (UN). Prenatal ultrasound was crucial for early detection and influenced management strategies. Additionally, SNP array analysis combined with short tandem repeats (STR) confirmed the biological relationship between the fetus and the mother, ensuring the integrity of the data. Exome sequencing identified candidate mutation sites, and whole genome sequencing provided insights into structural variations, facilitating personalized management approaches.
This study highlights the importance of early diagnosis and genetic counseling for at-risk families and emphasizes the need for further research to confirm genetic findings and investigate potential future therapies based on the identified mutations. Our research contributes valuable insights into the genetic and clinical characteristics of fetal skeletal dysplasia, paving the way for improved diagnostic accuracy and patient outcomes in affected families.
Keywords
- fetal skeletal dysplasia
- phenotype
- genetic reasons
- next-generation sequencing
- whole exome sequencing
Fetal skeletal dysplasia (FSD), as defined by the International Skeletal Dysplasia Registry (ISDR), is a heterogeneous group of congenital disorders characterized by abnormal bone and cartilage morphogenesis during fetal development, leading to skeletal deformities, growth restriction, and functional impairments. These conditions encompass over 400 distinct entities classified into major categories (e.g., chondrodysplasias, osteochondrodysplasias, and dysostoses) based on molecular, radiographic, and histopathological criteria established by ISDR. Diagnostic criteria for FSD, aligned with ISDR guidelines, include: (1) Prenatal ultrasonography: Detection of key abnormalities such as: Long bone shortening (e.g., femur length
Informed consents were acquired from the pregnant women and their relatives prior to their inclusion in this study. This study received approval from the Prenatal Diagnosis Ethics Committee at the Maternal and Child Health Hospital of Jiaxing, China (2025-Y-016) and followed the Helsinki Declaration of 1964 and its subsequent amendments. Participants were informed that the sequencing results from the exome sequencing cohort would be available for scientific reports after the study, and only results related to fetal abnormalities would be shared with parents.
We conducted a retrospective analysis of patients diagnosed with fetal skeletal dysplasia via prenatal ultrasound at the Fetal Medical Center of the Maternal and Child Health Hospital of Jiaxing, China. The patients were enrolled from January 2020 to December 2022. Chromosomal karyotyping and microarray analyses showed no abnormalities in the amniotic fluid or cord blood samples from the 28 fetuses. Among the cases, four involved one parent with skeletal dysplasia. One mother had a history of two pregnancies affected by skeletal dysplasia, while the remaining cases had no family history of hereditary diseases. Fig. 1 outlines the testing workflow.
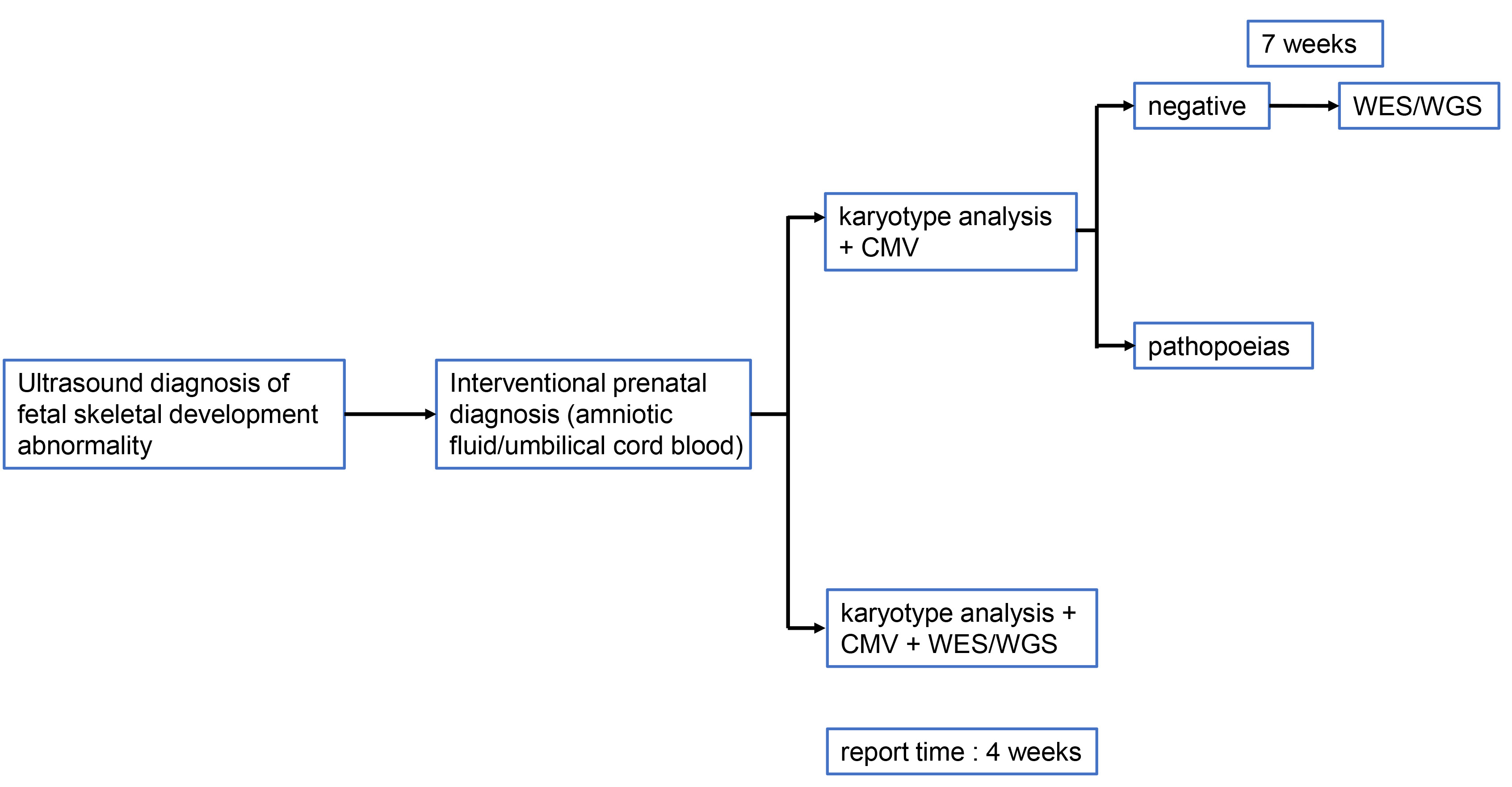 Fig. 1.
Fig. 1. The procedure of genetic testing for fetuses with suspected skeletal development abnormalities. WES, whole-exome sequencing; WGS, whole genome sequencing; CMV, cytomegalovirus.
This flowchart outlines the stepwise genetic testing pipeline for cases identified with fetal skeletal development abnormalities via ultrasound. The workflow proceeds as follows:
Initial Screening: Detection begins with ultrasound diagnosis of fetal skeletal development abnormality, which identifies cases requiring further prenatal genetic assessment.
Invasive Prenatal Sampling: For eligible cases, interventional prenatal diagnosis is performed, involving the collection of amniotic fluid or umbilical cord blood samples.
First-tier Genetic Analyses:
Two parallel initial testing pathways are employed based on clinical considerations:
Pathway 1: Karyotype analysis + cytomegalovirus (CMV) testing: Conventional cytogenetic analysis (karyotyping) assesses gross chromosomal abnormalities, while CMV testing rules out viral-associated fetal anomalies. If results from this pathway are negative (no pathogenic findings after 7 weeks of waiting for final karyotype and CMV results), the pipeline transitions to whole exome sequencing/whole genome sequencing (WES/WGS) for broader genetic interrogation. If pathogenic chromosomal changes or CMV-related pathologies (pathopoeias) are detected, this pathway concludes.
Pathway 2: Karyotype analysis + CMV + WES/WGS: This integrated pathway simultaneously performs karyotyping, CMV testing, and high-resolution exome/genome sequencing upfront, aiming to accelerate diagnosis for complex or high-risk cases.
Turnaround Time: The standard report time for results from the testing pipeline is 4 weeks for pathways incorporating concurrent WES/WGS, whereas the first pathway may extend to 7 weeks before potentially transitioning to additional sequencing.
Key transition: The move to WES/WGS occurs when initial karyotype and CMV testing (in Pathway 1) yields no definitive answers, enabling exploration of single-gene and submicroscopic genetic variants associated with skeletal dysplasias and other fetal anomalies.
All two-dimensional (2D) images were captured using the E8 ultrasound system (No.: YZB/AUS 1561-2015; GE Healthcare Austria GmbH & Co OG, Zipf, Upper Austria, Austria) with a transabdominal probe (No.: RAB2-5; GE Healthcare Austria GmbH & Co OG, Zipf, Upper Austria, Austria). All images were saved and analyzed offline with version 4.0 of the four-dimensional (4D) imaging software.
After gestational weeks had been verified by ultrasonography in the first trimester, it was found in the second and third trimesters that fetal femur lengths (FL) were less than the 5th quantile (P5) of the normal FL value in the corresponding gestational weeks. Regarding long bone morphologies, normal limb long bones exhibited straight, strong echoes with posterior sound shadows. Abnormalities were considered in cases of uniform thickening or local thickening of the long bone, bending deformation of the bone, and angular fractures. Finally, the ossification degree was determined via bone resonance and posterior bone shadows.
Routine fetal amniotic fluid or cord blood puncture was conducted. A total of 23 amniotic fluid samples (10 mL each), 5 cord blood samples (3 mL each), and 3 mL of venous blood samples from the couples were collected. The STR genotype detection revealed that all fetuses had biological parent-child relationships, and eliminated the contamination of fetal DNA by maternal genomic DNA.
Amniotic fluid, cord blood, and peripheral venous blood samples from pregnant women and their spouses were collected and subjected to G-band chromosomal karyotyping analysis. The chromosomes were prepared by conventional cell cultures, phytohemagglutinin stimulation and colchicine treatment. Cytogenetic analyses were conducted on the slides through G-band. The 25 mitotic phases were microscopically counted. Five of them were analyzed and described according to the International System for Human Cytogenetic Nomenclature (ISCN 2016), which standardizes the notation for chromosomal aberrations, copy number variants (CNVs), and genomic coordinates (Genome Reference Consortium Human Build 37 [GRCh37]/hg19 assembly).
Genomic DNA were extracted from amniotic fluid or cord blood samples and analyzed for single nucleotide polymorphisms using the Affymetrix CytoScan 750K (Affymetrix Inc., 901859, Santa Clara, CA, USA) array gene chip. Including 550,000 nonpolymorphic (NP) probes and 200,000 SNP probes (0.1Mb resolution) platform.
Extraction of DNA from cord blood and peripheral blood samples was performed using the DNA extraction kit (Tiangen Biochemical Technology Co., Beijing, China), as instructed by the manufacturer. Then, the purity and quantity of the DNA were assayed. The remaining samples were stored at –20 °C. Purification and sequencing of PCR amplification products were performed by Huada Gene Co., Ltd. (Shenzhen, Guangdong, China). Candidate mutation sites were determined after DNA extraction and quality testing, genome library construction, target region capture and sequencing, as well as bioinformatics analyses. Data analyses: sequenced fragments were aligned with the human genome version 19 (hg19) human reference genome on (University of California, Santa Cruz) UCSC through Burrows-Wheeler aligner (BWA) to remove duplicates. Then, Genome Analysis Toolkit (GATK) was used for base mass correction, single nucleotide variant (SNV), insertion-deletion (INDEL), and genotypic detection. Copy number variant (CNV) detection at the exon level was conducted by Exome Depth.
In this assay, about 500 ng of genomic DNA was broken by ultrasound into 250–300 bp fragments, mixed with the end repair buffer (T4 Polynucleotide Kinase, 10,000 U/mL, lot: 16041820; ENZYMATICS, Beverly, MA, US + Klenow fragment, 5000 U/mL, lot: 7121620; ENZYMATICS, Beverly, MA, US + TaKaRa Taq™, lot: AJ12290A; TAKARA Bio Inc., Kusatsu, Shiga, Japan) and repair enzyme mixture (10X T4 PNK Reaction Buffer, lot: EK0032; ENZYMATICS, Beverly, MA, US) (20 °C for 30 min; 65 °C for 30 min; 10 °C for 1 min). Then, the ligase, buffer and adaptor were added to the end repair system, and the mixture was kept at 22 °C for 30 min. After purification by magnetic beads, the sample was added to the PCR amplification reaction system (98 °C for 2 min; 98 °C for 20 s; 65 °C for 30 s; 72 °C for 40 s, a total of 8–10 cycles; 72 °C for 4 min). After magnetic bead purification, the quality of the library was tested. Briefly, the library had a, concentration
Based on high-throughput WES, Sanger sequencing was performed to verify the mutation sites of the fetus and parents. The corresponding PCR primers were designed for potential pathogenic sites, followed by PCR amplification and product purification. Sequencing was conducted on ABI3130 BigDye Terminator v3.1 Cycle Sequencing Kit, lot: 4337455 (Applied Biosystems, Foster, CA, US). Then, sequencing results were aligned with the standard sequence in GenBank using the Lush aligner share software version 1.13.1 Huada Gene Co., Ltd. (Shenzhen, Guangdong, China) to determine the mutation sites. The pathogenicity of mutation sites was determined based on genetic mutation classification standards formulated by the American college of medical genetics and Genomics (ACMG) in 2015. Mutation pathogenicity classification was based on ACMG and the Association for Molecular Pathology (AMP) Guidelines for Interpretation of Sequence Variations. This guideline was interpreted based on the ClinGen Sequence Mutation Interpretation Working Group and Association for Clinical Genomic Science (ACGS).
This study mainly adopts descriptive analysis.
According to the next-generation sequencing (NGS) results and genetic patterns, 28 patients were divided into 4 groups. All grouping information is shown in Table 1. The grouping criteria are as follows: (1) Parental dominant (PAD) Group (n = 5): Criteria: Cases with heterozygous pathogenic variants in autosomal dominant genes (e.g., FGFR3, ACAN) inherited from an affected parent, confirmed by parental Sanger sequencing (to rule out de novo mutations). Clinical correlation: Positive family history of skeletal dysplasia (e.g., achondroplasia) and typical radiographic features (e.g., rhizomelic limb shortening). (2) De novo Mutation Dominant (DNM) Group (n = 10): Criteria: Cases with novel heterozygous variants absent in both parents, validated by trio-based Whole Exome Sequencing (WES) (variant allele frequency
| Genetic pattern | PAD | DNM | AR | UN |
| Number of cases | 5 | 10 | 5 | 8 |
| Simple | 3 | 1 | 3 | 3 |
| Comorbidities | 2 | 9 | 2 | 5 |
Simple: only with skeletal system abnormalities; Comorbidity: Combined with other systemic abnormalities.
PAD, parental dominant; DNM, de novo mutation dominant; AR, autosomal recessive; UN, unresolved.
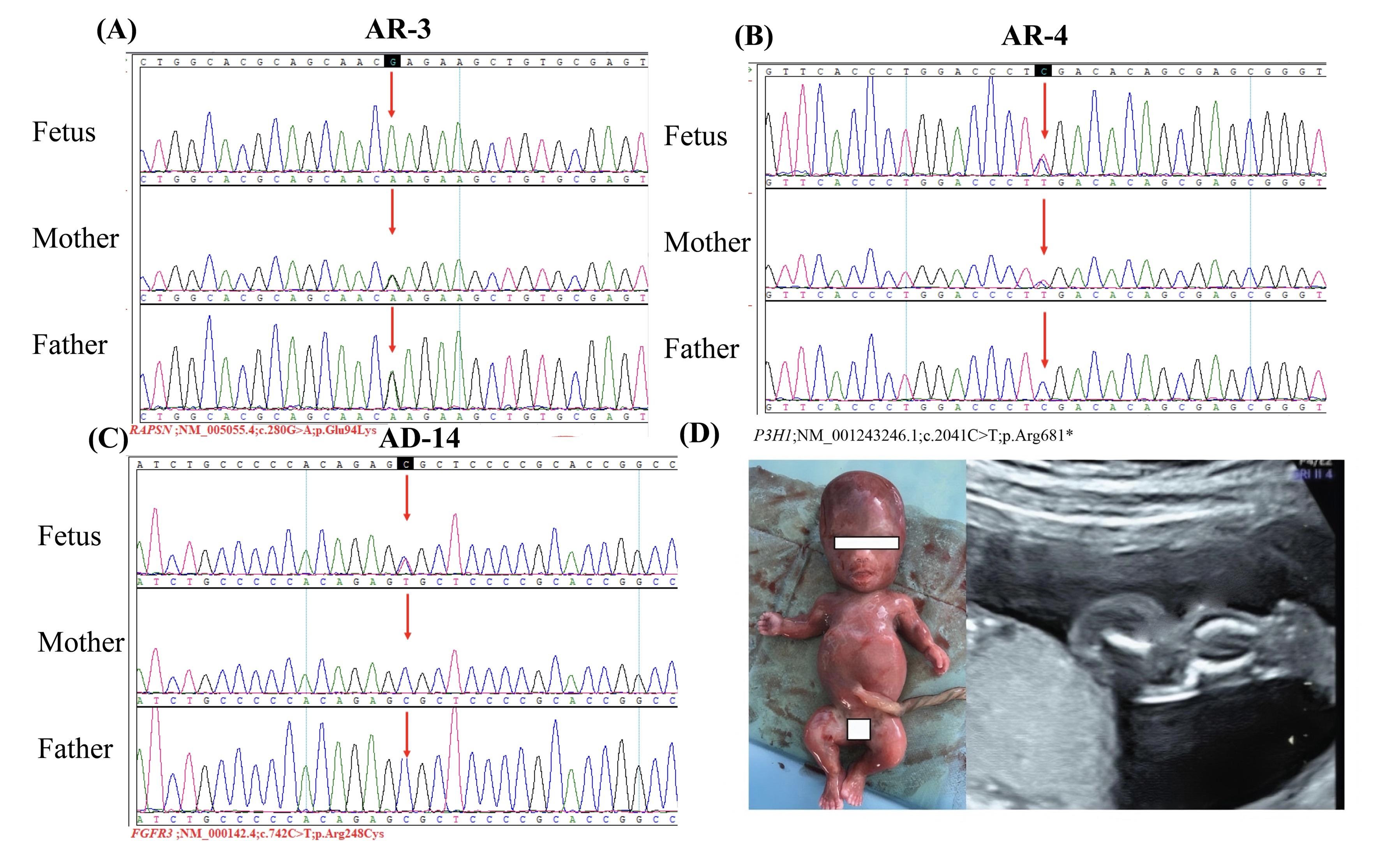 Fig. 2.
Fig. 2. Genetic mutations and induced labor with fetal bone abnormalities. (A) shows a homozygous mutation of RAPSN (inherited from both parents). (B) shows a homozygous mutation of P3H1 (inherited from both parents). (C) shows a heterozygous mutation of FGFR3 (newly developed mutation). (D) shows the appearance and ultrasound examination of induced labor with abnormal fetal bone development. RAPSN, receptor-associated protein of the synapse; P3H1, prolyl 3-hydroxylase; FGFR3, fibroblast growth factor receptor 3; AR, autosomal recessive; AD, autosomal dominant inheritance.
Gestational weeks: Gestational weeks for the PAD group ranged from 17 to 30 weeks (median = 20 weeks); 19 to 28 weeks (median of 25.5 weeks) for the DNM group; 16 to 33 weeks (median of 20 weeks) for the AR group and from 17 to 35 weeks (median of 28 weeks (the largest among the four groups)) for the UN group. Twenty-one cases were detected by ultrasound in the second trimester (pathogenic mutations were not found in 4 cases), while 7 cases were detected by ultrasound in the third trimester (pathogenic mutations were not found in 4 cases). The gestational weeks were determined by prenatal ultrasound. Differences in mean number of gestational weeks (24.4
Prenatal ultrasound performance: A total of 24 cases had shortness of long bones, 1 case had bent bones, 1 case had equinovarus, 2 cases had bone development deficiencies, 4 cases had retarded intrauterine growths and 17 cases had other abnormalities. Among the 8 cases without definite genetic etiologies, there was 1 case of multiple skeletal deformities at 18 weeks of pregnancy, 3 cases of femoral shortening with fetal growth restriction (FGR) (30–34 weeks of pregnancy), 1 case of gallbladder defects, 1 case of clubfoot (24 weeks of pregnancy), and 1 case who developed fetal edema and shortness of long bones around 18 weeks of two pregnancies. Twenty one cases developed fetal skeletal abnormalities in the second trimester (pathogenic mutations were not detected in 4 cases), while 7 cases had fetal skeletal abnormalities in the third trimester (pathogenic mutations were not detected in 4 cases).
The PAD group: One case had shortness of long bones with polyhydramnios; 1 case had shortness of long bones with hydrocephalus (
The DNM group: Eight cases had shortness of long bones combined with increased head circumference (
The AR group: One case had skeletal abnormalities with FGR; 1 case had diaphragmatic eventration and pleural effusion; 3 cases had simple shortness of long bones.
The UN group: Three cases had shortness of long bones with FGR; 1 case had edema; 1 case had cardiac malformations; 1 case had simple strephopodia; 2 cases had shorter femurs (
Fetal skeletal dysplasia represents a heterogeneous group of disorders characterized by abnormal growth and development of bones during fetal life. These conditions can lead to severe health complications, including intrauterine growth restrictions and severe anomalies, which create a significant burden on healthcare systems due to the need for extensive medical management and interventions. Diagnosis is complex due to the variety of skeletal dysplasias, each with varying phenotypic manifestations. Advances in imaging techniques, such as ultrasonography and fetal MRI, have improved the ability to identify skeletal abnormalities in utero, yet challenges persist in accurately diagnosing the specific type of dysplasia, particularly because some conditions shares, these clinical traits may not become apparent until the later stages of pregnancy [3, 7].
Moreover, this study seeks to bridge existing gaps in the literature regarding the genetic and clinical characteristics of fetal skeletal dysplasia. By examining various cases and correlating genetic findings with clinical presentations, we aspire to improve diagnostic capabilities and patient outcomes. The integration of advanced imaging techniques with genetic analyses may yield a more holistic understanding of fetal skeletal dysplasia, ultimately benefiting affected families through enhanced prenatal care and counseling [8, 9].
In summary, this research is poised to contribute significantly to the existing body of knowledge surrounding fetal skeletal dysplasia. By elucidating the genetic factors and phenotypic manifestations associated with these disorders, the findings could inform clinical practices and pave the way for targeted interventions. The ongoing evolution of genetic diagnostic methodologies presents an opportunity to refine our understanding of fetal skeletal dysplasia and improve the quality of care provided to families impacted by these challenging conditions [7].
This study aims to investigate the genetic underpinnings associated with fetal skeletal dysplasia through a comprehensive retrospective analysis. By employing advanced genetic testing methodologies, such as whole exome sequencing and chromosomal analysis, the research seeks to elucidate the phenotypic manifestations observed through prenatal imaging and their correlation with specific genetic mutations. Understanding the genetic basis of these disorders is paramount, as it not only enhances diagnostic accuracy but also informs the management and counseling of affected families. The key findings of this research offer insights into the inheritance patterns of skeletal dysplasia, which can significantly influence prenatal care and the clinical approach to managing pregnancies at risk of these conditions [10, 11].
This study significantly advances the understanding of fetal skeletal dysplasia by employing a comprehensive approach that integrates advanced genetic testing and prenatal imaging. Unlike previous studies that primarily focused on either imaging or genetic analysis, this research combines both modalities to provide a more holistic picture of the condition. For instance, while Rahemtullah et al. (1997) [2] highlighted the importance of the femur length-to-abdominal circumference ratio in predicting fetal outcomes in suspected skeletal dysplasia, our findings reveal distinct genetic patterns linked to various phenotypic presentations, thereby filling a critical gap in the literature regarding the genetic basis of these disorders. Moreover, our study provides evidence for the multiple inheritance patterns in human cases, contrasting with earlier research that primarily dealt with single-gene disorders, thereby extending the scope of genetic counseling and management strategies for affected families [1].
The implications of our findings for clinical practice are profound. By identifying specific genetic mutations associated with fetal skeletal dysplasia, healthcare providers can offer targeted counseling to families, enhancing their understanding of recurrence risks in future pregnancies. This aligns with the recommendations made by Lachman and Rappaport (1990) [4], who emphasized the necessity of genetic counseling in the management of skeletal dysplasias due to their high recurrence risk. Additionally, the integration of advanced prenatal imaging techniques with genetic insights could revolutionize prenatal management, allowing for earlier and more informed decision-making regarding potential interventions and parental expectations. The early detection of skeletal abnormalities not only aids in the timely preparation for possible medical interventions but also plays a crucial role in parental psychological readiness, as underscored in studies emphasizing the importance of informed parental counseling and support [3].
Despite the significant contributions of this study, several limitations must be acknowledged. The retrospective design and relatively small sample size constrain the generalizability of our findings, as highlighted in similar studies that called for larger, multi-center investigations to validate genetic findings in the context of fetal skeletal dysplasia [8]. Additionally, the absence of clinical validation for some genetic results limits the ability to fully characterize the phenotypic spectrum of skeletal dysplasia. Future research should aim to incorporate larger cohorts and longitudinal studies to assess the long-term implications of identified genetic mutations and to refine diagnostic protocols for prenatal skeletal dysplasia [12]. By addressing these limitations, subsequent studies can further enhance the understanding of the complex interplay between genetic mutations and phenotypic outcomes in fetal skeletal dysplasia.
The limitations of this study warrant careful consideration. The retrospective design inherently restricts the scope of conclusions that can be drawn, as it is based solely on previously collected data, which may introduce biases or gaps in information. Additionally, the relatively small sample size limits the generalizability of the findings to broader populations. Some genetic findings lacked clinical validation, which raises questions regarding their direct applicability in clinical settings. Future studies should aim to incorporate larger cohorts and prospective designs to enhance the robustness of the data. Moreover, integrating clinical validation of genetic findings will be crucial in establishing reliable diagnostic and management protocols for fetal skeletal dysplasia.
In conclusion, this research presents significant advancements in understanding the genetic and clinical characteristics of fetal skeletal dysplasia. The findings underscore the critical role of early diagnosis and genetic counseling in managing affected families. By elucidating distinct genetic patterns and their phenotypic manifestations, this study lays the groundwork for future investigations into targeted therapeutic strategies and improved prenatal care. The integration of advanced genetic testing methods with prenatal imaging offers a promising avenue for enhancing diagnostic accuracy and ultimately improving patient outcomes in fetal skeletal dysplasia.
All data reported in this paper will also be shared by the lead contact upon request.
XW, PT and JL designed this study. XW, JL, and JT conducted research. PT provided assistance in the design of the research plan and suggestions for the overall writing approach of the manuscript. XW and JL analyzed these data. All authors have contributed to the editing and revision of the manuscript. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was carried out in accordance with the guidelines of the Declaration of Helsinki. Received approval from the Prenatal Diagnosis Ethics Committee at the Maternal and Child Health Hospital of Jiaxing, China (Ethic Approval Number: 2025-Y-016), and all of the participants provided signed informed consent.
We sincerely appreciate Jiaxing Maternal and Child Health Hospital for providing technical assistance in sample sequencing. We also appreciate the constructive comments from the reviewers, which greatly improved the quality of this manuscript. Finally, we would like to express our gratitude to all the research participants for their collaboration.
This research was funded by the Research on People’s Livelihood Science and Technology Innovation of Jiaxing Science and Technology Program Project, grant number: 2021AD30088.
The authors declare no conflict of interest.
During the preparation of this work the authors used ChatGpt-3.5 in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG39131.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.