
 , Qihan Wu 1,*
, Qihan Wu 1,*1 Shanghai-MOST Key Laboratory of Health and Disease Genomics, NHC Key Lab of Reproduction Regulation, Shanghai Institute for Biomedical and Pharmaceutical Technologies, 200237 Shanghai, China
2 Department of Pathology, Xiangya Hospital, School of Basic Medical Sciences, Central South University, 410013 Changsha, Hunan, China
†These authors contributed equally.
Abstract
Background: Ovarian cancer has a poor prognosis, and DNA damage-response (DDR) genes are associated with both its occurrence and prognosis. However, previous studies have mostly focused on genetic mutations, with no clear conclusions on epigenetic factors such as DNA methylation. Methods: In this study, we comprehensively investigated the relationship between promoter methylation of DDR genes and ovarian cancer prognosis. We performed combined multidata analysis of the promoter methylation, expression, homologous recombination defieiency (HRD) score, and drug sensitivity of 377 DDR genes in ovarian cancer by utilizing The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) datasets. We then validated abnormal promoter methylation and its relationship with overall survival in clinical samples. Results: Our analysis identified 52 methylation-driven DDR genes that exhibited abnormal expression due to abnormal promoter methylation. These genes are mostly related to BRCA1-related DNA damage repair and cell cycle regulatory pathways. Further studies revealed six of these genes, BRCA1, PTTG1, TTK, AURKA, CDC6, and E2F1, to be significantly associated with HRD scores. Among them, E2F1, PTTG1, and CDC6 are associated with drug sensitivity. Finally, we verified in 81 ovarian cancer samples that methylation of the promoter of these three genes was significantly associated with patient survival. Conclusions: Our study identified a large number of methylation-driven aberrantly expressed DDR genes in ovarian cancer, some of which affect disease prognosis. Levels of methylation of these gene promoters may serve as potential prognostic markers.
Keywords
- ovarian cancer
- DNA damage response
- DNA methylation
- prognosis
- promoter
Ovarian cancer (OC) is an aggressive epithelial malignancy and the most lethal gynaecologic cancer, with a 5-year survival rate of only 49.7% [1]. Type I epithelial ovarian cancers (EOCs) are considered relatively stable tumors that usually arise from identifiable precursor lesions such as endometriosis or borderline tumors of low malignant potential. In contrast, type II EOCs are considered from the outset to be biologically aggressive tumors with a propensity to metastasize from small primary lesions. High-grade plasmacytoid ovarian cancer is the most common type of EOC, accounting for approximately 75% of EOCs, and develop according to the type II pathway and present p53 and BRCA mutations [2]. Ovarian cancer is usually diagnosed at a late stage after it has spread beyond the pelvis due to the lack of effective screening tools and specific early diagnostic biomarkers [3, 4]. The cost-effectiveness of early detection and prevention of ovarian cancer over the past decade is also reflected in the economy, with the cost of treatment per ovarian cancer patient remaining the highest of all cancer types. For example, the average initial cost in the first year is approximately USD 80,000, while the cost in the final year may increase to USD 100,000 [5]. Moreover, ovarian cancer has a poor prognosis, mainly involving a high recurrence rate and easy drug resistance, with three-quarters of patients at advanced stages relapsing and eventually dying from drug-resistant disease [6]. Therefore, exploring biomarkers for early diagnosis and drug-resistant therapeutic targets is a major way to improve cancer prognosis. Genomic analyses fail to capture the complexity of the intrinsic effects of the tumor ecosystem, with proteins being the primary effector molecules of cellular function. Proteomic studies utilizing reversed-phase protein arrays can identify proteins associated with prognosis, chemotherapy tolerance, and progression in ovarian cancer patients. The use of mass spectrometry allows subclassification of ovarian cancer and characterization of homologous recombination defieiency (HRD). Proteomic analysis of ovarian cancer can reveal how ovarian cancer cells and tumor ecosystems adapt to treatment and potentially improve patient prognosis by developing and implementing effective drug combinations [7].
Genomic instability is a hallmark of cancer. Eukaryotic cells have formed an effective mechanism called the DNA damage response (DDR) during evolution that is invoked to repair DNA damage and accurately replicate and inherit genetic material [8]. DDR is a kinase-based signalling cascade [9] that refers to a complex network composed of multiple signalling pathways and proteins involved in cell cycle control, apoptosis, and DNA repair. Through these pathways, the genome maintains its integrity even when cells are exposed to various exogenous or endogenous stresses [10, 11]. The combination of high genome replication rates and defective DNA repair mechanisms in cancer cells leads to genomic instability, especially in ovarian cancer [6]. The phosphatidylinositide 3-kinases (PI3K) pathway, which is frequently upregulated in EOC, is implicated in multiple processes in DNA replication and cell cycle regulation, and plays an important role in chemoresistance and preservation of genomic stability. Inhibition of PI3K may decrease the activity of the spindle assembly checkpoint protein Aurora kinase B, leading to genomic instability and catastrophic mitotic consequences, which in turn increase the incidence of lagging chromosomes in prophase [12]. Multiple genes of the DDR, such as BRCA1 and BRCA2, have been found to be related to genome instability and susceptibility to ovarian cancer [13]. Dysregulation of DDR may also lead to increased sensitivity or drug resistance in ovarian cancer, and this knowledge can be used to improve cancer treatment. At present, treatment of ovarian cancer mainly includes surgery, chemotherapy and targeted therapy; the two modes of action of the latter two approaches mainly damage DNA. Abnormalities in DDR function are important determinants of ovarian cancer risk, progression, and treatment response [14]. Genomic instability is associated with homologous recombination (HR) defects and prognosis. DNA double-strand breaks (DSBs) as one of the most harmful forms of DNA damage, posing a threat to genomic stability and integrity. To maintain genomic stability and integrity, cells have evolved sophisticated DNA repair mechanisms. There have been six primary pathways of DDR identified, which are variably used to address DSB and signgle-strand DNA breaks (SSB) damage from a variety of mechanisms of injury. Non-homologous end joining (NHEJ) and HR are two major pathways involved in repairing DSBs. HR pathways become active in the S/G2 phase due to the availability of a sister chromatid, whereas NHEJ repairs DSBs throughout all cell cycle [15]. HRD is common in many cancers and compromises DNA replication stress and genomic stability [16]. The HRD score is significantly associated with progression-free survival (PFS) in ovarian cancer and is a key determinant of response to platinum-based chemotherapy (causing DNA damage) and poly adenosine diphosphate ribose polymerase (PARP) inhibitors (inhibiting DNA damage repair) [17]. In ovarian cancer, mutations in BRCA1/2 are known to be the most important factor for high HRD scores, but even in the absence of BRCA1/2 mutations, tumours with high HRD scores still show strong sensitivity to platinum and PARP inhibitors [18]. In addition to BRCA1/2, the relationship between other DDR pathway genes and HRD needs to be further investigated.
Epigenetic silencing phenocopies DNA events [19]. DNA methylation is a covalent modification of cytosines; it is the most studied epigenetic mechanism and is associated with regulation of mRNA and miRNA expression [18]. In general, promoter methylation is thought to alter chromatin conformation by recruiting methyl-binding domain proteins (MBDs), which prevent transcription factor binding and reduce gene expression [19]. It has been suggested that DNA methylation is a more reliable marker than gene expression [20] and that the combination of both is more beneficial for explaining cancer onset and progression. In recent years, an increasing number of studies have shown that abnormal gene methylation in a promoter region is associated with chemotherapy and targeted therapy in ovarian cancer [21]. It has been found that reduced BRCA1 expression due to BRCA1 promoter hypermethylation can lead to radiosensitization of ovarian cancer [13], and patients with BRCA1 hypermethylation have similar progression-free survival compared to patients with BRCA1 mutations. Some DNA damage response genes, such as BRCA1, FANCF, GST01, MGMT, and MLH1, have been found to have abnormal promoter methylation in ovarian cancer [22], but the promoter methylation of other DDR genes affecting their expression and their clinical significance have not been analysed relatively completely.
In this study, we conducted comprehensive multidata analysis of promoter methylation, gene expression, HRD score, and drug sensitivity of 377 genes involved in DDR using The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) datasets. We identified methylation-driven DDR genes in ovarian cancer, some of which correlated significantly with HRD and sensitivity to cisplatin/paclitaxel. Furthermore, we validated the prognostic significance of promoter methylation of three genes, PTTG1, CDC6, and E2F1, in 81 ovarian cancer clinical formalin-fixed paraffin-embedded (FFPE) samples. Our findings suggest that these genes may serve as new prognostic markers in ovarian cancer patients.
A list of 377 genes involved in DNA damage and DNA damage response pathways is available at PathCards (https://pathcards.genecards.org/). The details of all genes are summarized in Supplementary Table 1.
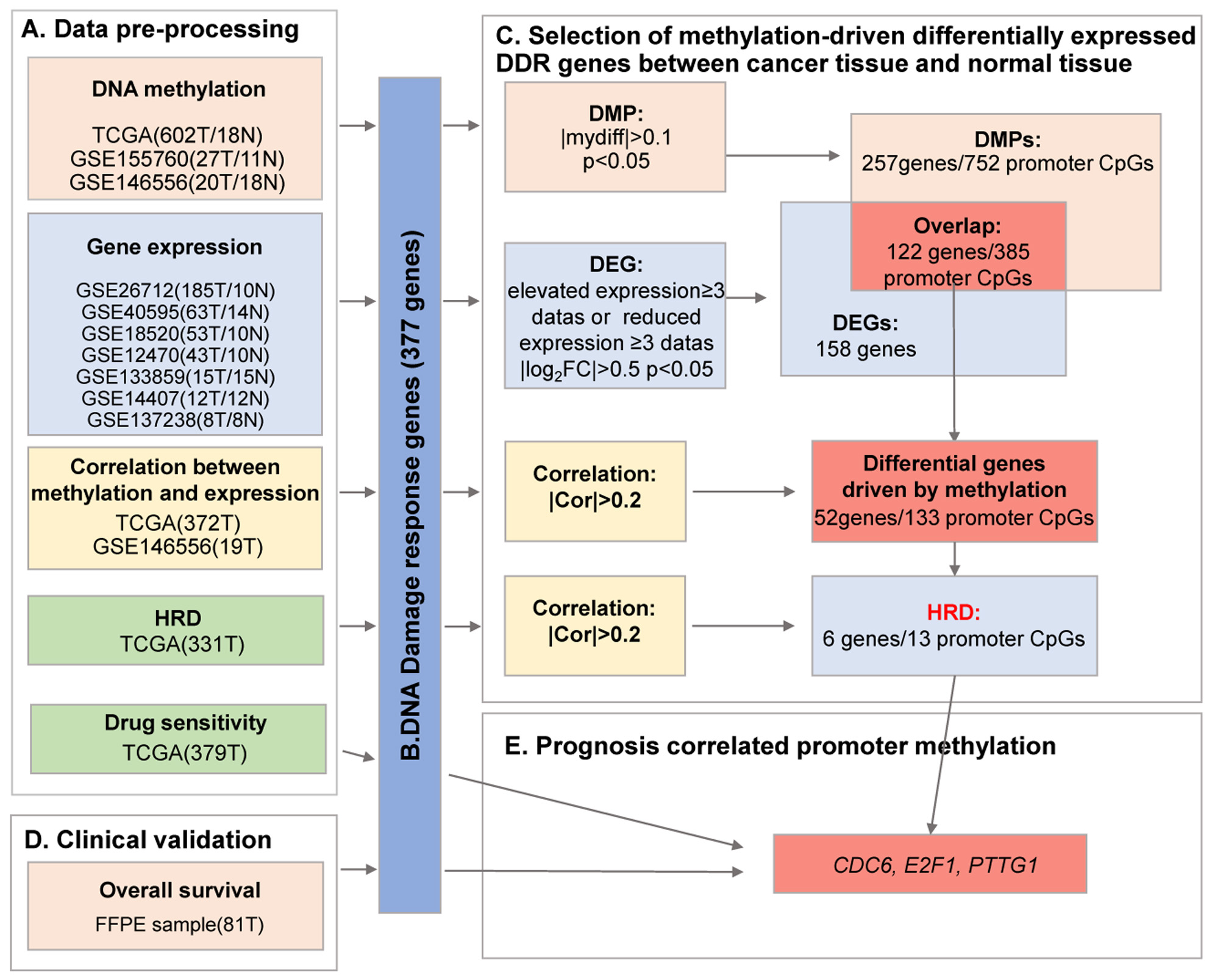
Bioinformatics analysis of DNA methylation and gene expression data of ovarian cancer tumour samples was performed using publicly available databases, including TCGA and GEO. The workflow involved in the analysis is shown in Fig. 1.
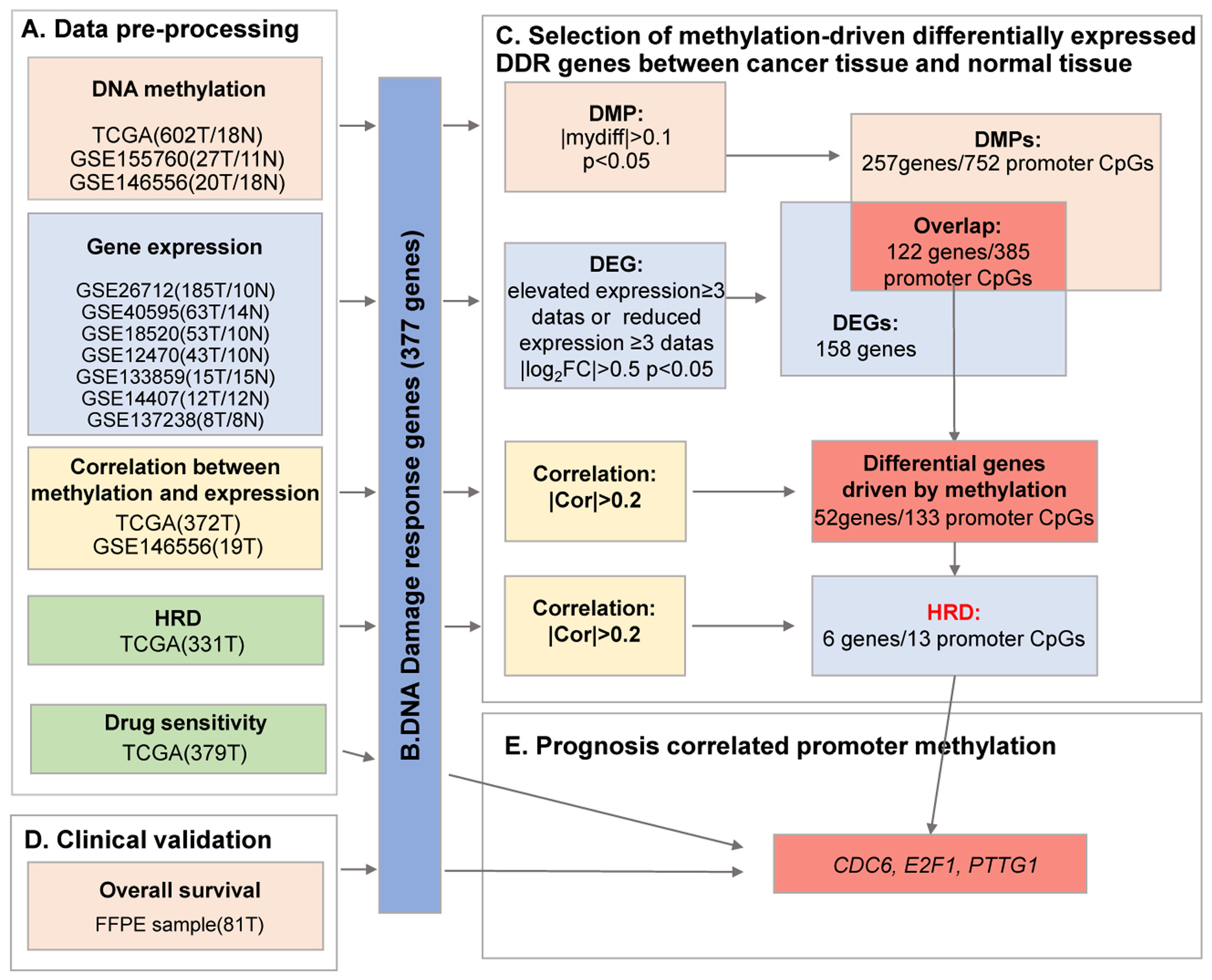 Fig. 1.
Fig. 1.A flow chart of exploration of DNA damage-response (DDR)
methylation-driven genes in ovarian cancer. Ovarian cancer data from The Cancer
Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases (A) were
preprocessed to extract information on 377 DNA damage repair genes (B) for
methylation-driven differential expression analysis between cancer tissues and
normal tissues (C), which were combined with clinical samples (D) to screen for
promoter methylation associated with prognosis (E). T, tumour tissue; N, normal
control tissue; HRD, homologous recombination deficiency; DMP, differentially
methylated promoter; DEG, differentially expressed gene; FFPE, formalin-fixed paraffin-embedded;
DNA methylation data were downloaded from TCGA (602 cancer samples/18 normal
samples) and GEO (https://www.ncbi.nlm.nih.gov/geo/, GSE155760 27 cancer
samples/11 normal samples and GSE146556 20 cancer samples/18 normal samples).
Methylation sites from 2500 bp upstream to 2000 bp downstream of the gene
transcription start site were analysed to identify the differential level of
methylation in gene promoters [23]. The ChAMP R package (version 1.10.0,
https://bioconductor.org/packages/ChAMP/) [24] was used to analyse the
differential level of DDR gene promoter methylation between normal and ovarian
cancer samples. The sites that met the absolute value of methylation difference
Seven gene expression datasets, GSE26712 (185 cancer samples/10 normal samples),
GSE40595 (63 cancer samples/14 normal samples), GSE18520 (53 cancer samples/10
normal samples), GSE12470 (43 cancer samples/10 normal samples), GSE133859 (15
cancer samples/15 normal samples), GSE14407 (12 cancer samples/12 normal
samples), and GSE137238 (8 cancer samples/8 normal samples), were used in gene
expression analysis. “Affy” (version 1.50.0,
https://bioconductor.org/packages/affy) and “limma” (version 3.28.14,
https://bioconductor.org/packages/limma/) R packages were used to detect
differentially expressed genes (DEGs) between normal and ovarian cancer samples.
The parameters for DEGs were set as an absolute value of
The R package MethylMix was designed to identify gene expression that
correlated with methylation events. For the data from TCGA (372 samples) and
GSE146552 (19 samples), we used MethylMix [25] to perform integrated analysis of
DDR gene promoter methylation and expression, and an absolute value of
correlation
STRING (https://string-db.org/) was used to build a protein‒protein interaction
(PPI) network, with the minimum required interaction score set at 0.4. The
resulting data were imported into Cytoscape (version 3.7.2, Institute of Systems
Biology (ISB), Seattle, WA, USA) for visualization; subsequently, the app MCODE
(Molecular Complex Detection) was applied to perform module analysis (node score
cut off
The 331 ovarian cancer samples with available HRD and gene expression data in TCGA were used to identify DDR pathway genes associated with HRD scores. The R package scarHRD, which is based on next-generation sequencing data, was employed to calculate three HRD metrics: telomere allelic imbalance (TAI), loss of heterozygosity (LOH), and large-scale state transitions (LST). Subsequently, the limma R package was utilized to correlate HRD scores with gene expression data using Spearman’s correlation coefficient. Allele-specific copy numbers of TCGA-OV (ovarian cancer in TCGA) were obtained from a whole-exome sequencing (WES) database using Sequenza. Paired tumour and normal sample bam files were downloaded from the GDC data portal. The allele-specific copy number was then calculated using scarHRD, except for the HRD scores TAI, LST, and LOH. Finally, HRDsum was calculated as the sum of TAI, LST, and LOH scores.
We calculated the half inhibitory concentration (IC50) of cisplatin and paclitaxel using the “pRRophetic” [26] package and 379 tumour samples from TCGA. The IC50 values were Z-score normalized. Expression of DDR-related genes was then correlated with IC50 using Pearson correlation analysis in R software.
In this study, we collected a set of 81 ovarian cancer FFPE samples for
validation analysis; the patients had a mean age at diagnosis of 50.6
DNA for FFPE samples was extracted using GeneRead DNA FFPE Kit (QIAGEN, Hilden,
Nordrhein-Westfalen, Germany), and methylation levels of candidate genes were
analysed by sulfite amplification and next-generation sequencing (NGS). Briefly,
10 ng genomic DNA extracted from FFPE material was bisulfite transformed with EZ
DNA Methylation-Lightning Kit (Zymo Research, Irvine, CA, USA). The
bisulfite-transformed DNA was used to amplify the target fragment with TaKaRa EX
Taq Hot Start Version Kit (TaKaRa, Kyoto, Japan). The primers used for
amplification were as follows: BRCA1 forward: 5
The library was 2
The survival rates of PTTG1, E2F1, and CDC6
methylation were estimated using the Kaplan‒Meier (K-M) method by the survminer
(version 0.4.9, https://cran.r-project.org/) R package. This package applies the
log-rank test for survival time comparisons. A value of p
To investigate epigenetic aberrations of DDR genes and their impact on ovarian cancer development, we analysed promoter methylation and gene expression of 377 DDR genes using data from both the GEO and TCGA databases. The promoter region was defined as spanning from 2500 bp upstream to 2000 bp downstream of the transcriptional start site (TSS).
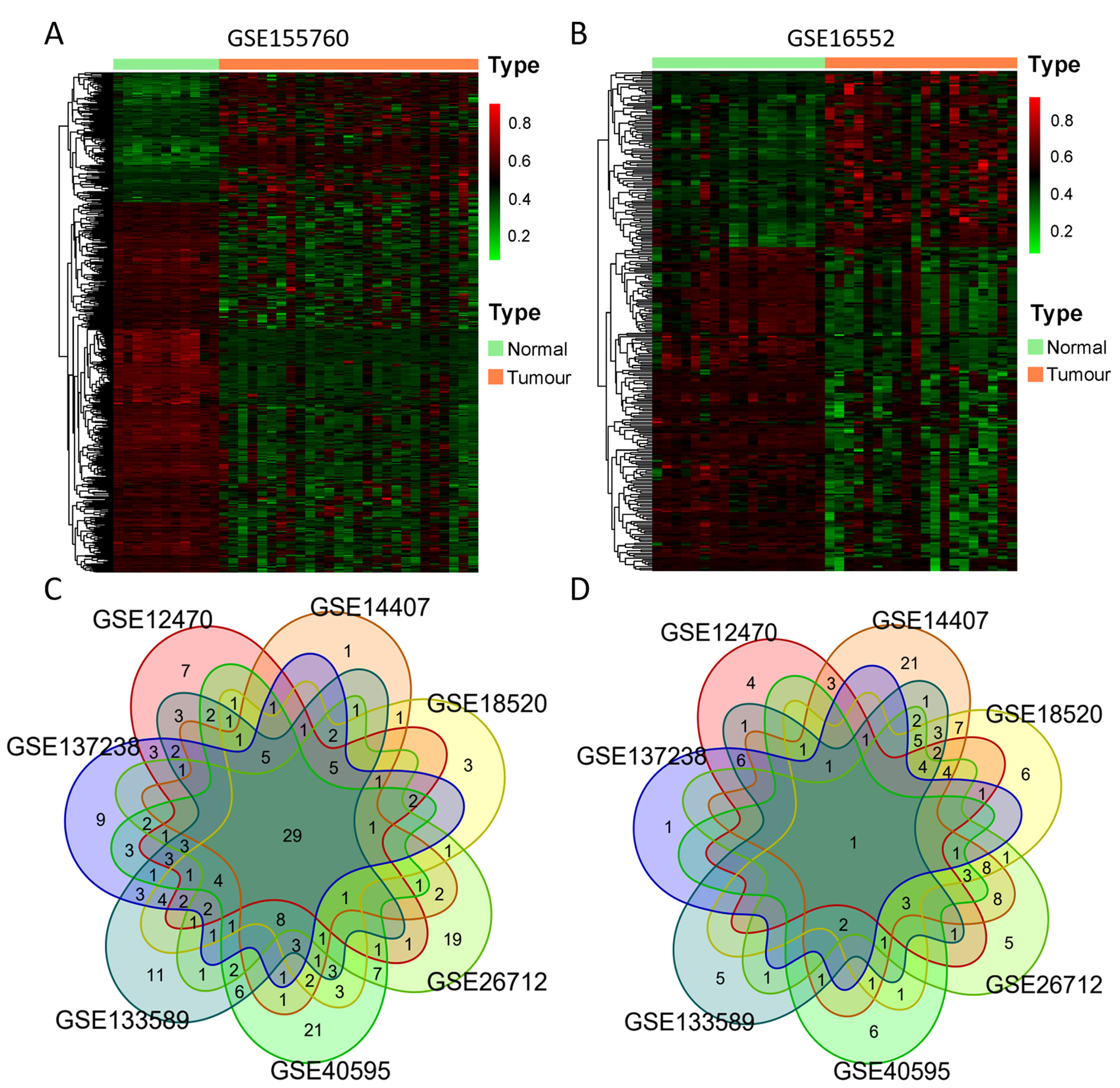
We chose three DNA methylation datasets and seven gene expression datasets that contained both ovarian cancer and normal tissues. Using a standardized approach described in the Materials and Methods section, we identified 249 DNA damage response (DDR) gene promoter methylation abnormalities in ovarian cancer within the GSE155760 dataset (Infinium MethylationEPIC 850K), with 174 CpG sites being hypermethylated and 496 CpG sites hypomethylated. The results of differentially methylated CpG sites are displayed in Fig. 2A. Within GSE146556 (Infinium MethylationEPIC 450K), we found 121 DDR genes (with 105 hypomethylated and 193 hypermethylated CpG sites), as shown in Fig. 2B. Another 33 DDR genes (with 6 hypomethylated and 35 hypermethylated CpG sites) were identified in TCGA (Infinium MethylationEPIC 27K) datasets. Combining the results of the three databases, we identified 257 genes and 752 CpG sites that were differentially methylated between tumour and normal tissues across the three datasets.
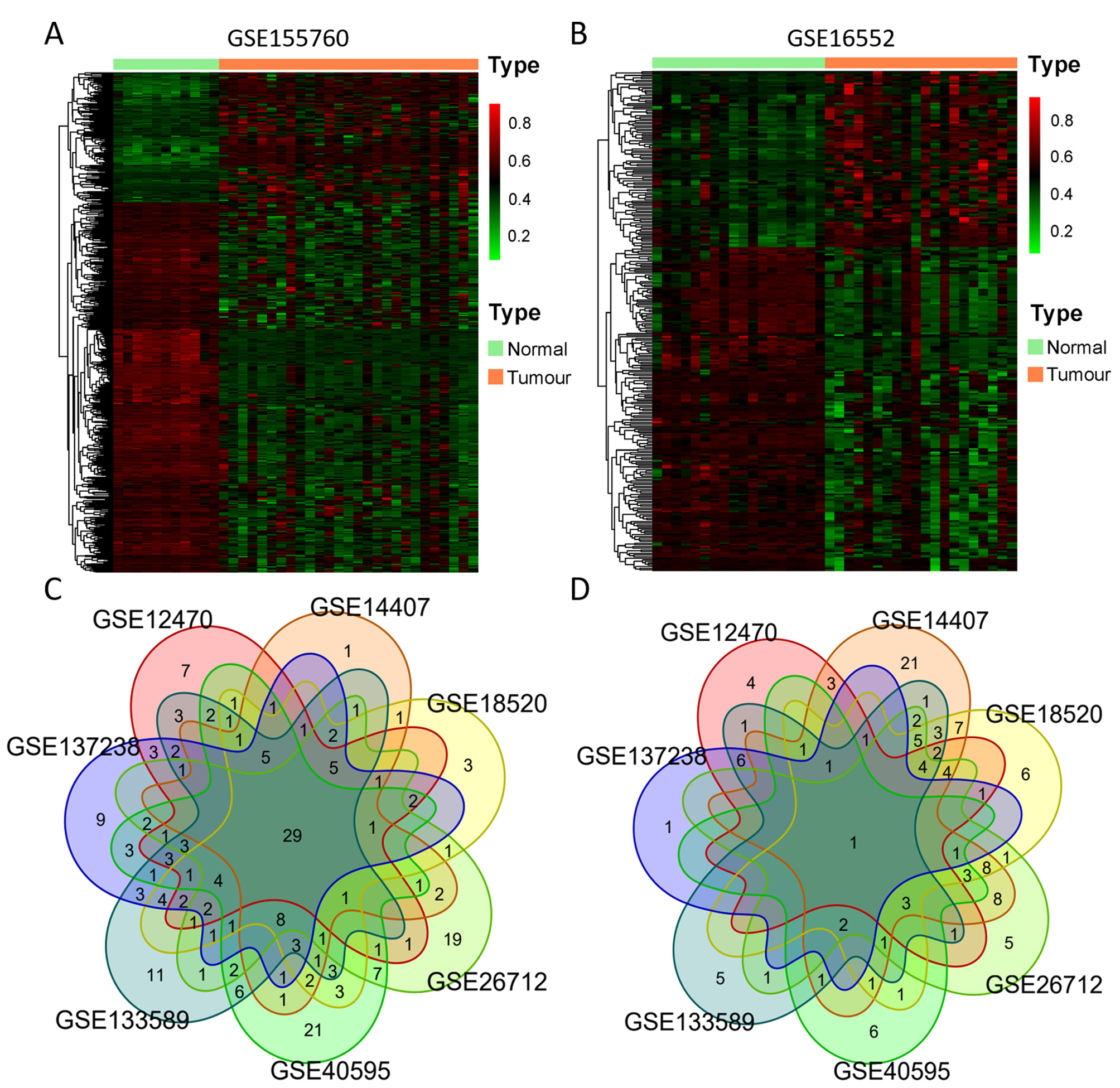 Fig. 2.
Fig. 2.Differential DNA methylation and expression of 377 DDR genes. (A,B) Heatmap of differentially methylated promoter (DMP) CpGs between normal and ovarian tumour tissues from GSE155760 (760 CpGs) and GSE146556 (298 CpGs). Red represents highly methylated CpG, and green represents CpG with low methylation. (C,D) Venn diagram of aberrantly upregulated expression (C) and downregulated expression (D) of DDR genes in 7 GEO datasets (GSE26712, GSE40595, GSE18520, GSE12470, GSE133859, GSE14407 and GSE137238).
In each of the seven gene expression datasets, we compared tumour tissues with normal control tissues and identified differentially expressed genes: 119 genes (86 upregulated and 33 downregulated, GSE12470), 154 genes (70 upregulated and 84 downregulated, GSE14407), 149 genes (92 upregulated and 57 downregulated, GSE18520), 150 genes (100 upregulated and 50 downregulated, GSE26712), 146 genes (124 upregulated and 22 downregulated, GSE40595), 158 genes (116 upregulated and 42 downregulated, GSE133589), and 119 genes (105 upregulated and 14 downregulated, GSE137238). We then obtained a list of upregulated or downregulated genes by identifying those that were found in more than three datasets, as indicated in the Venn diagrams in Fig. 2C,D. A total of 158 genes were selected, including 106 upregulated and 52 downregulated. By overlapping the results of the promoter methylation and expression analysis, we identified 122 DDR genes exhibiting aberrant promoter methylation (385CpGs) and expression in ovarian cancer.
We conducted correlation analyses of DDR gene promoter methylation and
expression in ovarian cancer tissue samples. Of the 122 genes with abnormal DNA
methylation and expression, we observed that 133 CpGs showed methylation levels
correlating with gene expression (
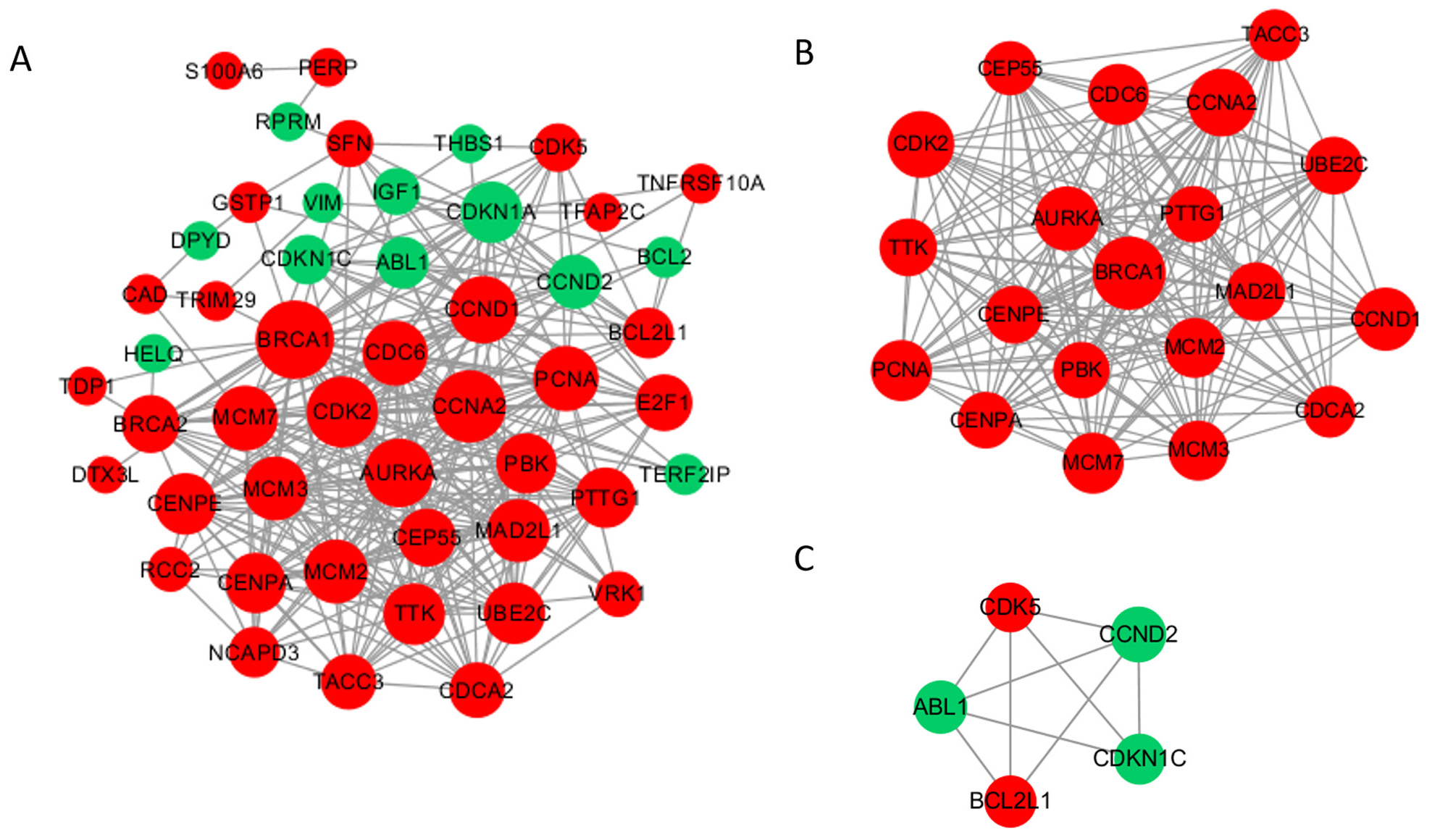
To investigate the function of methylation driver genes with aberrant expression in the damage repair response pathway, we conducted PPI analysis. The PPI network diagram is presented in Fig. 3A, consisting of 49 nodes and 331 edges. The 6 most highly connected pivotal genes identified were BRCA1, CCNA2, CDK2, AURKA, CCND1, and CDC6. We detected two significant modules using the MCODE application (Fig. 3B,C). The first module comprises 20 nodes with 175 edges, all of which are upregulated genes, mostly involved in DNA damage repair and mitosis-related processes, with BRCA1 as the core. The second module comprises 5 nodes and 9 edges, with 2 upregulated genes and 3 downregulated genes, mainly associated with the cell cycle and anti-apoptosis functions.
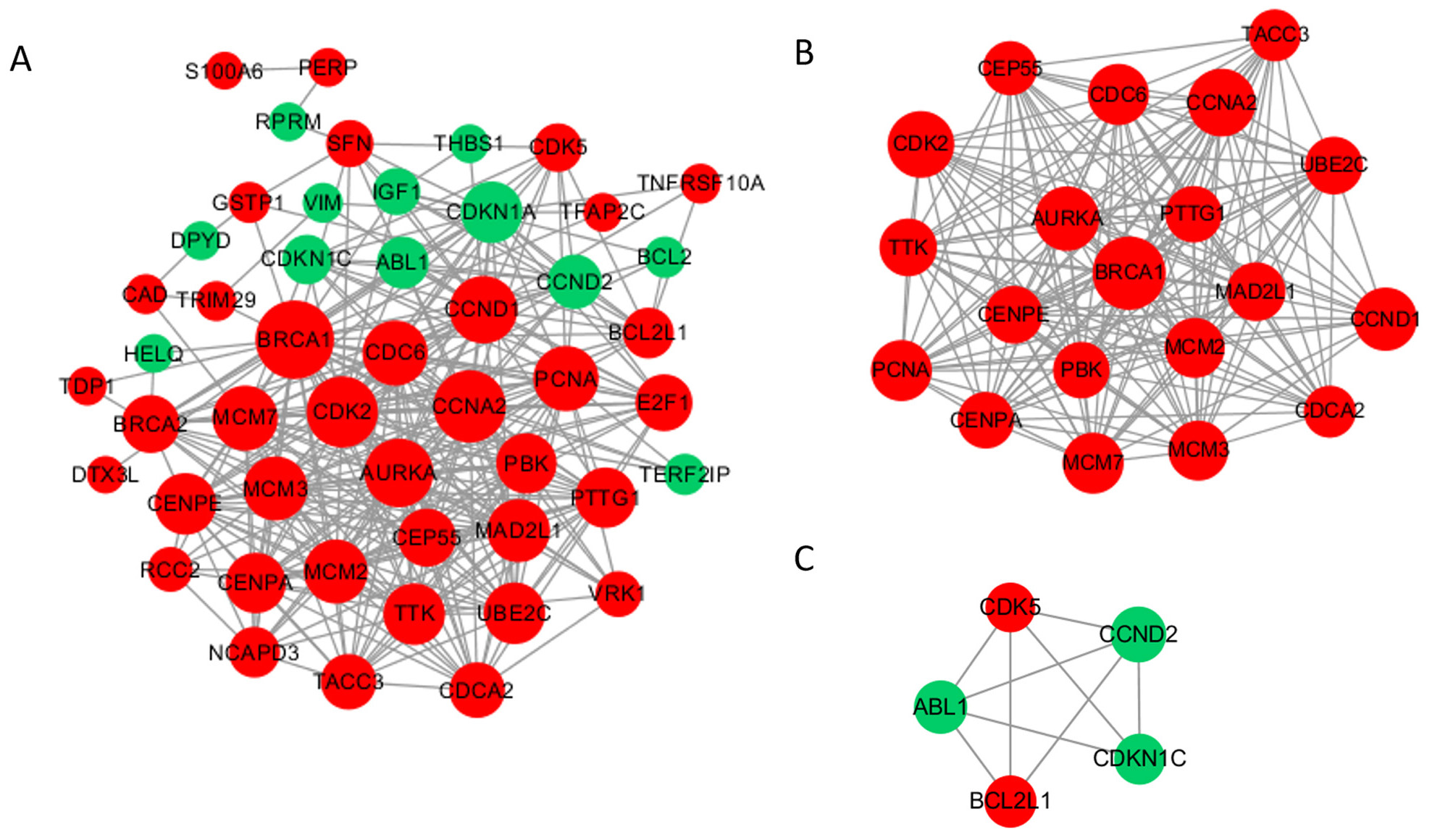 Fig. 3.
Fig. 3.Protein‒protein interaction (PPI) network and top 2 modules of the hypomethylated-upregulated (HOUP) genes. (A) PPI networks. Dark nodes represent hub genes. The size of the node indicates the connection degree value. Red represents upregulated genes, and green represents downregulated genes. (B,C) Module 1 consists of 20 nodes, and module 2 consists of 5 nodes.
To investigate whether the 52 methylation-driven DDR genes are associated with
ovarian cancer prognosis, we first analysed the relationship between gene
expression and homologous recombination defects (HRD). In ovarian cancer, HRD
scores have been found to be strongly associated with drug sensitivity and
survival. Through HRD analysis of 331 ovarian cancer samples in TCGA, we
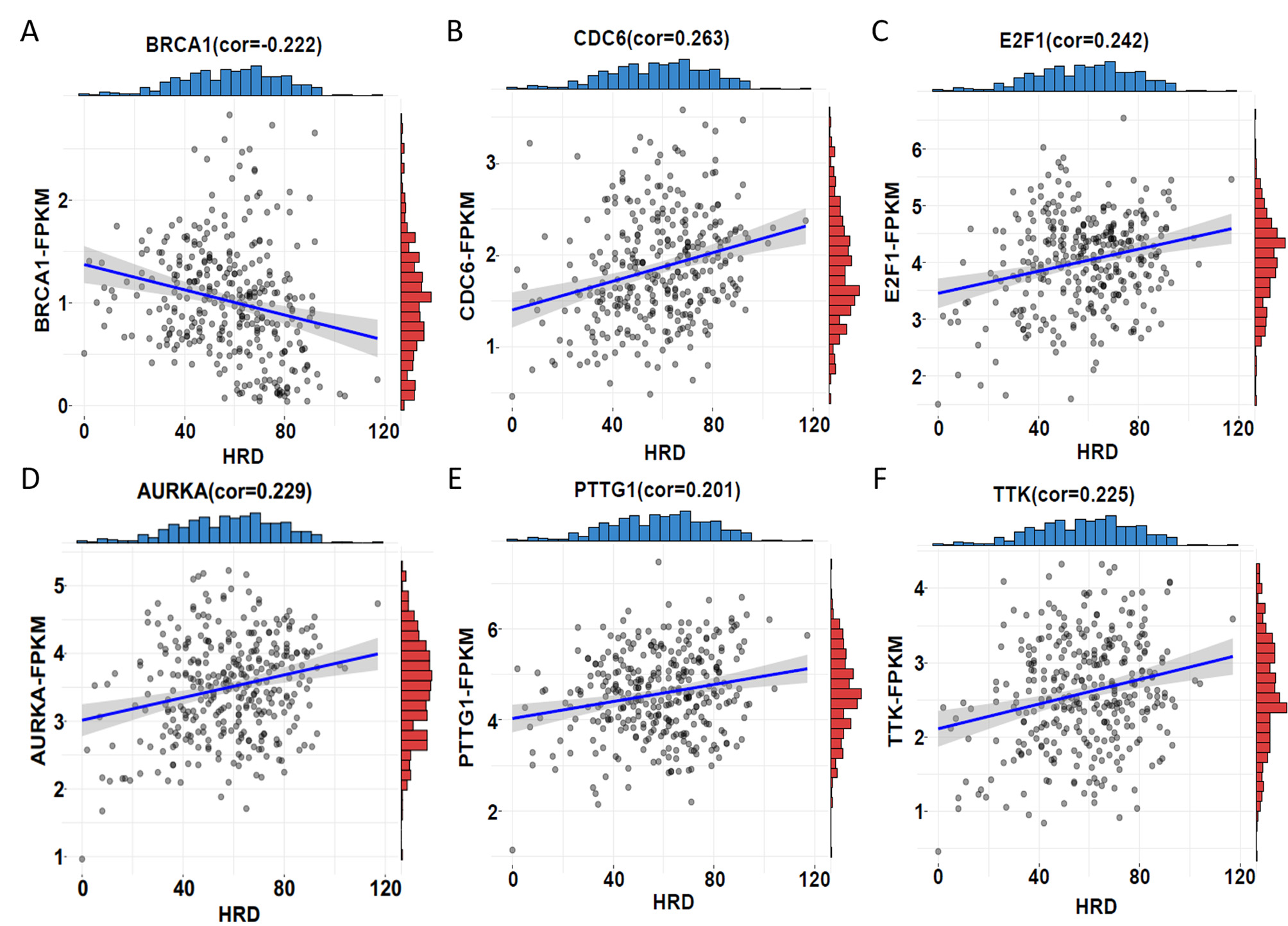
identified that expression of 6 genes, including AURKA, BRCA1,
CDC6, E2F1, PTTG1, and TTK, correlated with
the HRD score (
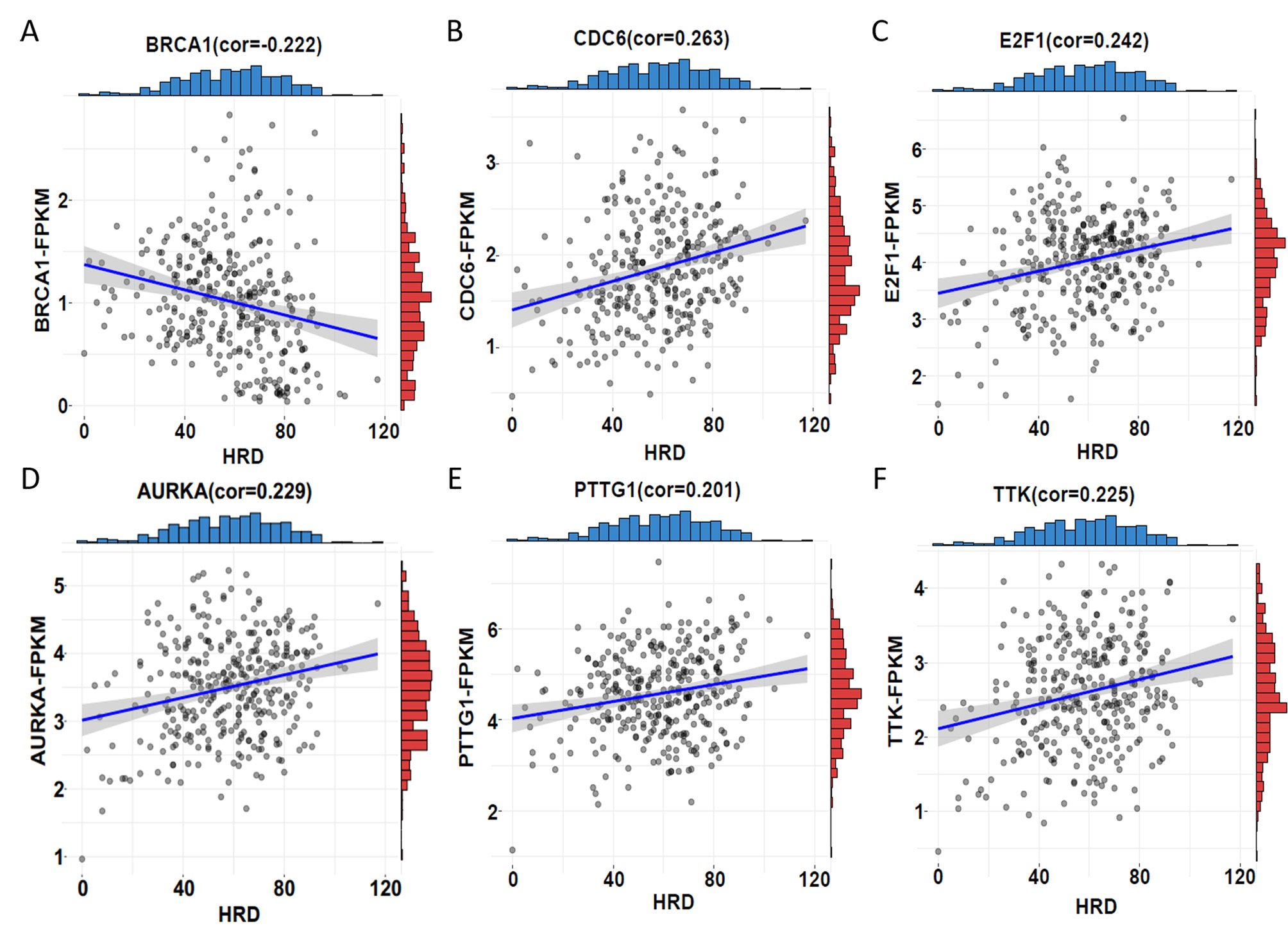 Fig. 4.
Fig. 4.Correlation between HRD score and expression. (A)
BRCA1 showed a significant negative correlation between HRD score and
expression (correlation
| CpG sites | Methylation | Gene | Expression | Meth-Exp cor |
HRD-Exp cor | ||||
| Normal | Tumour | Mydiff |
p value | logFC | p value | ||||
| cg05471521 | 0.303 | 0.136 | –0.167 | 2.92 |
AURKA | 2.625 | 2.79 |
–0.301 | 0.229 |
| cg19088651 | 0.092 | 0.214 | 0.122 | 1.90 |
BRCA1 | 1.163 | 6.20 |
–0.567 | –0.222 |
| cg16006004 | 0.646 | 0.378 | –0.268 | 1.07 |
BRCA1 | 1.163 | 6.20 |
–0.274 | –0.222 |
| cg27581762 | 0.692 | 0.510 | –0.182 | 9.78 |
BRCA1 | 1.163 | 6.20 |
–0.488 | –0.222 |
| cg14947218 | 0.615 | 0.388 | –0.226 | 1.76 |
BRCA1 | 1.163 | 6.20 |
–0.332 | –0.222 |
| cg24900425 | 0.890 | 0.526 | –0.364 | 1.92 |
BRCA1 | 1.163 | 6.20 |
–0.471 | –0.222 |
| cg25288140 | 0.844 | 0.551 | –0.293 | 7.48 |
BRCA1 | 1.163 | 6.20 |
–0.496 | –0.222 |
| cg19291926 | 0.215 | 0.333 | 0.118 | 6.19 |
CDC6 | 1.944 | 3.25 |
–0.324 | 0.263 |
| cg00509772 | 0.456 | 0.298 | –0.158 | 9.18 |
E2F1 | 3.146 | 5.97 |
–0.359 | 0.242 |
| cg26775866 | 0.325 | 0.070 | –0.254 | 2.20 |
PTTG1 | 3.378 | 1.03 |
–0.362 | 0.201 |
| cg17367077 | 0.911 | 0.561 | –0.350 | 1.43 |
PTTG1 | 3.378 | 1.03 |
–0.504 | 0.201 |
| cg24433016 | 0.672 | 0.377 | –0.295 | 1.15 |
TTK | 2.283 | 2.13 |
–0.285 | 0.225 |
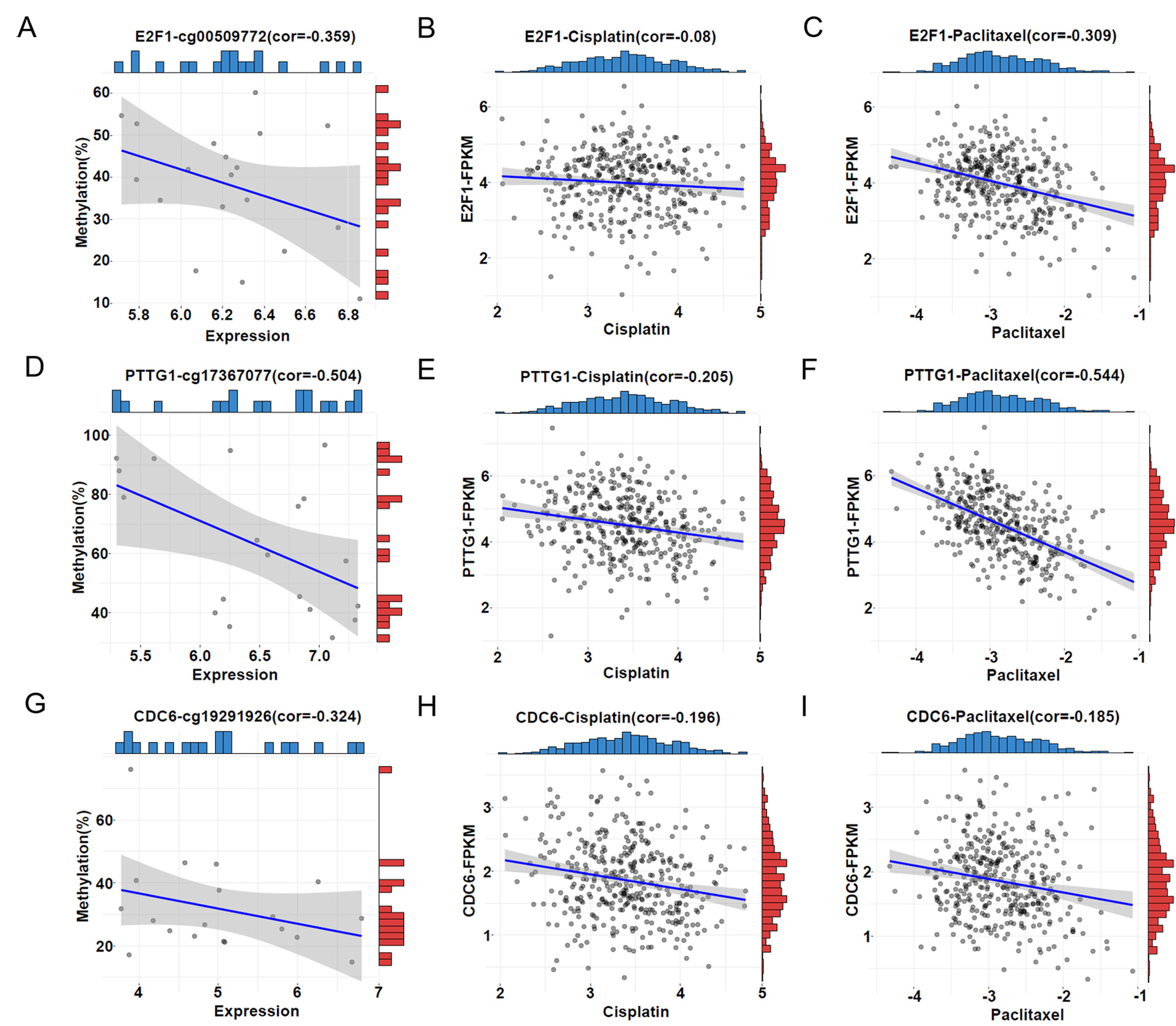
Moreover, we investigated whether the 6 HRD score-related genes are associated with drug sensitivity. Cisplatin and paclitaxel are the primary drugs used in first-line clinical treatment of ovarian cancer. Therefore, we first obtained the half-inhibitory concentrations (IC50) of cisplatin and paclitaxel for 379 TCGA tumour samples using the “pRRophetic” package and then correlated them with expression of the six genes. The results revealed a robust association between increased expression of PTTG1 and CDC6 and decreased IC50 for cisplatin (Fig. 5E,H), E2F1 expression did not significantly correlate with cisplatin IC50 values (Fig. 5B), indicating that increased expression of PTTG1 and CDC6 leads to increased sensitivity of patients to cisplatin. Similarly, elevated expression of E2F1, PTTG1, and CDC6 exhibited a robust correlation with increased sensitivity to paclitaxel (Fig. 5C,F,I). These findings are consistent with upregulation of gene expression leading to elevated HRD score. In addition, analyzing the GSE146556 dataset, we found that aberrant promoter methylation of E2F1, PTTG1 and CDC6 genes altered gene expression (Fig. 5A,D,G). Therefore, it can be hypothesized that aberrant promoter methylation of E2F1, PTTG1 and CDC6 genes altered gene expression and thus affected the sensitivity of patients to cisplatin and paclitaxel, which constituted a potentially effective prognostic indicator in ovarian cancer.
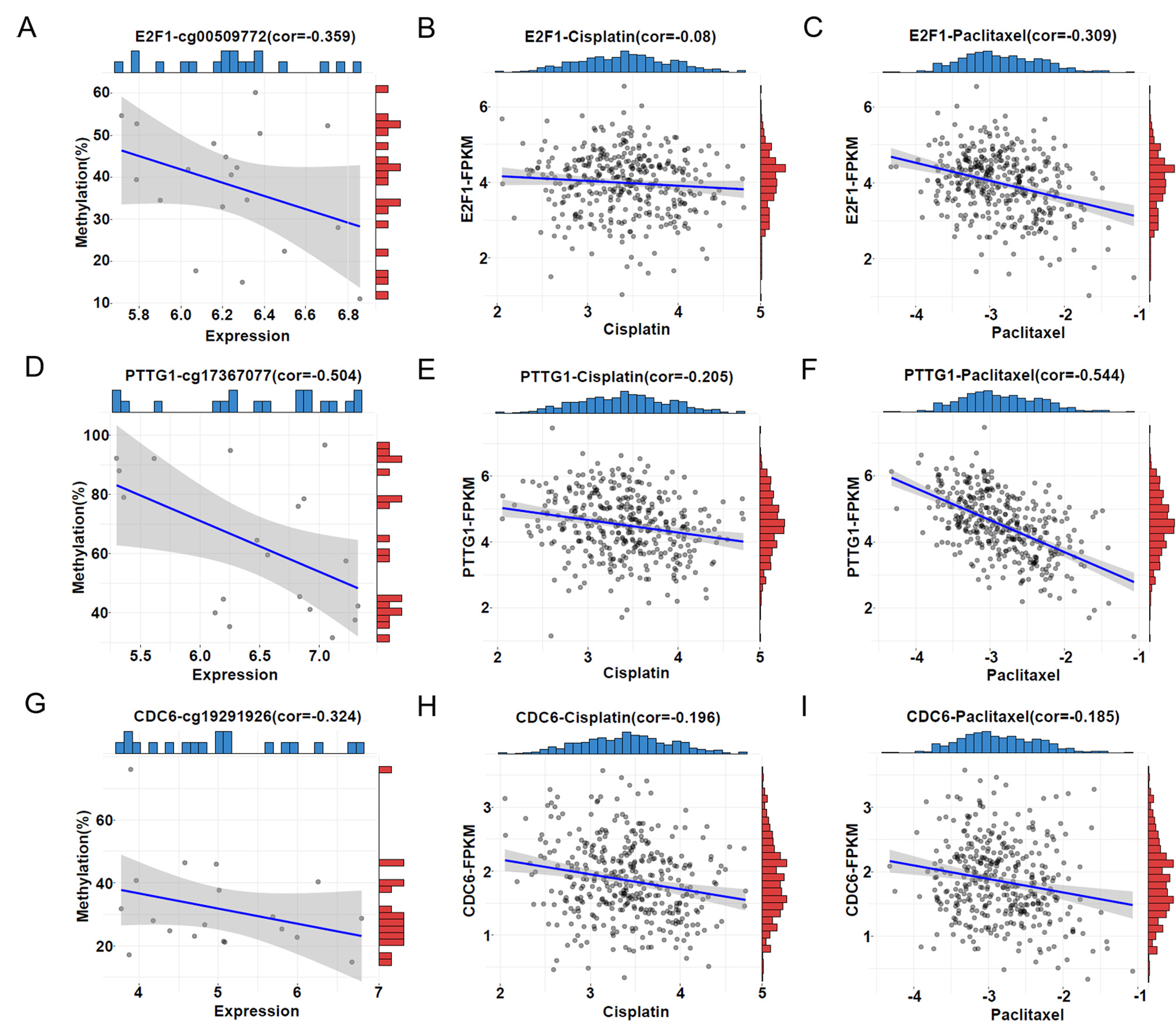 Fig. 5.
Fig. 5.Correlations between DDR gene methylation, expression, and the half-inhibitory concentrations (IC50). (A,D,G) Correlation between promoter methylation level and expression level of E2F1, PTTG1, and CDC6. The horizontal coordinate represents the FPKM value of the samples, and the vertical coordinate represents the methylation level of the promoters. The blue bar represents the sample distribution of expression and the red bar the sample distribution of promoter methylation. (B,E,H) Correlation between IC50 of cisplatin and expression of E2F1, PTTG1, and CDC6. The horizontal coordinate represents the IC50 of cisplatin; the vertical coordinate represents the expression level of the genes. The blue bar represents the sample distribution of the IC50 of cisplatin; the red bar represents the sample distribution of expression. (C,F,I) Correlation between the IC50 of paclitaxel and expression of E2F1, PTTG1, and CDC6. The horizontal coordinate represents the IC50 of paclitaxel; the vertical coordinate represents the expression level of the genes. The blue bar represents the sample distribution of the IC50 of paclitaxel; the red bar represents the sample distribution of expression.
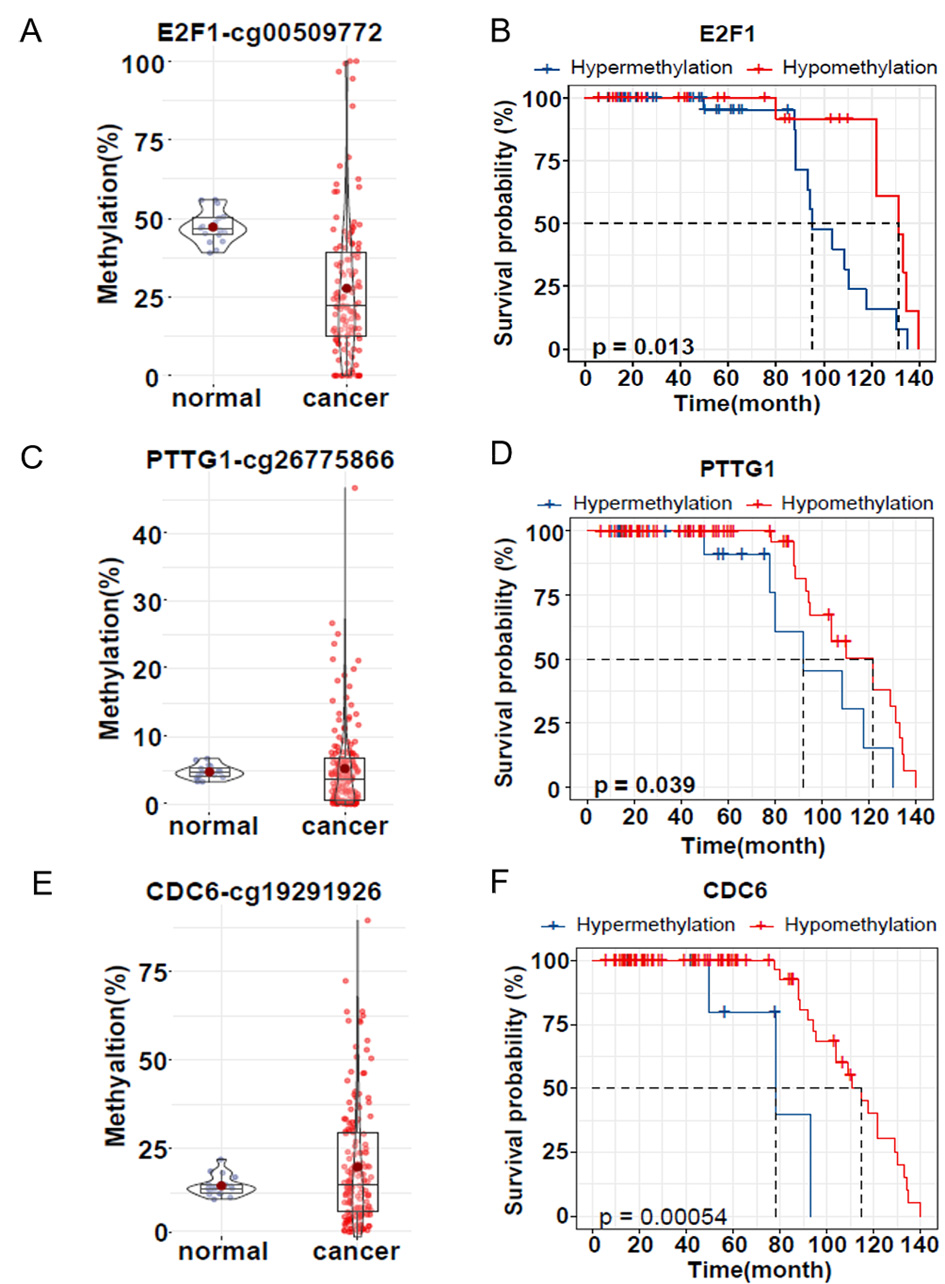
To assess whether promoter methylation of these three genes is associated with
prognosis, we collected 160 ovarian cancer samples (81 of these samples had
survival information) and 17 normal ovarian samples and used bisulfite amplicon
sequencing to detect the same CpG sites in the promoter as in the dataset. The
mean methylation of E2F1 promoter cg00509722 in ovarian cancer tissues
(27.74%) was 19.48% lower than in normal tissues (47.22%), p
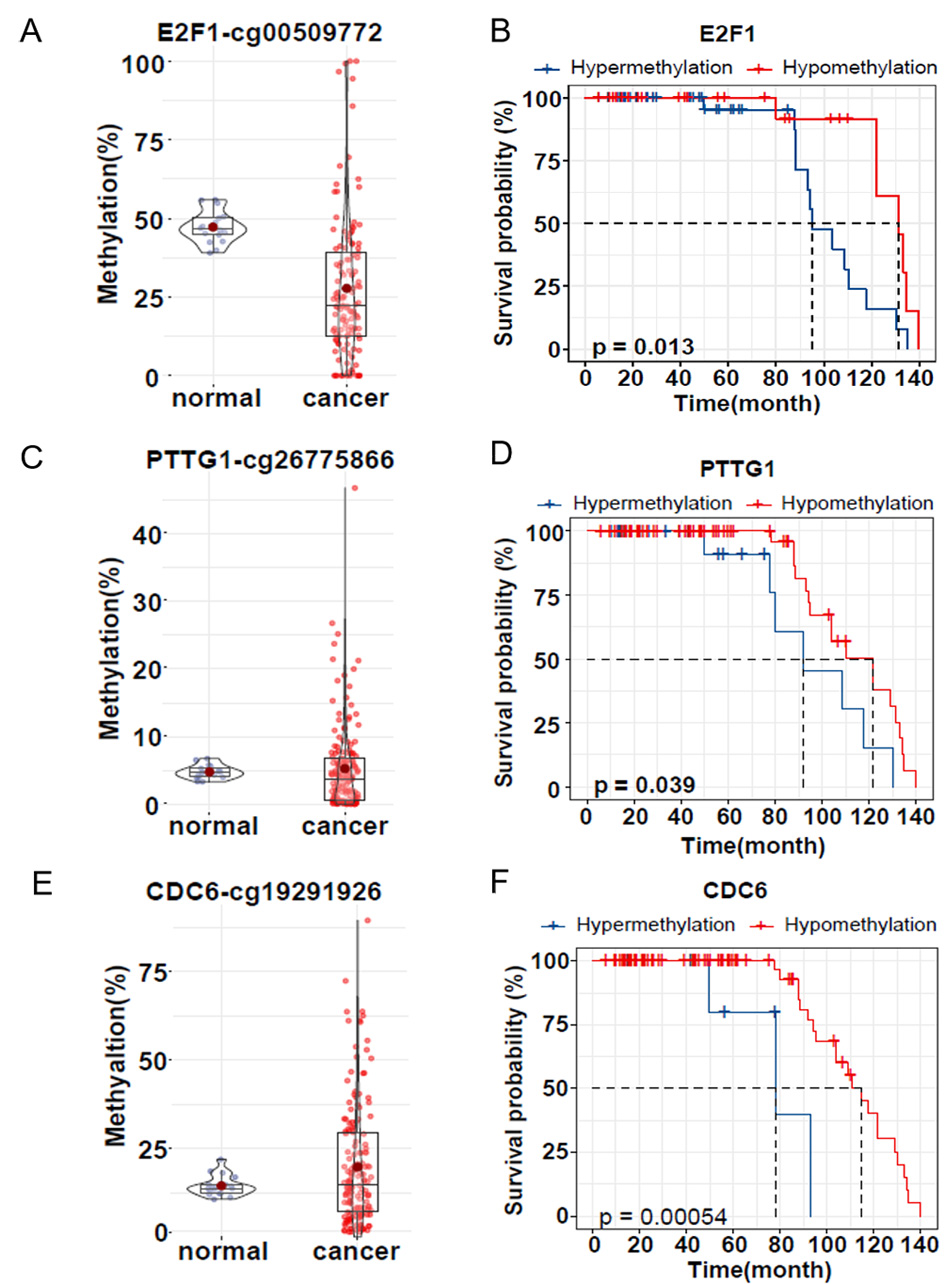 Fig. 6.
Fig. 6.DNA methylation level and survival analysis of clinical
samples. (A,C,E) Violin plot and box plot showing validation of E2F1,
PTTG1 and CDC6 promoter methylation in FFPE samples from normal
ovaries (purple, n = 17) and ovarian cancer patients (red, n = 160). (B,D,F)
Kaplan‒Meier survival analysis of hypermethylation (blue) and hypomethylation
(red) of the three genes E2F1, PTTG1 and CDC6 in
clinical samples. Survival rates are shown in months after surgery. p
The occurrence and prognosis of ovarian cancer are intricately linked to DDR genes [13]. However, prior studies have primarily focused on genetic mutations, with limited conclusive evidence on epigenetic factors such as DNA methylation. In this study, we identified 52 methylation-driven DDR genes using ovarian cancer data from publicly available databases. These genes, which are primarily associated with BRCA1-related DNA damage repair and cell cycle regulatory pathways, displayed abnormal expression due to abnormal promoter methylation. Subsequent analysis revealed that six of these genes have significant associations with HRD, indicating their potential relevance to ovarian cancer prognosis. Additionally, our investigation demonstrated that three genes, E2F1, PTTG1, and CDC6, are linked to drug sensitivity. Finally, we confirmed the association between promoter methylation of these three genes and survival in clinical ovarian cancer samples.
Cancer cells are characterized by the genome-wide hypomethylation, as well as hypermethylation at CpG islands of certain promoters, which can reversibly or irreversibly alter gene function, thereby contributing to cancer progression [27]. Promoter hypermethylation is a crucial epigenetic alteration that leads to gene inactivation and can have similar effects to inactivating mutations [28]. In ovarian cancer, BRCA1 is the most extensively studied gene and plays a crucial role in the DDR signalling pathway [29]. In recent years, poly adenosine diphosphate ribose polymerase inhibitors (PARPis) have emerged as a promising option for treating cases with BRCA mutations. However, the resistance observed in BRCA1/2 mutated tumours to PARPis suggests that this therapy may not be as effective as initially anticipated [30]. Combining PARPis with immunotherapies, such as anti-CTLA-4 and PD-1/PD-L1, could potentially be a more suitable therapeutic strategy. There is evidence suggesting that BRCA deficiency may induce a STING-dependent innate immune response by promoting the production of type I interferon and pro-inflammatory cytokines [31, 32]. In addition to BRCA1 mutation, the promoter hypermethylation of BRCA1 in OC displays similar clinicopathological features [33]. This finding suggests that epigenetic markers, including DNA methylation, have the potential to be used as prognostic markers for treatment selection. However, there is a lack of consensus on this issue due to the limited number of studies and varying research techniques. Therefore, our study aims to provide evidence of the association between DNA methylation of DDR genes and prognosis in ovarian cancer patients.
Our study involved methylation analysis of 377 genes in the DDR pathway using a
total of 696 samples from databases. Surprisingly, we found abnormal methylation
at the 752 CpG sites in the promoter regions of 257 genes in ovarian cancer.
These findings suggest that DNA methylation-dependent epigenetic alterations of
DDR genes in ovarian cancer are more common than previously thought. We further
analysed 7 GEO ovarian cancer expression datasets and identified 158 genes that
were consistently underexpressed or overexpressed in more than 3 datasets. By
overlapping our methylation and expression results, we identified 122 DDR genes
(385CpGs) with aberrant promoter methylation and expression in ovarian cancer
tumours. We defined genes with a correlation between promoter methylation and
gene expression (
In recent years, research on HRD, a crucial feature of ovarian cancer, has increased significantly [36]. HRD is most commonly observed in ovarian cancer, and higher HRD scores are linked to better clinical outcomes and overall survival rates [37]. Several DDR genes, including BRCA1, BRCA2, and RAD51C, have been linked to HRD when mutated [38]. However, the underlying cause of approximately 50% of HRD cases is still unknown, and epigenetic abnormalities may play a significant role [39]. Our study identified six DDR genes with epigenetic changes that are linked to HRD: BRCA1, AURKA, CDC6, E2F1, PTTG1, and TTK. Interestingly, except for BRCA1, the other five genes were all hypermethylated in the promoter region, exhibited increased expression and were associated with high HRD scores. This indicates that the mechanism of function of these five genes may differ from that of BRCA1, which requires further investigation. Furthermore, our findings showed that E2F1, PTTG1, and CDC6 are associated with sensitivity to cisplatin or paclitaxel and overall survival in clinical samples. These three genes are potential prognostic markers and the first to be linked to ovarian cancer prognosis.
Although these three genes show potential as prognostic markers, there is currently a lack of clear evidence linking their methylation status with prognosis in ovarian cancer. PTTG1 is an oncogene that is highly expressed in various tumours, including ovarian cancer [40]. It plays a significant role in chemosensitivity, chromosome stability, and DNA repair [41]. Our study discovered promoter hypomethylation of PTTG1 to be associated with better prognosis, increased HRD score, platinum and paclitaxel sensitivity, and longer survival. These results are in contrast to previous studies involving some other tumours [40, 42, 43]. For instance, PTTG1 overexpression has been linked to lower overall survival in laryngeal cancer [40], and PTTG1 levels correlate negatively with survival in hepatocellular carcinoma (HCC) [42]. Deletion of PTTG1 in cells increases their sensitivity to doxorubicin [43]. These inconsistencies may arise due to PTTG1’s multiple functions in the DNA damage response. PTTG1 interacts with the protein Ku-70, the regulatory subunit of DNA-dependent protein kinase (DNA-PK), and Ku-70 releases PTTG1 when DNA damage occurs. Subsequently, DNA-PK phosphorylates PTTG1, blocking cell cycle progression to maintain chromosomal stability [44]. PTTG1 also interacts with p53, blocking its specific binding to DNA and inhibiting its transcriptional activity [45], thus suppressing apoptosis. However, overexpression of PTTG1 stimulates Bax, a downstream target of p53 [46], and activates apoptosis. Therefore, in ovarian cancer cells with high PTTG1 expression levels, PTTG1 inhibits the cell cycle when drug-induced DNA damage occurs. Although regulation of apoptosis by PTTG1 is possible in both directions, expression of PTTG1 may lead to apoptosis when a certain threshold is reached [45, 46, 47]. PTTG1 may also regulate selection of HR or NHEJ after DNA double-strand breaks through regulation of p53, but the mechanism is unclear. Our study also found that PTTG1 is a methylation-driven gene in ovarian cancer, consistent with reports for endometrial cancer (EC) and lung adenocarcinoma (LUAD) [48, 49].
Interestingly, E2F1 specifically binds to the hPTTG1 promoter and increases endogenous hPTTG1 mRNA and protein levels [45], thereby activating the E2F1/PTTG1 signalling pathway to promote the G1/S transition of the cell cycle in cancer cells [50]. The E2F family of transcription factors regulates transcription of S-phase cell cycle proteins, DNA replication, DNA repair, and apoptosis [51]. However, E2F1 reportedly has conflicting roles in DNA damage in tumour cells. In some cases, E2F1 is important for DNA damage-induced apoptosis through transcriptional activation of p73 and possibly other proapoptotic target genes [51]. In other cases, E2F1 stimulates DNA repair and promotes survival in response to DNA damage [52]. Different survival states resulting from these two contradictory outcomes have been reported in both ovarian and hepatocellular carcinoma (HCC) [53]. We found that downregulation of promoter methylation of E2F1 in ovarian cancer resulted in increased expression and that increased expression correlated positively with HRD score and increased sensitivity to cisplatin. Clinical sample validation further showed promoter hypomethylation of E2F1 in ovarian cancer to be associated with better overall survival. Nevertheless, the mechanism of action of E2F1 in OC has not been fully elucidated, and research has been reported that single-site methylation combinations of multiple genes, including E2F1, are associated with OC prognosis [54]. The prognostic impact of single gene DNA methylation has not been reported, and the contribution of E2F1 to the DNA replication stress phenotype in OC remains unclear.
E2F1 is a regulator of the CDC6 gene [55]. CDC6 is a key factor in chromosomal replication licensing during the cell cycle, and it is essential for replication initiation, as well as for the ataxia telangiectasia and Rad3-related protein and its downstream effector kinase, checkpoint kinase 1 (ATR-Chk1) checkpoint [56], which plays a significant role in cell proliferation, migration, invasion, and tumour metastasis in various cancers [57]. Numerous recent studies have investigated the association between CDC6 expression and prognosis of different tumours, including lung cancer, estrogen receptor (ER) positive breast cancer, prostate cancer, bladder cancer, gastric cancer, and glioma [57]. Abnormally high CDC6 expression is often significantly associated with poor prognosis. In addition, recent research has revealed that CDC6 expression is higher in epithelial ovarian cancer (EOC) tissues than in normal ovarian tissues and that patients with high CDC6 levels have poor survival rates [58]. Although CDC6 methylation has not been well studied, CDC6 hypermethylation has been observed only in breast cancer, and the combination of CDC6 mRNA expression and the CpG island methylation phenotype (CIMP) may predict survival [59]. Although abnormal CDC6 DNA methylation has not been reported in ovarian cancer, our research found that abnormal CDC6 promoter methylation and expression in ovarian cancer correlated significantly with HRD scores. Furthermore, high CDC6 expression increased patient sensitivity to cisplatin and paclitaxel, and clinical sample validation further demonstrated that promoter hypomethylation of CDC6 in ovarian cancer is associated with better survival. Overall, these findings suggest that CDC6 promoter methylation may serve as a promising prognostic indicator for clinical applications.
In this study, we demonstrated that aberrant promoter methylation and expression of several genes, including PTTG1, E2F1, and CDC6, affected HRD scores and were significantly associated with survival in ovarian cancer. Moreover, all three genes are functionally directly related to the G1/S phase transition and induction of apoptosis after DNA damage. However, this study has some limitations. First, due to the scarcity of complete ovarian cancer data in the TCGA and GEO databases, our analysis relied on merging multiple data analyses, and bias may have been caused by confounding factors. Second, the absence of control leukocyte data prevented acquisition of HRD scores for clinical samples, and only overall survival data were used to assess the effect of gene methylation on prognosis, with no validation of intermediate processes. Therefore, further experimentation with more clinical samples is needed to better understand the biological functions and intrinsic molecular mechanisms of PTTG1, E2F1, and CDC6 methylation affecting HRD and prognosis, as are in vitro assays.
Overall, our study provides important insights into the role of epigenetic regulation in ovarian cancer and highlights the potential importance of DNA methylation as a biomarker for treatment selection and prognosis determination. These findings may lead to improved, personalized treatment strategies for ovarian cancer patients in the future.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
WH, MW and QW designed the research study. HZ collected the data from databases and performed the data analysis. WH, HZ, and XN constructed the figures. WH and SZ performed the experiments and the formal analysis. GS, HL and GY collected tissue samples. WH and HZ wrote the manuscript. WH, HZ, MW and QW reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors contributed to the article and approved the final version.
The studies involving human participants were reviewed and approved by the Xiangya School of Medicine, Central South University (approval no. 2019030306). The patients/participants provided their written informed consent to participate in this study. The study was conducted in accordance with the Declaration of Helsinki.
We thank the TCGA working groups for generously sharing their data.
This research was supported by Shanghai Municipal Health Commission Funds (Grant No. 20204Y0274 and 202140339) and Innovation-oriented Science and Technology Grant from NHC Key Lab of Reproduction Regulation (Grant No. CX2022-03) and Shanghai Natural Science Foundation (Grant No. 22ZR1456300).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.