

 , Emil Birch Christensen 2,3,4, Niels Abildgaard 1,2,3, Torben Barington 2,3,4, Thomas Lund 1,2,3,5, Jakub Krejcik 1,2,*
, Emil Birch Christensen 2,3,4, Niels Abildgaard 1,2,3, Torben Barington 2,3,4, Thomas Lund 1,2,3,5, Jakub Krejcik 1,2,*
1 Department of Haematology, Odense University Hospital, 5000 Odense, Denmark
2 Centre for Cellular Immunotherapy of Haematological Cancer Odense (CITCO), Odense University Hospital, 5000 Odense, Denmark
3 Department of Clinical Research, University of Southern Denmark, 5230 Odense, Denmark
4 Department of Clinical Immunology, Odense University Hospital, 5000 Odense, Denmark
5 Centre for Innovative Medical Technology (CIMT), Odense University Hospital, 5000 Odense, Denmark
Abstract
Multiple myeloma (MM) is a haematological malignancy originating from terminally differentiated B cells, resulting in significant morbidity and mortality. Currently, MM is regarded as an incurable disease, often exhibiting a relapse-remitting pattern that necessitates multiple lines of therapy. It is now well-established that ineffective immunosurveillance plays a critical role in the progression of MM. Consequently, strategies that redirect immune effector cells against MM have emerged as effective treatment modalities, particularly in cases where standard care therapies fail. T cell-based immunotherapy has gained considerable attention in ongoing clinical trials; however, natural killer (NK) cells, known for their ability to execute cytotoxicity against infected and malignant cells with precision, may offer complementary therapeutic advantages over T cells and possess untapped therapeutic potential. This review seeks to introduce readers to the significance of NK cell-mediated immunosurveillance in the context of MM, explore the potential benefits of redirecting NK cells against MM, and illustrate how current treatment strategies are often reliant on the functionality of NK cells. Most importantly, new promising mechanisms of harnessing NK cell-based immunity against MM are reviewed and put into a clinical perspective to highlight their implications for patient treatment and outcomes.
Keywords
- Multiple myeloma
- NK cells
- allogenic NK cells
- adoptive cell therapy
- CAR NK cells
- monoclonal antibodies
- immunomodulatory drugs
Multiple myeloma (MM) is classified as a plasma cell neoplasm, marked by the unregulated expansion of monoclonal plasma cells (PC) in the bone marrow (BM) [1]. Most MM cases arise from an asymptomatic, pre-malignant clonal proliferation of PCs known as monoclonal gammopathy of undetermined significance (MGUS) [2]. However, not all patients diagnosed with MGUS progress to develop MM [3, 4]. MM is a heterogeneous disease with manifestations of multiple organ damage [2, 5], and despite advancements in therapeutic strategies, the disease remains incurable.
The pathogenesis of MM is not fully understood. Various mechanisms contribute in disease development and progression, primarily characterized by evasion of immune surveillance and alterations in the bone marrow microenvironment that favor the proliferation of malignant plasma cells (PC) [6, 7]. The promotion of angiogenesis through cytokines such as vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2) is critical, as these factors stimulate the formation and growth of new blood vessels, thereby increasing microvascular density in the bone marrow. Elevated microvascular density has been correlated with poor clinical outcomes [8]. Other implicated mechanisms in MM pathogenesis include impaired dendritic cell functions [9, 10, 11, 12], accumulation of dendritic cells (DC) in the bone marrow [12], expansion of myeloid-derived suppressor cells (MDSC) [10, 11], increase in pro-inflammatory subsets of T cells [9], cytotoxic T cell dysfunction [9, 10], tumor associated macrophages [10, 11, 12], and upregulation of regulatory T cells, which collectively enhance immunosuppression and support carcinogenesis [9, 10, 11, 13]. Furthermore, dysfunctional natural killer (NK) cells, characterized by both quantitative and functional abnormalities, has been shown to significantly influence MM development and prognosis [9, 10, 13].
Natural killer cells are a subset of lymphocytes that primarily execute two key
functions: recognition and lysis of stressed and transformed cells, and the
production of cytokines [14]. They are classified as group 1 innate lymphoid
cells due to their ability to secrete interferon-gamma (IFN-
The release of cytokines from NK cells is crucial for promoting or restraining immune responses through communication with dendritic cells, as well as promoting adaptive immune responses by the enhancement of antigen-specific T cell responses [16, 17, 19]. NK cells possess the ability to rapidly attack and eliminate abnormal cells that lack expression of major histocompatibility complex class I (MHC-I) [15, 19]. This is common in neoplastic and virally infected cells. Upon activation through NK cell-activating receptors, NK cells induce apoptosis by the release of granzyme B, perforin, TNF-related apoptosis-inducing ligand (TRAIL), and Fas Ligand (FasL) [20, 21].
NK cell functions are contingent upon the interplay of activating and inhibitory
receptors [20, 22]. Upon contact with other cells, NK cell responses are mediated
by integration of signals, with no single receptor predominantly influencing
activation [23]. Among the activating receptors are the natural cytotoxicity
receptors (NKp30, NKp44, and NKp46) belonging to the immunoglobulin family, and
play a critical role in NK cell-mediated tumor immunosurveillance [24]. Other
activating receptors in NK cells include natural killer group 2 member D (NKG2D),
receptors of the signaling lymphocytic activation molecule (SLAM) family, and the
Ig-like domain containing receptor DNAX accessory molecule-1 (DNAM-1), among
others [20, 22]. Furthermore, NK cells can recognize the Fc portion of antibodies
that are bound to tumor cells, infected cells, or pathogens via the Fc receptor
Fc
NK cells can avoid activating signals through concomitant signaling from MHC-I specific inhibitory receptors which recognize certain classical and non-classical MHC molecules preventing attacks on healthy cells, while attacking abnormal cells lacking these ligands. This last phenomenon is called recognition of “missing self”. Major inhibitory receptors include killer cell immunoglobulin-like receptors (KIRs) and natural killer group 2 member A (NKG2A), which recognize a variety of human leukocyte antigen (HLA) class I alleles [15, 20, 25]. In addition to these receptors, several other critical pathways mediate inhibitory signals to NK cells. Notably, programmed cell death protein-1 (PD-1) becomes activated upon engagement with its ligand, programmed cell death ligand 1 (PD-L1) [27]. Other pathways include lymphocyte activation gene 3 (LAG3), T cell immunoglobulin and mucin-domain containing-3 (TIM3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT) [28].
NK cells play a significant role in the development and progression of various haematological malignancies, including MM [29, 30, 31, 32]. A reduction in NK cell cytotoxicity has been associated with advanced stage MM [33], while chemotherapy-treated patients exhibiting higher NK cell activity at presentation have better cumulative survival rates compared to those with low NK cell activity [34]. The quality and quantity of NK cells may be particularly pertinent in patients treated with novel therapies that are heavily reliant on NK cell function (discussed later in this review) [35, 36, 37]. In addition, several alterations related to NK cells are associated with the progression from precursor conditions, such as MGUS and smoldering multiple myeloma (SMM), to symptomatic MM [33, 34]. Alteration in the activating NKG2D pathway and downregulation of FasL have been observed in patients with MM compared to those with MGUS, indicating insufficient NK cell activation and resistance to FasL-induced apoptosis in patients who progress to MM [38]. Unsurprisingly, the phenotype and numbers of circulating NK cells, along with additional markers, might serve as predictive indicators for the risk of progression from SMM to MM, potentially forming the basis of a minimally invasive risk stratification model utilizing blood immunophenotyping [39].
It is established that the number of NK cells is decreased in the bone marrow of patients with MM, leading to diminished cytotoxic activity [40, 41, 42]. In the context of MM, NK cells experience chronic stimulation, which results in an increase in terminally differentiated NK cells, accompanied by a loss of their effector functions. These NK cells exhibit an exhausted phenotype characterized by the upregulation of inhibitory receptors and downregulation of activating receptors [40, 43].
In MM, the expression of NK cell activating receptors including DNAM-1, NKp30, NKp44, and NKp46 is significantly diminished [41, 42, 44, 45, 46]. DNAM-1 induces myeloma cell killing through ligands Nectin-2 and the poliovirus receptor (PVR), which are expressed on myeloma cells [46]. Notably, low expression of DNAM-1 is found in patients with active disease, while elevated levels of DNAM-1 are found in patients who have achieved complete response to therapy [46]. In contrast to the reduced expression of activating receptors, there is an increase in the expression of inhibitory receptors TIM3, TIGIT, and glucocorticoid-induced tumor necrosis factor receptor (GITR) on NK cells from patients with MM. This increase contributes to the downregulation of NK cell cytotoxicity [44]. Additionally, high levels of the TIGIT ligand Nectin-2 and PVR have been found in the bone marrow of MM patients [40, 41, 42, 47]. Importantly, while healthy NK cells express low levels of PD-1, its expression is upregulated in the context of MM, regulated by interleukin 2 (IL-2) [27, 48]. Similar to other tumors, myeloma cells also express the ligand PD-L1, which interacts with PD-1 found on NK cells. This interaction leads to the downregulation of NK cell cytotoxicity, facilitation immune escape of myeloma cells [41].
Various cell-bound and soluble factors contribute to the suppression of NK cell
immunity within the protective tumor microenvironment (TME) induced by MM [40].
Myeloma cells exhibit low levels of MHC-I and high levels of MHC class I-related
chain A (MICA), making them recognizable to NK cells, particularly in the early
stages of the disease [41, 49]. However, as the disease progresses from MGUS to
MM, the expression of MICA on myeloma cells decreases, while high levels of the
protein Erp5 increase, promoting the shedding of MICA from myeloma cells [50, 51].
The activation of NK cells through the recognition of MICA on myeloma cells
relies on the activating receptor NKG2D. Circulating MICA, the product of MICA
shedding, downregulates NKG2D receptor expression on NK cells, thereby
compromising the cytotoxic effect of NK cells against myeloma cells and
facilitating immune escape by MM [41, 49, 50, 51]. Furthermore, myeloma cells
overexpress surface HLA class I antigen E (HLA-E) which binds to the inhibitory
receptor NKG2A, thereby mediating NK cell inhibition [52, 53]. The cytokine
transforming growth factor
Allogenic haematopoietic stem cell transplantation (HSCT) has provided the first demonstrable evidence that MM can be effectively cured through the transfer of immune cells from a donor to a recipient. The graft-versus-myeloma (GvM) effect primarily relies on the alloimmune response mounted by donor immune cells against MM cells. Notably, donor T cells typically target host specific antigens rather than tumor specific antigens [64]. Consequently, allogeneic HSCT is frequently associated with T cell-mediated graft-versus-host disease (GvHD), a condition that necessitates the application of immunosuppressive therapies for its prevention and management. In the context of MM, NK cells might may contribute to the GvM effect observed following allogenic HSCT [65]. As previously mentioned, NK cell activity is modulated by a combination of activating and inhibitory signals derived from target cells [20, 22, 23]. Inhibitory signals are mediated through KIRs, which recognize allotype determinants presented by various human leukocyte antigen (HLA) class I alleles. Unlike other immune cells, inhibitory signals predominantly influence NK cells, preventing them from targeting and killing autologous cells. HLA class I KIR ligands exhibit variability among individuals, allowing for NK cell alloreactivity in transplantation scenarios. This phenomenon occurs when the recipient lacks the KIR ligands that are present in the donor, as evidenced by studies on acute myeloid leukemia patients undergoing haploidentical HSCT, which demonstrated effective NK cell-mediated responses across histocompatibility barriers [66, 67].
The application of KIR ligand mismatched NK cells has been investigated in the treatment of MM. Transfusion of T-cell depleted KIR ligand mismatched NK cells, followed by autologous HSCT, has shown variable NK cell alloreactivity, yet retains the capability to effectively kill myeloma cells within the alloreactive NK cell products (Fig. 1). This approach has been implemented without GvHD or failure of the engraftment of the autologous stem cells [68]. An in vitro study has reported enhanced NK cell cytotoxicity and ADCC against myeloma cells when employing allogeneic KIR-mismatched NK cells as opposed to autologous NK cells [69]. A recent phase I assessing the efficacy of allogenic umbilical cord blood derived NK cell (CB-NK) transplantation followed by autologous HSCT in MM patients indicated that CB-NK cells exhibit higher expression levels of activating receptors compared to endogenous NK cells in vivo [70]. Importantly, these findings suggest that adoptive NK cell therapy can be effectively implemented using an allogeneic source without a significant risk of GvHD, distinguishing it from T-cell-based therapeutic strategies.
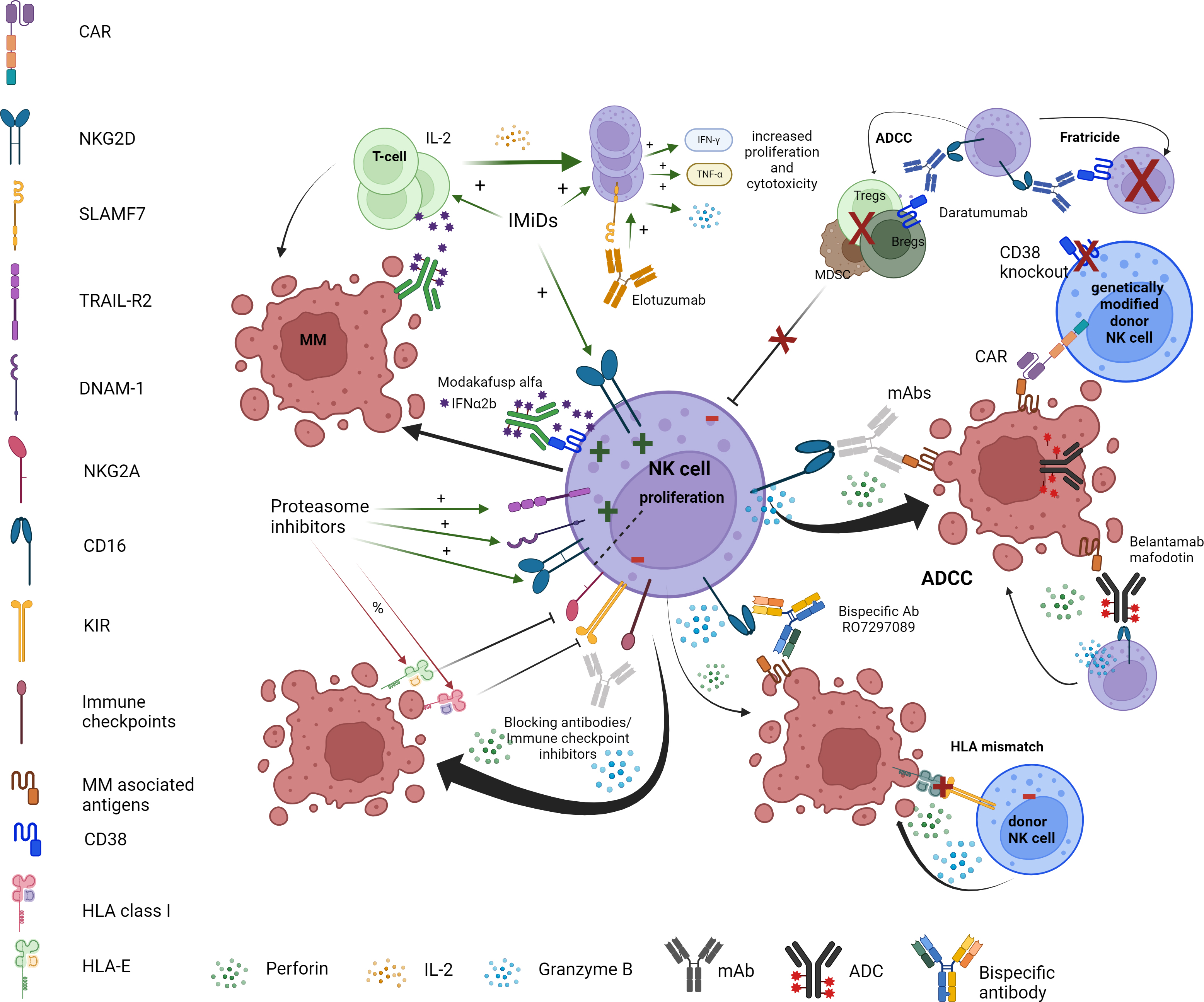 Fig. 1.
Fig. 1.
Harnessing the power of NK cells against multiple myeloma. This figure presents the different ways in which each type of myeloma therapy utilizes NK cell functions against MM cells. Abbreviations: MM, multiple myeloma (representing malignant plasma cell); IL, interleukin; IMiDs, immunomodulatory drugs; IFN, interferon; TNF, tumor necrosis factor; ADCC, antibody dependent cellular cytotoxicity; Tregs, regulatory T cells; Bregs, regulatory B cells; MDSC, myeloid derived suppressor cells; CAR, chimeric antigen receptor; mAbs, monoclonal antibodies; Ab, antibody; HLA, human leukocyte antigen; ADC, antibody drug conjugate; NKG2D, natural killer group 2 member D; KIR, killer cell immunoglobulin-like receptor; SLAMF7, signaling lymphocytic activation molecule family member 7; DNAM-1, DNAX accessory molecule-1. + = positive effect on, i.e., increase, stimulate or activate. % = negative effect on, i.e., inhibition or decreased activation. Created using BioRender.com. Agreement number: LS276QITRP.
The effectiveness of current MM therapies, including immunomodulatory drugs and monoclonal antibodies, is heavily dependent on the presence and functionality of endogenous NK cells [49, 71], which are often functionally deficient in the setting of MM [35, 36, 37]. As such, allogeneic NK cells may serve as a promising resource for adoptive immune cell therapies, offering potential advantages through genetic manipulation and the development of true off-the-shelf products. Several ongoing clinical trials are exploring the allogeneic transfer of NK cells (Table 1).
| Investigational drug | Source | Patients | Phase | Objectives | Trial ID and status |
| NK cells genetically engineered with NY-ESO-1 TCR/IL-15 cell receptor | Allogenic | RRMM and RR PCL | 1 and 2 | NCT06066359 | |
| Recruiting | |||||
| CIML NK cells with KP1237 and IL-2 | Autologous | MRD positive MM post autologous HSCT | 1 | NCT04634435 | |
| Recruiting | |||||
| Cyclophosphamide, TGFbi NK cells and isatuximab | Allogenic | RRMM | 1 | NCT06203912 | |
| Recruiting | |||||
| Investigational drugs: NK cells with IL-2 and TGFbi (conditioning regimen of Flu and Cy) | Allogenic | Advanced cancers including RRMM | 1 | NCT05400122 | |
| Non-HLA matched | Recruiting | ||||
| NK cells with or without isatuximab as maintenance after autologous HSCT | Autologous | NDMM eligible for autologous HSCT | 2 | NCT04558931 | |
| Recruiting | |||||
| CB derived NK cells after myeloablative or non-myeloablative CB transplantation | Allogenic | Haematologic malignancies including MM | 2 | NCT02727803 | |
| Recruiting | |||||
| GIC-102 (off the shelf allogenic NK cells) | Allogenic | Advanced cancers including RRMM | 1 and 2 | NCT05880043 | |
| Non-HLA matched | Recruiting | ||||
| IDP-023 (off the shelf allogenic NK cells) with or without daratumumab and with or without IL-2 | Allogenic | NHL and RRMM | 1 and 2 | NCT06119685 | |
| Non-HLA matched | Recruiting | ||||
Abbreviations: NK, natural killer; TCR, T-cell receptor; IL, interleukin; RRMM, relapsed or refractory multiple myeloma; PCL, plasma cell leukemia; DLT, dose limiting toxicity; OR, overall response; PFS, progression free survival; DOR, duration of response; CIML, cytokine induced memory like; MRD, minimal residual disease; MM, multiple myeloma; HSCT, haematopoietic stem cell transplantation; OS, overall survival; ORR, overall response rate; TGFbi, transforming growth factor beta inhibitor; Flu, fludarabine; Cy, cyclophosphamide; HLA, human leukocyte antigen; NDMM, newly diagnosed multiple myeloma; CB, cord blood; GvHD, graft versus host disease; NHL, Non-Hodgkin lymphoma; NY-ESO-1, New York Esophageal Squamous cell carcinoma 1.
In addition, the transfer of autologous NK cells is under investigation in multiple clinical trials as well (Table 1). These studies focus on various approaches, including combination therapies with existing myeloma treatments (NCT04558931) and NK cell-based therapies utilizing autologous NK cells in the form of cytokine-induced memory-like NK cells in conjunction with bispecific antibodies (NCT04634435).
Chimeric antigen receptor (CAR) T cell therapy has emerged as a significant
advancement in the treatment of haematological malignancies, including MM. This
approach involves the genetic modification of T cells to express CARs that
specifically recognize tumor-associated antigens, such as B-cell maturation
antigen (BCMA) (Fig. 1). Currently, only autologous CAR T cells are approved for
clinical use, and anti-BCMA CAR T cell therapy has shown promising outcomes in
patients with relapsed or refractory multiple myeloma (RRMM) [72]. However, CAR T
cell therapy has certain limitations, including toxic side effects [73], he
challenges associated with using previously treated autologous T cells [74],
manufacturing constraints [73], and the potential risk of GvHD in allogenic CAR T
cells [75]. To address these concerns, CAR NK cells are being explored as a
viable alternative. In addition to their CAR-dependent functions, CAR NK cells
retain the ability to mediate tumor cell killing through their inherent NK cell
functions, potentially enhancing the efficacy of CAR-based treatments for myeloma
[76]. Furthermore, CAR NK cells may provide patients with improved accessibility
to a therapeutic option that carries a reduced risk of adverse side effects,
thereby enhancing patients’ quality of life. It is important to note that most
current CAR NK cell constructs are based on designs tailored for T cell
activation, which may limit the effectiveness of CAR NK cells in vivo.
CAR T constructs have predominantly utilized CD3
While most studies on CAR NK cells remain preclinical, a Phase I and II study
involving allogeneic CAR NK cells expressing an anti-CD19 CAR and IL-15 in
heavily pretreated lymphoid malignancies demonstrated encouraging responses
without incidents of cytokine release syndrome or GvHD [78, 79]. Importantly, the
quality of the cord blood unit used was identified as a predictor of therapeutic
response, with the persistence of CAR NK cells correlating with response rates. A
phase I and II trial utilizing IL-15 and nicotinamide-enhanced CAR NK cells
(GDA-201) showed promising results in lymphoma treatment when combined with the
monoclonal antibody rituximab, although the same CAR NK cell did not yield
clinical efficacy in MM when combined with elotuzumab [80]. Current clinical
trials investigating various CAR NK cell models for MM treatment are ongoing
(Table 2). A recent study has indicated that a CD70 CAR NK cell exhibited
increased activity compared to non-transduced NK cells, effectively clearing
tumor cells more rapidly, and demonstrated tumor control and prolonged survival
in mouse models [81]. This has led to the initiation of an ongoing clinical trial
assessing a CD70 CAR NK cell (NCT05092451). A SLAM family member 7 SLAMF7 CAR NK
cell showed heightened cytolytic activity against human myeloma cells and
increased production of IFN-
| Investigational drug | Source | Patients | Phase | Objectives | Trial ID and status |
| BCMA CAR NK (conditioning regimen: Flu and Cy) | Allogenic | RRMM | 1 | NCT05008536 | |
| Unknown status | |||||
| BCMA CAR NK92 | Unknown | RRMM | 1 and 2 | NCT03940833 | |
| Unknown status | |||||
| CAR.70/IL15-transduced CB-NK | Allogenic | CD70+ haematologic malignancies including MM | 1 and 2 | NCT05092451 | |
| Recruiting | |||||
| FT576: BCMA CAR NK with or without daratumumab (conditioning regimen: Flu, Cy and Benda) | Allogenic NK cell derived from a clonal, CD38-knockout, human-induced pluripotent stem cell line | RRMM | 1 | NCT05182073 | |
| Active, not recruiting | |||||
| BCMA CAR NK | Allogenic | RRMM | 1 | NCT05652530 | |
| Recruiting | |||||
| LUCAR-B68: BCMA CAR NK | Unknown | RRMM | 1 | NCT05498545 | |
| Not yet recruiting | |||||
| BCMA CAR NK (conditioning regimen: chemotherapy not otherwise specified) | Allogenic | RRMM and RR PCL | 1 | NCT06045091 | |
| Recruiting | |||||
| BCMA CAR NK (conditioning regimen: Flu and Cy) | Unknown | RRMM | 1 and 2 | NCT06242249 | |
| Not yet recruiting | |||||
| KN1102: NKG2D CAR NK | Unknown | RRMM | 1 | NCT06379451 | |
| Not yet recruiting | |||||
Abbreviations: BCMA, B-cell maturation antigen; CAR, chimeric antigen receptor; NK, natural killer; Flu, fludarabine; Cy, cyclophosphamide; RRMM, relapsed or refractory multiple myeloma; DLT, dose limiting toxicity; CB, cord blood; MM, multiple myeloma; Benda, bendamustine; PCL, plasma cell leukemia; OS, overall survival; ORR, overall response rate; PFS, progression free survival; DOR, duration of response; NKG2D, natural killer group 2 member D.
Monoclonal antibodies (mAb) daratumumab and isatuximab targeting the CD38 molecule overexpressed by MM cells, have demonstrated efficacy as standalone treatments and now constitute a cornerstone of MM therapy across various treatment combinations [89]. By binding to CD38 on MM cells, these antibodies induce immune-mediated cytotoxicity by various mechanisms of action. Notably, they can elicit NK cell-mediated antibody dependent cytotoxicity (ADCC) through an Fc receptor-dependent mechanism in a dose dependent and primarily effective towards CD38high myeloma cells [71] (Fig. 1). Furthermore, it has been demonstrated that targeting CD38 in MM depletes highly immunosuppressive CD38+ positive cells in TME, thereby alleviating the state of immune suppression and contributing to the upregulating of NK cell cytotoxicity against MM cells [71, 90].
Clinical evidence underscores the critical role of NK cells in mediating daratumumab’s therapeutic effects. Fc receptor polymorphisms in MM patients might have an impact on response and progression free survival (PFS) after daratumumab treatment [35]. Notably, the expression of specific NK cell surface antigens correlates with a response to daratumumab monotherapy. Patients with primary refractory disease show a higher frequency of NK cells in the BM with an exhausted and activated phenotype at baseline, with decreased expression of CD16 and granzyme B, and increased expression of TIM3 and HLA DR isotype (HLA-DR) compared to patients who respond to treatment. This exhausted or activated NK cell phenotype is also associated with significantly impaired PFS and overall survival (OS) [36].
Correspondingly, a recent single-cell analysis of immune microenvironment in newly diagnosed MM patients treated with a potent daratumumab combination therapy revealed that the proportion of NK cells in BM is the only immune cell population linked to sustained minimal residual disease (MRD) negativity. The study suggests that failure to achieve sustained MRD negativity is associated with a reduction in baseline NK cell numbers [37]. The same study also suggested marked CD14+ monocyte expansion seen in the MRD-negative cases. Interestingly, it has been previously reported that the targeting of CD38+ NK cells by daratumumab promotes monocyte activation, leading to increased expression of T cell costimulatory molecules (such as CD86 and 80) and enhancing anti-MM phagocytic activity both ex vivo and in vivo [91].
Importantly, most of NK cell subpopulations express high levels of CD38 leading to reduced NK cell counts in patients treated with daratumumab. This reduction occurs due to the rapid elimination of CD38+ NK cells through NK cell fratricide [91]. Despite this, NK cells of daratumumab treated patients are not completely depleted and may still contribute to ADCC, clinical efficacy and infection control [92]. However, NK cells surviving daratumumab-induced fratricide have a lower CD16 and granzyme B expression which might limit their cytotoxicity against MM. Interestingly, ex vivo killing assays with BM samples from daratumumab refractory patients demonstrated that daratumumab-mediated elimination of MM cells can be significantly improved by the addition of healthy donor-derived NK cells [36]. This suggests that daratumumab induced NK cell dysfunction and depletion may be contributing to treatment failure. To address the challenges posed by daratumumab-induced NK cell fratricide and dysfunction, several strategies have been developed. These approaches include engineering NK cells to express a non-cleavable version of CD16, which results in enhanced ADCC activity [93], generating NK cells with CD38 deletion to eliminate daratumumab-induced fratricide [94], or expanding specific NK cell subsets with superior ADCC activity [94].
A subset of NK cells known as adaptive NK cells exhibit lower CD38 expression compared to conventional NK cells. It has been suggested that response to daratumumab treatment is dependent on the composition of adaptive NK cell present in the bone marrow [95]. The presence of these low CD38-expressing adaptive NK cells may facilitate evasion of daratumumab-induced NK cell fratricide. Adaptive NK cells in NDMM have an effective killing of myeloma cells in the presence of daratumumab, and patients with high risk MM have a lower proportion of adaptive NK cells compared to other myeloma patients, which might contribute to treatment resistance towards daratumumab in this group of patients [95].
Interestingly, another CD38 targeting antibody, isatuximab, primarily induces NK cell depletion through exhaustion rather than fratricide in an ex vivo setting [96]. When assessing NK cell count and activity in patients treated with isatuximab, there was no observed reduction in CD38+ NK cell counts among responders in a clinical trial involving heavily pretreated myeloma patients, unlike the findings in daratumumab-treated patients [97]. The reason for this may be attributed to isatuximab’s ability to sustain anti-myeloma effects through ADCC, suggesting that isatuximab may be better tolerated by NK cells.
Anti-SLAMF7 antibody, elotuzumab, is an IgG1 antibody targeting a surface protein expressed on myeloma cells, which is also naturally present on NK cells. A preclinical study has demonstrated that elotuzumab effectively induces strong NK cell-mediated ADCC against myeloma cells both in vitro and in vivo [98]. Furthermore, elotuzumab directly activates NK cells by binding to SLAMF7 on both NK cells and myeloma cells, thus mediating NK cell cytotoxicity against myeloma cells independent of ADCC [99, 100, 101] (Fig. 1). Unlike daratumumab, elotuzumab treatment results in minimal NK cell fratricide despite the expression of SLAMF7 on NK cells. However, in a clinical setting, no response has been observed to elotuzumab monotherapy [102].
A rare subset of NK cells, termed g-NK cells, is characterized by the absence of
the FcR
Importantly, the combination of mAbs with immunomodulatory drugs (lMiDs) has demonstrated higher response rates in RRMM compared with treatment with IMiDs and dexamethasone alone [105, 106, 107]. Preclinical studies have indicated that lenalidomide treatment enhanced mAbs-induced NK cell-mediated ADCC, which may account for improved treatment responses with these combination therapies [108, 109, 110]. Recently, the phase III MAIA study compared the efficacy of lenalidomide and dexamethasone against the combination treatment of daratumumab, lenalidomide, and dexamethasone in newly diagnosed transplant ineligible MM patients. The results confirmed the superiority of triplet combination therapy. Interestingly, the combination of daratumumab, lenalidomide and dexamethasone in this group of older patients showed unprecedented long PFS close to PFS observed for younger transplant eligible patients, a group of patients which typically has a significantly better treatment outcome [111, 112]. These clinical results suggest a potent synergism between IMiDs and mAbs.
IMiDs, such as pomalidomide and lenalidomide along with their newer generation counterparts known as cereblon E3 ligase modulators (CELMoDs), are an important class of drugs in the treatment of MM across all disease stages. IMIDs and CELMoDs mediate antiproliferative effects on tumor cells through various mechanisms, some of which are specifically derected at enhancing the activity of NK cells. IMiD and CELMoD activity in MM is dependent on the protein cereblon [113, 114]. By recruiting cereblon, these drugs facilitate the targeted degradation of transcription factors Ikaros and Aiolos. This process not only results in directs antitumor effects ob myeloma cells but also enhances the activation of surrounding immune cells, thereby promoting an anti-tumor immune response [115].
IMiDs enhance the proliferation of T and NK cells while increasing the
expression of key activating receptors, including CD16 and NKG2D, on NK cells.
These effects lead to heightened NK cell activation, a lowered activation
threshold of NK cells, and improved ADCC [49, 116]. Specifically,
lenalidomide-induced enhancement of NK cell ADCC is believed to be associated
with increased levels of FasL and granzyme B, as well as enhanced production of
cytokines like IFN-
Comparing these existing effective therapies that utilize NK cell function with the earlier mentioned allogenic NK cell transfer and CAR NK cell treatments, the existing treatments present no risk of serious side effects such as GvHD or the development of CRS which are potential complications associated with NK cell-based therapies.
Proteasome inhibitors are crucial in the treatment of MM and are recommended in both first line and subsequent lines of therapy for patients eligible for HSCT as well as those inelegible [2]. Currently, three types of proteasome inhibitors are available: bortezomib, carfilzomib, and ixazomib, all of which are utilized in various combinations and settings resulting in improved treatment outcomes [127, 128, 129, 130]. Proteasome inhibitors induce myeloma cell death through multiple NK cell-dependent mechanisms. Bortezomib and carfilzomib decrease the expression of HLA class I on myeloma cells, which increases their susceptibility to lysis by NK cells in a dose-dependent manner [131, 132]. In in vitro expanded human NK cells, bortezomib sensitizes tumor cells to NK cell cytotoxicity by upregulation the expression of TRAIL-R2 on NK cells through death receptor 5 (DR5) [133, 134]. Moreover, bortezomib contributes to a reduction in NKG2A-mediated inhibition of NK cells due to decreased expression of HLA-E on myeloma cells [134]. Bortezomib enhances expression of PVR and nectin-2 on myeloma cells, which facilitate greater recognition and activation of the activating receptors DNAM-1 and NKG2D on NK cells [135] (Fig. 1). Together, these mechanisms illustrate how proteasome inhibitors can augment NK cell-mediated responses against myeloma cells, improving therapeutic efficacy in the management of MM.
Belantamab mafodotin is one of the newer therapeutic agents for the treatment of MM, functioning as a dual-acting antibody-drug conjugate (ADC). It consists of an anti-B cell maturation antigen afucosylated, humanized IgG1 conjugated with the tubulin polymerization disrupting agent maleimidocaproyl monomethyl auristatin F. This drug exhibits multiple antitumor effects, some of which leverage NK cell functions, such as increased NK cell infiltration and enhancement of NK cell-mediated ADCC demonstrated in mouse models [136] (Fig. 1). Belantamab mafodotin induces enhancement of NK cell-mediated ADCC against myeloma cells in vitro in human models, without altering growth and survival of NK cells [137].
Modakafusp alfa (formerly known as TAK-573) is a novel first-in-class humanized
anti-CD38 IgG4 monoclonal antibody designed to deliver IFN
Bispecific killer cell engagers (BiKE) are antibodies designed to simultaneously bind to a specific receptor on NK cells and a tumor antigen expressed on malignant cells. This dual binding facilitates activation of NK cells, leading to tumor cell killing through the formation of a synapse. BiKEs are primarily being tested in preclinical settings with CD16 or NKG2D as the NK cell targets as well as BCMA or SLAMF7 as the target on myeloma cells. A preclinical study of a BCMA/CD16 bispecific antibody RO7297089 demonstrated a selective binding to CD16 on NK cells, which induced lysis of BCMA positive myeloma cells in cynomolgus monkeys with a favorable safety profile [142] (Fig. 1). In a phase I study, RO7297089 was demonstrated to be well tolerated in patients with RRMM and showed to induce some anti-myeloma activity [143]. A novel bispecific antibody targeting SLAMF7 on myeloma cells and NKG2D on immune cells utilizes the presence of the activating receptor NKG2D on multiple immune cells [144]. Another BiKE targets BCMA on myeloma cells and carries MICA molecules to stimulate NK cell activation through biding to NKG2D on NK cells [145].
Checkpoint inhibitors have been used in treatment of solid cancers for many years, and some checkpoint inhibitors have found their place in treatment of haematological malignancies. In MM, checkpoint inhibitors have garnered interest in recent years. In MM, the PD-1/PD-L1 axis is upregulated and leads to inhibition of NK cell activation, which presents a possible treatment target (Fig. 1). In a preclinical study, a PD-1 antibody enhanced NK cell function, specifically against PD-L1 positive human MM cells in vitro [48]. The combination of IMiDs and PD-1 inhibitors increased NK cell activation and apoptosis of myeloma cells, indicating the potential of a synergistic effect between IMiDs and PD-1 inhibitors [48, 146]. In studies of monotherapy, PD-1 inhibition in newly diagnosed MM or in RRMM treated with combination therapy of lenalidomide or pomalidomide and dexamethasone with PD-1 inhibitors, no significant response to treatment was found, and some studies demonstrated lack of tolerance due to high rates of adverse events [147, 148, 149, 150].
KIR inhibition through anti-KIR antibody IPH2101 increases NK cell-mediated
tumor cell lysis in a preclinical stetting [151] (Fig. 1). IPH2101 as monotherapy
in patients with SMM showed lack of response, and the study was prematurely
terminated [152]. In the same study, IPH2101 induced hyporesponsiveness in NK
cells in vitro and in vivo, leading to a reduced ability to
secrete IFN-
Immunosuppressive alterations in the TME and the adaptation of MM cells to immune pressure present both opportunities and challenges in immunotherapeutic approaches to MM. Emerging evidence indicates that myeloma cells can evade immunotherapeutic pressure through various mechanisms. Antigen escape and dysfunctional T cells are well-characterized mechanisms contributing to resistance against T cell-redirecting therapies. It is plausible that the evasive properties of the MM cells, the suppressive TME, and the dysfunctional population of NK cells will represent significant obstacles to effective NK cell therapy for MM. As such, future treatment strategies may be more successful if they involve genetic manipulation of effector cells, enhancing their capacity to overcome the adverse TME and prevent antigen escape [157].
Given that the TME acts as a barrier to the efficacy of adoptive cell transfer, it is imperative to develop strategies facilitating the persistence and infiltration of effector cells into this hostile TME. Notably, the crosstalk between immune cells and vasculature influences the nature of anti-tumor responses [158], and disorganized vessels in TME hinder the trafficking and functionality of effector cells [159]. Interestingly, TME often manipulate the balance between pro- and anti-angiogenic factors [160]. NK cells can either inhibit or promote angiogenesis depending on the specific context, such as within a hypoxic TME [161]. In addition, excessive production of VEGF, a critical mediator of angiogenesis in MM [162], negatively regulates adaptive immune responses in TME [158]. Correspondingly, preclinical models have confirmed that dual targeting of immune responses and angiogenesis represents a promising strategy for improving aberrant vascular-immune crosstalk and enhancing the efficacy of immunotherapy [163]. The combination of anti-angiogenic treatment with immunotherapy has already proven to be a successful strategy in treating solid cancers [164].
Another aspect limiting the efficacy of current immunotherapy in MM is the limited number of targetable antigens. Given the significant challenges posed by antigen escape and on-target off-tumor toxicity [165, 166], there is a pressing need to develop strategies that target MM-specific antigens crucial to MM cell oncogenesis. Engaging antitumor responses against neoantigens derived from oncogenes [167] may provide a viable approach to address these current challenges. Achieving this will require collaborative efforts among basic, translational scientists, and industry partners to expedite the application of novel NK cell-based immunotherapies in clinical practice. Currently, NK cell-based therapies are going through their “first wave”, facing numerous limitations such as the lack of standardized regulations for quality assessment and the release of NK cell products for clinical application. Moreover, the future development of scalable manufacturing protocols for clinical-grade production will be essential to facilitate the translation of novel NK cell-based immunotherapies into clinical practice.
Multiple myeloma is a disease driven by immune dysfunction and escape that worsen as the disease progresses. NK cells play a pivotal role in maintaining anti-tumor immunity, and the dysfunction of NK cells significantly contributes to the development, progression, and treatment resistance observed in MM. Therefore, restoring NK cell functions is crucial for achieving disease control and ultimately curing MM. Through current therapeutic approaches NK cell-based strategies are being used in a multifaceted way. This is demonstrated through immunomodulatory drugs (IMiDs), proteasome inhibitors, monoclonal antibodies, and most importantly strategies combining these drugs together. A prime example is the combination of lenalidomide and daratumumab, which represents the most effective treatment option for newly diagnosed transplant ineligible MM patients. This combination therapy exemplifies synergistic NK cell activation, resulting in prolonged progression free survival. Importantly, NK cell fitness in MM patients is significantly inferior to that of NK cells harvested from healthy individuals. Therefore, combining NK cell-dependent therapies with the administration of donor-derived NK cells presents a promising strategy to mitigate the limitations of existing treatments. Furthermore, the adoptive transfer of HLA-mismatched NK cells has been associated with a graft-versus-myeloma effect, further enhancing the cytotoxic capacity of donor-derived NK cells. Importantly, unlike T cells, NK cell transfer between unrelated individuals is not associated with significant alloreactivity against healthy tissues. This selective alloreactivity towards malignant cells renders NK cells a unique source of immune effector cells, allowing for genetic manipulation and the potential development of true off-the-shelf cellular therapy products. An increasing number of clinical trials are exploring genetically modified NK cell therapies, which have yielded promising results. Future studies combining these new living drugs with established treatments will further elucidate the most effective strategies for achieving synergistic drug effects, ultimately benefiting patients with MM.
Writing — original draft preparation, IC and JK. Writing — content, review, and editing, EBC, NA, TB, and TL. Supervision, JK. Conception and design of the review — IC, JK, EBC, NA, TB, and TL. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.