






 , Yi Li 1,2, Lingbing Meng 1, Chenguang Yang 1,2, ChenXi Xia 1,3, Xiang Wang 1,2,*
, Yi Li 1,2, Lingbing Meng 1, Chenguang Yang 1,2, ChenXi Xia 1,3, Xiang Wang 1,2,* , Fang Wang 1,2,*
, Fang Wang 1,2,*1 Department of Cardiology, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, 100730 Beijing, China
2 Graduate School of Peking Union Medical College, Chinese Academy of Medical Sciences, 100730 Beijing, China
3 Peking University Fifth School of Clinical Medicine, 100730 Beijing, China
Abstract
Heart failure with preserved ejection fraction (HFpEF) is a systemic syndrome primarily associated with fibrosis, oxidative stress, inflammation, and cellular apoptosis. Growth differentiation factor 15 (GDF15), a biomarker commonly used in clinical studies, exhibits protective effects on the myocardium. Therefore, the focus of the present study is to determine the mechanism by which GDF15 protects cardiac function in HFpEF.
We conducted functional enrichment analysis and protein-protein interaction network analysis on genes highly expressed in HFpEF but lowly expressed in normal samples. We established an HFpEF rat model by feeding the rats with a high-fat diet and administering N-omega-nitro-l-arginine-methyl ester (L-NAME) in their drinking water and silenced GDF15 by tail vein injection of lentivirus (L3110). After 12 weeks of feeding, echocardiographic examinations were performed. Following euthanasia of the rats, blood and heart tissue samples were collected. Heart tissue sections were stained using Masson’s trichrome and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining methods. Western blot (WB) analysis was employed to determine the concentrations of relevant proteins.
The echocardiographic results showed that compared with the HFpEF + MOCK group, the HFpEF+silencing GDF15 (siGDF15) group exhibited more severe cardiac dysfunction, with significant decreases in ejection fraction (p < 0.05) and E/A ratio (p < 0.001). WB results demonstrated that, compared with the HFpEF + MOCK group, the HFpEF+siGDF15 group exhibited increased expression of cardiac fibrosis-associated proteins, including collagen I (p < 0.01), collagen III (p < 0.01), and α-smooth muscle actin (α-SMA) (p < 0.01). Additionally, oxidative stress-associated biomarkers such as myeloperoxidase (MPO) (p < 0.01) and oxidized low-density lipoprotein (ox-LDL) (p < 0.01), inflammation-associated biomarkers, including interleukin-1 beta (IL-1β) (p < 0.01), interleukin-6 (IL-6) (p < 0.01), interleukin-8 (IL-8) (p < 0.01), and tumor necrosis factor α (TNFα) (p < 0.01), and apoptosis-associated biomarkers like cleaved caspase-3 (p < 0.01) and BCL2-associated X (BAX) (p < 0.01) were also upregulated in HFpEF+siGDF15 group.
Our research indicates that GDF15 preserves cardiac function by inhibiting myocardial fibrosis, reducing myocardial cell oxidative stress, alleviating cardiac inflammation, and suppressing myocardial cell apoptosis.
Keywords
- growth differentiation factor 15
- heart failure with preserved ejection fraction
- cardiac fibrosis
- oxidative stress
- inflammation
- apoptosis
Heart failure (HF) is a significant public health challenge worldwide. HF with preserved ejection fraction (HFpEF) constitutes more than half of all HF instances, whose prevalence and mortality are continuously increasing along with the aging population and an escalating comorbidity burden [1, 2]. HFpEF is a systemic syndrome primarily associated with mechanisms such as fibrosis, oxidative stress, inflammation, and cellular apoptosis [3, 4]. Several studies have found that inhibiting myocardial fibrosis in HFpEF can improve cardiac function, accompanied by alleviation of oxidative stress, inflammation, and cellular apoptosis [5, 6, 7]. Currently, due to the complexity of pathophysiological mechanism and very limited therapeutical options of HFpEF, it presents a major challenge for clinical practice.
Growth differentiation factor 15 (GDF15) is a peptide hormone that is a member
of the transforming growth factor beta (TGF
Our research demonstrates that GDF15 is upregulated in HFpEF, and it protects cardiac function by inhibiting myocardial fibrosis, reducing myocardial cell oxidative stress, alleviating cardiac inflammation, and suppressing myocardial cell apoptosis.
The dataset GSE126062, which relates to HFpEF, was obtained from the Gene Expression Omnibus (GEO)
database available at http://www.ncbi.nlm.nih.gov/geo/ and was generated using
the GPL11154 platform. This dataset comprises 3 HFpEF tissue samples and 3 normal
tissue samples, utilized for pinpointing differentially expressed genes (DEGs)
associated with HFpEF. We utilized the R package “limma” (version R4.2.0) to conduct probe
summarization and background correction on the gene expression data in GSE126062.
To adjust the initial p-values, we applied the Benjamini-Hochberg method, and
calculated the fold change (FC) to assess the significance of differences in gene
expression. The false discovery rate (FDR) served as the criterion for
determining significance. DEGs were defined as those with a
p-value
By conducting functional enrichment analysis and building and examining a protein-protein interaction (PPI) network, we identified 5 hub genes that exhibited elevated expression in HFpEF samples compared to normal samples, which showed lower expression levels of these genes. To further explore diseases linked to these hub genes, we entered them into the Comparative Toxicogenomics Database (CTD) database and found that all 5 genes are related to HF, cardiac diseases, coronary artery disease, and inflammation.
The Animal Ethics Committee of Beijing Hospital granted approval for this study
(No. 00001857). A model of HFpEF was established in eight-week-old Dextran
Sulfate Sodium (DSS) rats [SPF Biotechnology Co., Ltd., Beijing, China] by
administering a high-fat diet and N-omega-nitro-l-arginine-methyl ester (L-NAME,
N5751-1G, Shanghai North Connaught bio technology Co., Ltd, Shanghai,
China)-infused drinking water. A total of 40 rats were utilized for the creation
of this HFpEF model. Following a week of adaptive feeding, the rats were randomly
assigned into four groups, and the experiments were carried out for a period of
12 weeks: (1) blank control group (n = 10), (2)
HFpEF group (n = 10): a high fat diet (HFD, 60%
calories from lard) + L-NAME (0.5 g/L in drinking water), (3) HFpEF + silencing
GDF15 group (HFpEF+siGDF15 group, n = 10): HFD
(60% calories from lard) + L-NAME (0.5 g/L in drinking water), received
lentivirus tail vein injection (2
After euthanasia of the rats, their heart samples were isolated and immersed in 10% phosphate-buffered formalin (PBD999, Scytek, Shanghai, China) for a duration of 24 hours for fixation. Following this, the samples were embedded in paraffin and sectioned into slices of 4–5 micrometers in thickness. The histologic sections of the tissues underwent staining procedures using Masson’s trichrome stains (G1006-1 to G1006-6, Servicebio, Wuhan, China). Imaging of these sections was conducted using the NanoZoomer Digital Pathology RS system (E800, Nikon, Tokyo, Japan). Computerized planimetry was employed to quantify the mean cross-sectional area of the cardiomyocytes and to assess the degree of fibrosis present.
Upon euthanasia, heart tissue samples of rats were excised and fixed in 10% phosphate-buffered formalin for 24 hours. Subsequently, the fixed samples were embedded in paraffin and sectioned into slices with a thickness of 4–5 µm. Terminal deoxynucleotidyl transferase-mediated dexoxyuridine triphosphate nick-end labeling (TUNEL) staining was performed using the Tetramethylrhodamine (red) Tunel Cell Apoptosis Detection Kit (G1502-100T, Servicebio, Wuhan, China) following the manufacturer’s instructions. In this procedure, apoptotic nuclei were labeled with red fluorescein, and all cardiomyocyte nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, G1012, Servicebio, Wuhan, China). Visualization of the heart tissue sections was achieved through confocal microscopy (LSM700, Zeiss, Jena, Germany). The apoptosis rate was determined by calculating the ratio of TUNEL-positive nuclei to the total number of DAPI-stained nuclei.
Proteins were extracted from rat heart tissue, and the total protein
concentration was measured using Bicinchoninic Acid (BCA, G2026-200T, Servicebio, Wuhan, China). Following protein
denaturation, gel formation, sample application, electrophoresis, gel excision,
membrane transfer, blocking, and antibody incubation, the membrane was visualized
and imaged. The primary antibodies utilized included rabbit anti-GDF15 antibody
(1:2000; 27455-1-AP; Proteintech; Wuhan, China), mouse anti-NT-proBNP antibody
(1:1000; ab13115; abcam; Cambridge, UK), rabbit anti-Collagen Ⅰ antibody (1:1000;
14695-1-AP; Proteintech; Wuhan, China), rabbit anti-Collagen Ⅲ antibody (1:300;
22734-1-AP; Proteintech; Wuhan, China), rabbit anti-
After a 12-week feeding period, echocardiography was conducted using the Vevo 1100 High-Resolution Imaging System (Visual Sonics Inc.; Toronto, Canada). The animals were anesthetized with isoflurane (RWD Life Science Co., Ltd; Shenzhen, China) at a concentration of 1.5% to 2.0% and promptly placed on a thermostat set to 37 °C to maintain normal body temperature. The ultrasound beam’s position and orientation were meticulously adjusted to acquire clear echocardiographic images of the left ventricle. Subsequently, M-mode images were captured for the assessment of left ventricular functional parameters.
The data are reported as mean
In our proteomics analysis of HFpEF rat cardiac tissue, we identified 3625 DEGs, which were primarily enriched in functions related to immune response, inflammatory response, and cell death, as determined by functional enrichment analysis. We analyzed the intersection of hub genes by maximum clique centrality (MCC), edge percolated component (EPC) and density of maximum neighborhood component (DMNC) algorithms to extract more core targets, which revealed significant involvement of cyclin-dependent kinase 1 (CDK1), centromere protein F (CENFP), aurora kinase B (AURKB), kinesin family member 2C (KIF2C), and notably growth differentiation factor 15 (GDF15) (Fig. 1A). Subsequently, the reference value of the core target for different diseases (including heart failure) was analyzed (Fig. 1B,C). Given its strong association with cardiac diseases and its significantly elevated expression in the hearts of HFpEF rats, GDF15, which is recognized for its cardioprotective effects, is implicated as a key protective mechanism in the pathogenesis of HFpEF.
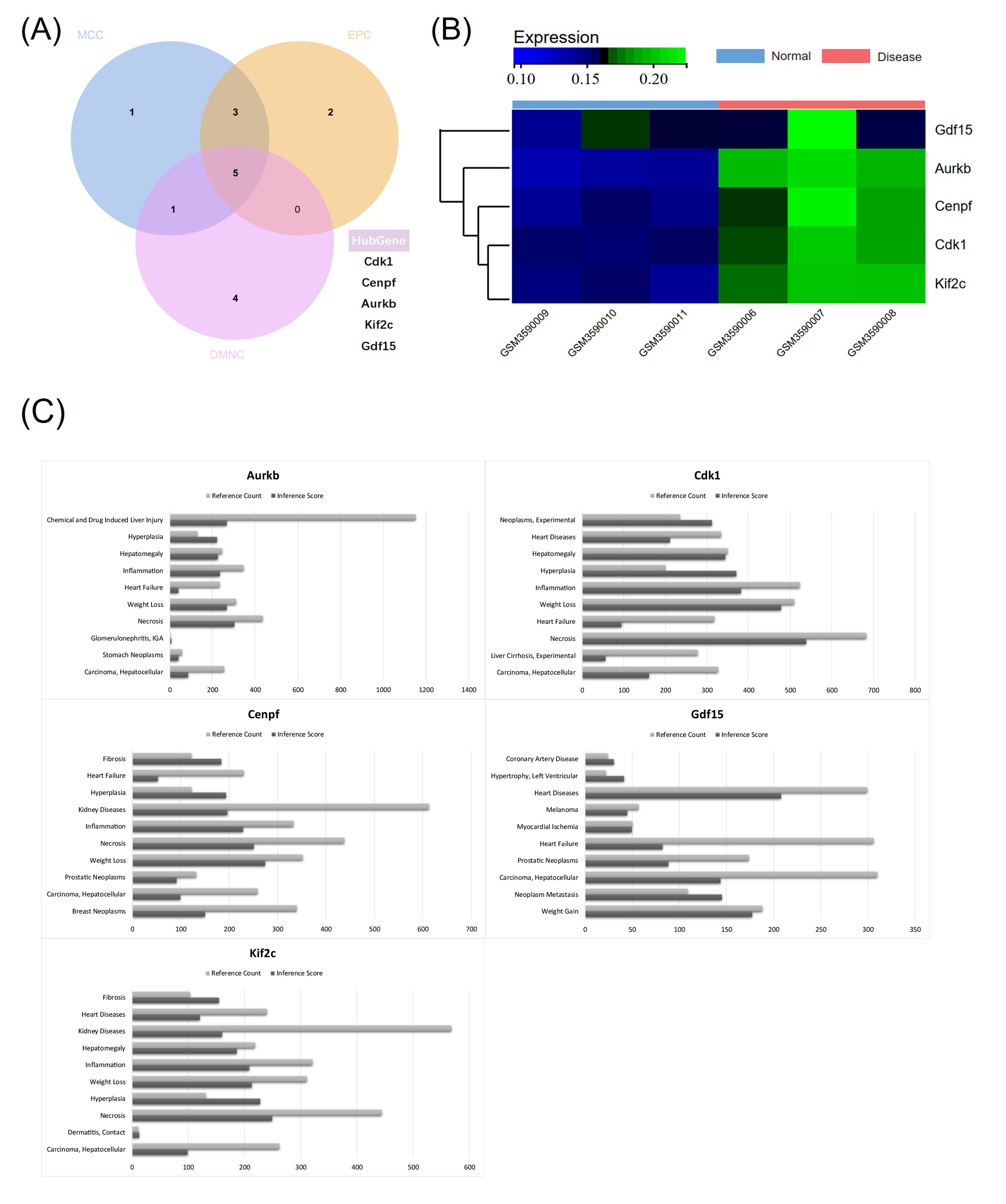 Fig. 1.
Fig. 1.
Network module genes of heart failure with preserved ejection fraction (HFpEF). (A) Identify hub genes. (B) The hub genes were all highly expressed in HFpEF samples and exhibited low expression in normal samples. (C) Reference count and inference score of hub genes in different diseases.
To verify the cardioprotective effects of GDF15 in HFpEF, a model of HFpEF in rats was established, and the expression level of GDF15 was regulated through lentivirus transfection. Compared with the blank control group, the ejection fraction (EF) of the HFpEF group decreased significantly (Fig. 2A), and plasma NT-proBNP levels were significantly elevated (Fig. 2B), confirming the successful establishment of the HFpEF model. In comparison to the HFpEF + MOCK group, GDF15 expression levels in the cardiac tissue of si-GDF15 rats were significantly reduced, indicating the successful establishment of the si-GDF15 rat model (Fig. 2B). Upon the silencing of GDF15, the EF and E/A ratio in HFpEF rats further decreased (Fig. 2A), and plasma NT-proBNP levels dramatically increased (Fig. 2B). These findings suggest that cardiac GDF15 plays a protective role in the cardiac function of HFpEF rats, particularly in preserving diastolic function.
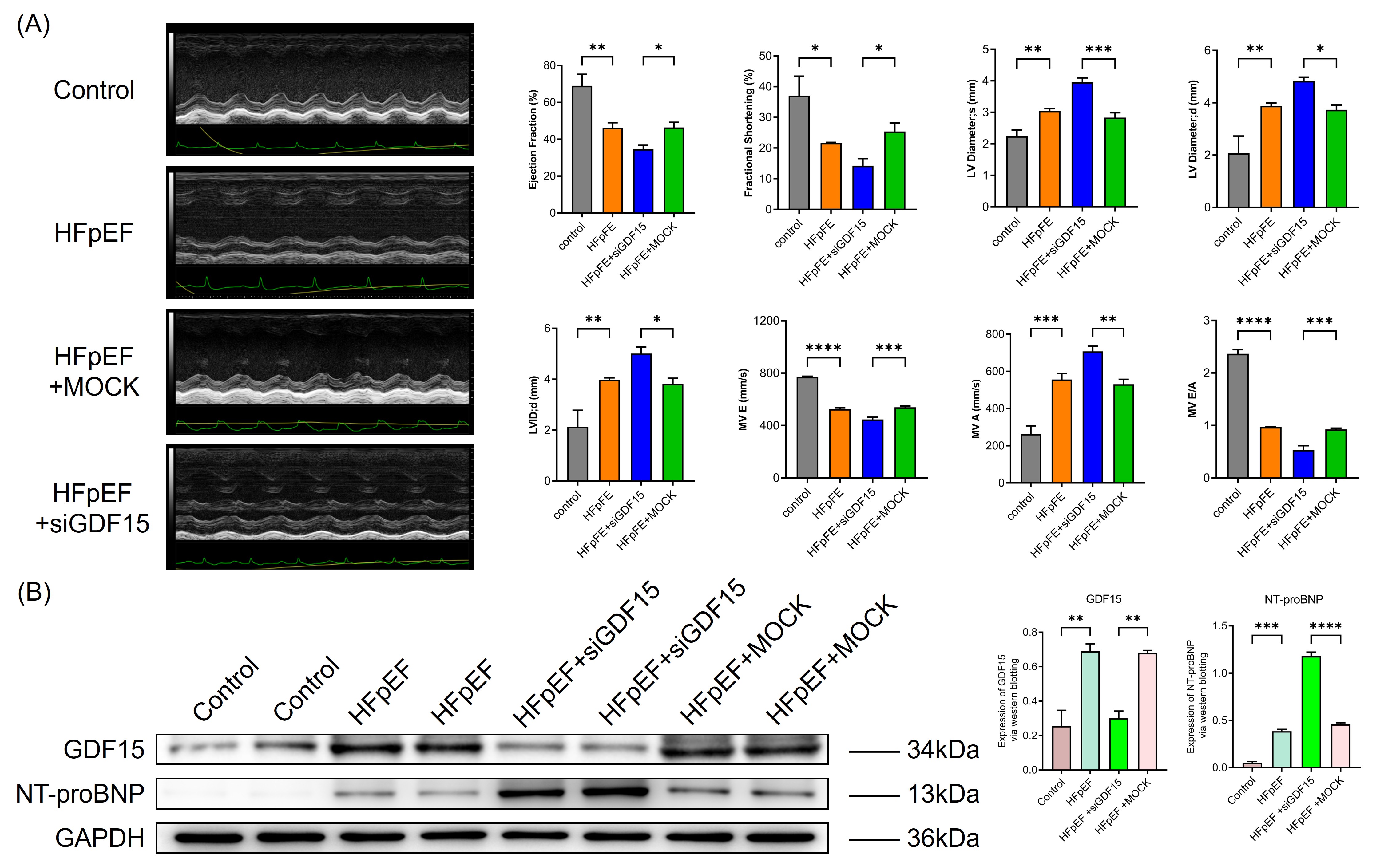 Fig. 2.
Fig. 2.
Growth differentiation factor 15 (GDF15) had cardioprotective
effects in HFpEF. (A) Silencing GDF15 further exacerbates cardiac structural and
functional damage in HFpEF. Typical echocardiogram images are shown in the
figure. Quantified of ejection fraction, fraction shortening, left ventricular
(LV) dimension systole, LV dimension diastole, LV internal dimension diastole
(LVIDd), mitral orifice peak E blood flow velocity (MV E), mitral orifice peak A
blood flow velocity (MV A) and MV E/A are shown in the bar graph. *p
Due to HFpEF, primarily characterized by diastolic dysfunction, cardiac tissue fibrosis is markedly evident, and GDF15 has been reported to possess antifibrotic effects. Therefore, the extent of cardiac fibrosis in rats from each group was assessed. Consistent with previous literature [18, 19, 20, 21], HFpEF rats exhibited significantly increased cardiac fibrosis and elevated expression of fibrosis-related proteins compared with the blank control group (Fig. 3A,B). Rats in the HFpEF + si-GDF15 group demonstrated more pronounced cardiac fibrosis and significantly higher expression of fibrosis-related proteins compared with the HFpEF + MOCK group (Fig. 3A,B). These findings suggest that the improvement in diastolic function observed in HFpEF rats treated with GDF15 is associated with its antifibrotic effects.
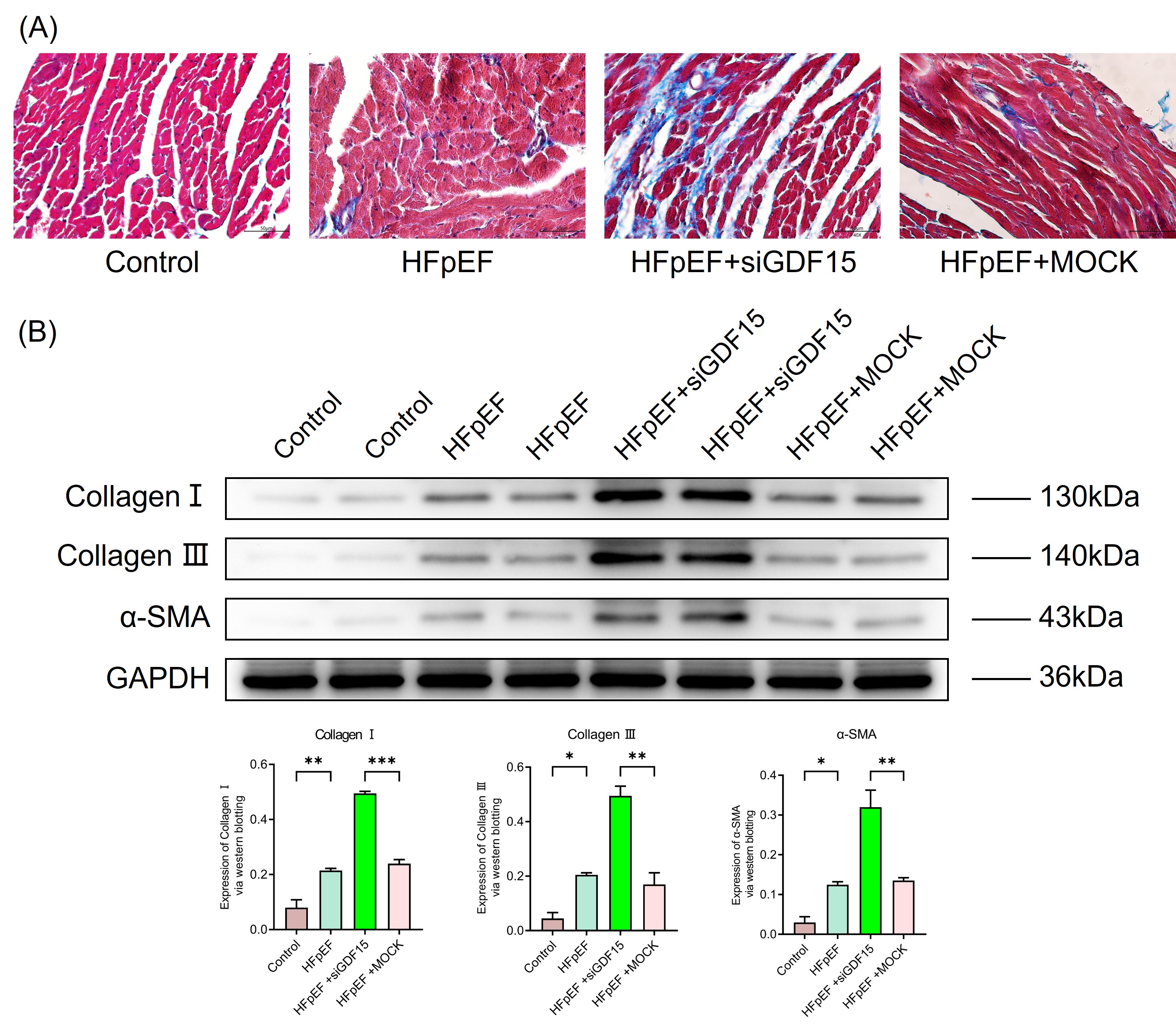 Fig. 3.
Fig. 3.
GDF15 inhibited cardiac fibrosis in HFpEF. (A) Masson’s
trichrome staining for collagen deposition in the cardiac tissue (scale bar: 50
µm). (B) Expression of cardiac fibrosis-associated biomarkers, including
Collagen Ⅰ, Collagen Ⅲ, and
Severe oxidative stress in cardiac tissue has been reported in association with HFpEF, and GDF15 has been shown to possess antioxidant properties. Therefore, we assessed the levels of oxidative stress markers in the cardiac tissue of rats from each group. Similar to the previous findings [22, 23, 24], HFpEF rats exhibited markedly elevated levels of ox-LDL and MPO in their cardiac tissue (Fig. 4). Following the silencing of GDF15 in cardiomyocytes, these oxidative stress markers further increased, indicating that the protective role of GDF15 in HFpEF may be related to its ability to reduce oxidative stress, particularly lipid oxidation.
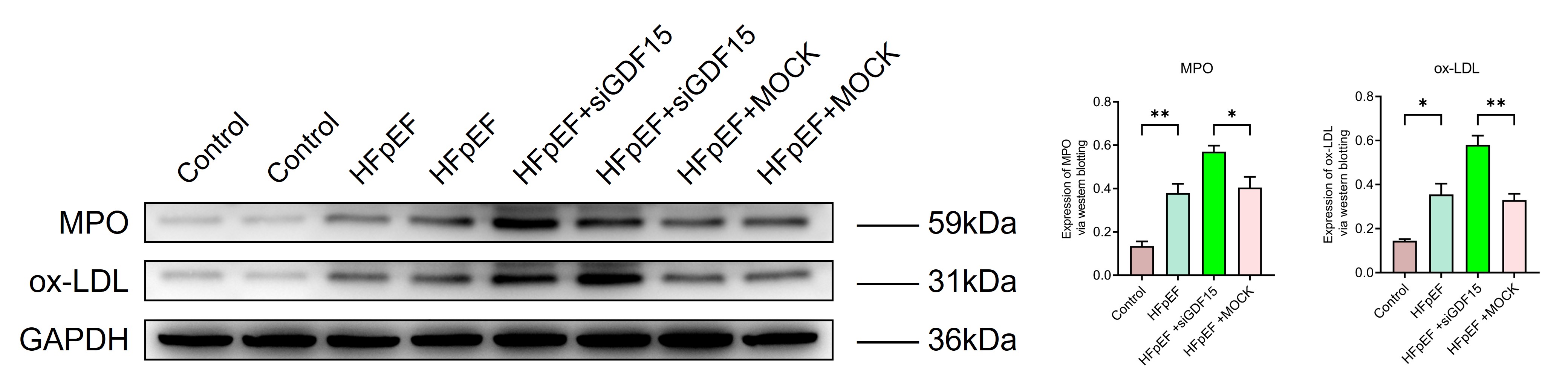 Fig. 4.
Fig. 4.
GDF15 decreased oxidative stress in HFpEF. Expression of
oxidative stress-associated biomarkers, Myeloperoxidase (MPO) and oxidized
low-density lipoprotein (ox-LDL). The original western blot (WB) images are organized in Supplementary Western Blot Raw Image. *p
In further detection of inflammatory cytokines in cardiac tissues revealed a significant increase in IL-1
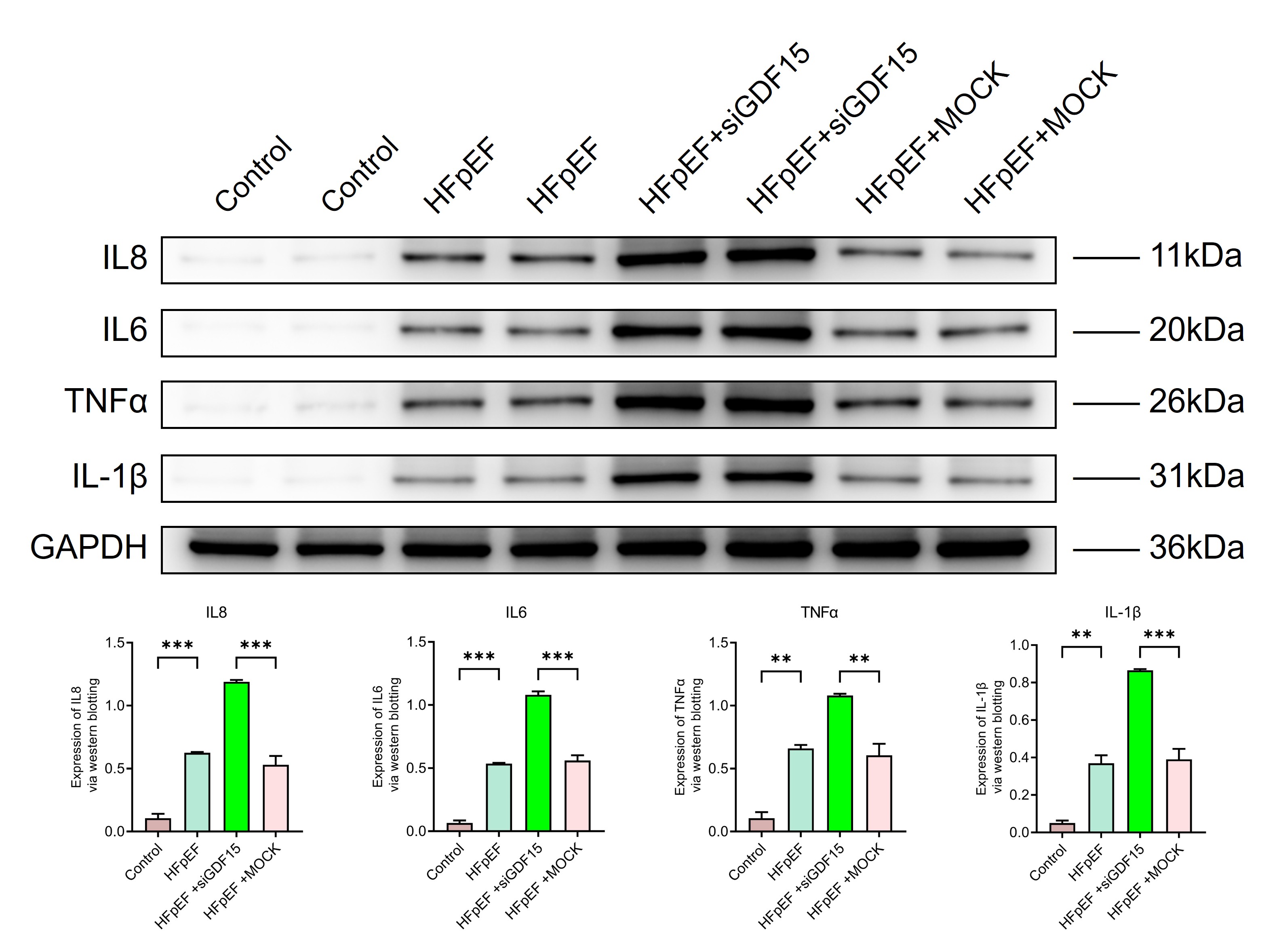 Fig. 5.
Fig. 5.
GDF15 reduces cardiac inflammatory responses in HFpEF.
Expression of
inflammation-associated biomarkers, Interleukin-1 beta (IL-1
To further explore whether the protective effect of GDF15 on HFpEF is associated with its anti-apoptotic function, TUNEL staining was also performed on cardiac tissues from each group of rats (Fig. 6A). The results indicated a significant increase in TUNEL-positive cells in the heart tissue of HFpEF rats compared to the blank control group. This increase was even more pronounced after GDF15 was silenced, suggesting that GDF15 exerts an anti-apoptotic effect in the context of HFpEF. Further WB analysis revealed that apoptotic proteins such as Cleaved Caspase-3 and Bax in the cardiomyocytes of HFpEF rats were significantly upregulated (Fig. 6B), and this upregulation was exaggerated after GDF15 silencing. These findings confirm that the cardioprotective effect of GDF15 is mediated, at least in part, through its anti-apoptotic action.
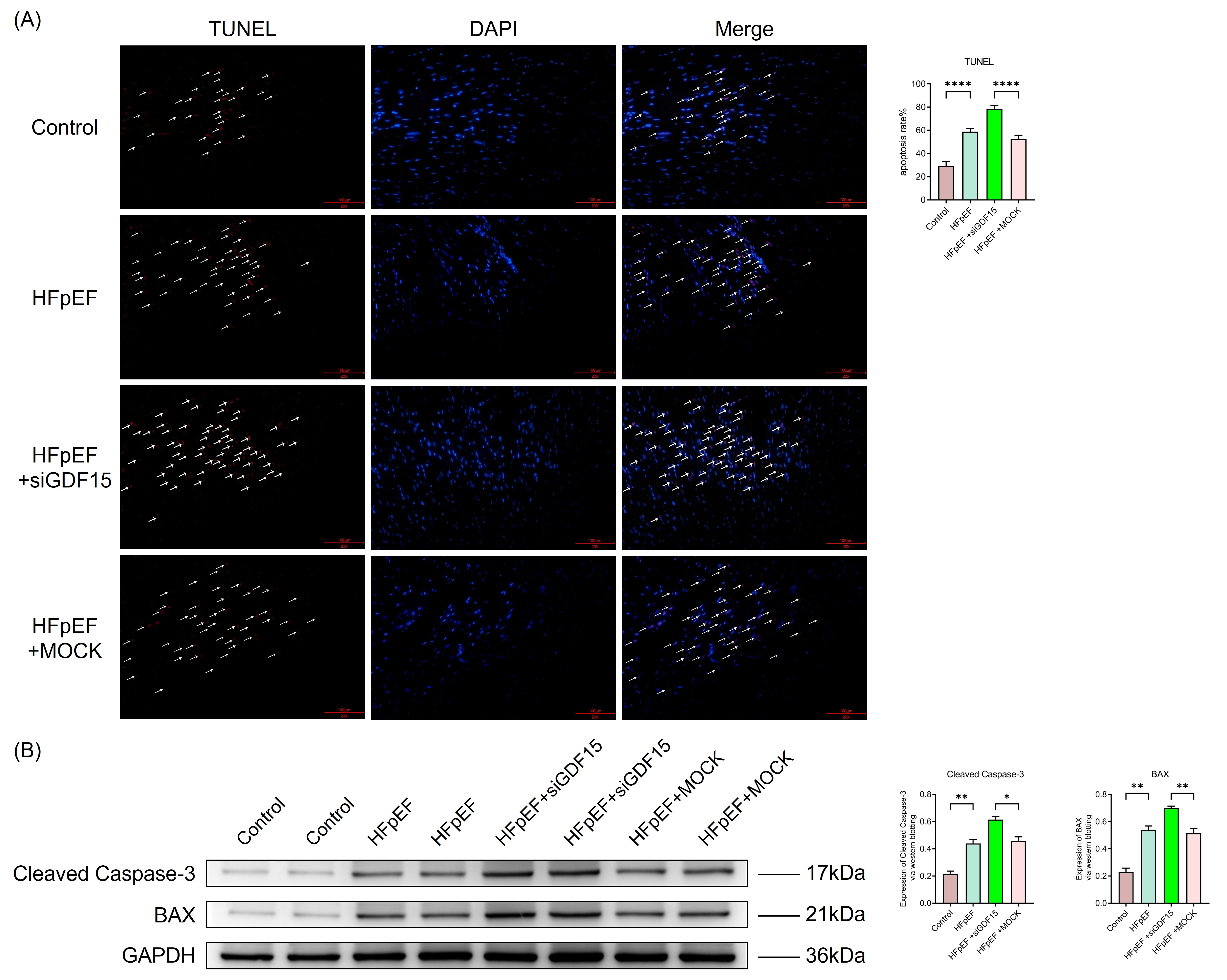 Fig. 6.
Fig. 6.
GDF15 suppresses apoptosis in cardiomyocytes in HFpEF. (A)
Representative photographs of TUNEL staining in heart sections (scale bar: 100
µm). The arrows are used for positive marking. ****p
In this study, we demonstrated that GDF15 was up-regulated and protected cardiac function in HFpEF rats. We also found that the mechanism by which GDF15 preserves cardiac function in HFpEF is related to inhibiting fibrosis, reducing oxidative stress, regulating the inflammatory response, and alleviating cardiomyocyte apoptosis.
Myocardial fibrosis, marked by the presence of perivascular and interstitial
fibrosis, constitutes a crucial pathological hallmark of HFpEF and underlies LV
diastolic dysfunction in patients with this condition [28]. Recent studies have
explored diverse anti-fibrotic therapies for HFpEF, encompassing angiotensin receptor-neprilysin inhibitor (ARNI), sodium-dependent glucose transporters 2 (SGLT-2)
inhibitors, and statins [6, 29, 30]. The cardiac extracellular matrix (ECM) is
predominantly structured by cardiac collagen, which consists mainly of 85%
Collagen I and 11% Collagen III [31]. Myofibroblasts, a distinct subset of
activated fibroblasts characterized by
The onset of HFpEF has been confirmed to be linked to metabolic reprogramming and mitochondrial dysfunction, leading to severe oxidative stress [35]. Consistent with previous finding, our study also observed excessive oxidative stress in patients with HFpEF. This oxidative stress is, in part, a consequence of mitochondrial dysfunction, which induces GDF-15 expression through increased reactive oxygen species (ROS) production [36]. GDF15 is believed to possess antioxidative effects in various cell types, including cardiomyocytes, osteoblasts, and tumor cells [37, 38, 39]. Further supporting its protective role, research by Gao et al. [11] demonstrated that GDF15 can protect cardiac function by inhibiting oxidative stress in a myocardial ischemia-reperfusion injury (MIRI) animal model. In line with these findings, our study revealed that GDF15 significantly reduces inappropriate oxidative stress in cardiomyocytes in HFpEF.
Heart failure has been considered a systemic inflammatory response [40]. Some
studies suggest that abnormal inflammatory responses also contribute to the
pathogenesis of HFpEF [41, 42]. In our study, we observed significant infiltration
of inflammatory cells in the cardiac tissues of HFpEF rats compared to normal
myocardium, and further WB analysis revealed significant upregulation of various
pro-inflammatory cytokines including IL-1
Due to the involvement of various forms of programmed cell death in the
pathogenesis of HFpEF, including cardiomyocyte apoptosis considered as a major
mechanism [46], our study revealed a significant upregulation of cardiomyocyte
apoptosis levels in HFpEF compared to healthy hearts. Research on GDF15’s role in
inhibiting programmed cell death is well-documented. It has been shown to
restrain ferroptosis in cardiomyocytes in a myocardial ischemia-reperfusion
injury rat model [11], protect airways by inhibiting necroptosis in obese
asthmatic mice models [47], and preserve pancreatic
In summary, our research indicates that GDF15 can protect cardiac function in HFpEF through various mechanisms. However, there are corresponding shortcomings that deserve consideration. First, we have only completed experiments after silencing GDF15, and the next step is to conduct rescue experiments after silencing GDF15, as well as experiments involving GDF15 overexpression. Second, we did not obtain human tissue samples for experimental research.
In conclusion, this current study not only confirms the involvement of myocardial fibrosis, oxidative stress, excessive inflammatory responses, and cardiomyocyte apoptosis in the pathogenesis of HFpEF but also demonstrates that GDF15 can protect against HFpEF through multiple mechanisms including inhibition of myocardial fibrosis, oxidative stress, excessive inflammatory responses, and cardiomyocyte apoptosis. This may provide new strategies for the treatment of HFpEF.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
FW, XW and XM designed the research study. YL, LM, CX, and CY performed the research. YL and LM analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were approved by the Animal Experimentation Committee of Beijing Hospital, China, and conducted in accordance with ethical guidelines (protocol number: No. 00001857).
Not applicable.
This study was funded by the National High Level Hospital Clinical Research Funding (No. BJ-2024-180), National Natural Science Foundation of China (No. 82470363), National High-Level Hospital Clinical Research Funding (No. BJ-2023-170), National High Level Hospital Clinical Research Funding (No. BJ-2022-117) and National Key R&D Program of China (grant number 2020YFC2008100).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL26857.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.