

 , Shan Ye 1,2, Tie-Lun Yin 1,2, Shuo Zhang 1,2, Dan-Feng Zheng 4, Jia-Yu Fu 1,2, Guang-Wei Ma 5, Dong-Sheng Fan 1,2,6,*
, Shan Ye 1,2, Tie-Lun Yin 1,2, Shuo Zhang 1,2, Dan-Feng Zheng 4, Jia-Yu Fu 1,2, Guang-Wei Ma 5, Dong-Sheng Fan 1,2,6,*
1 Department of Neurology, Peking University Third Hospital, 100191 Beijing, China
2 Beijing Municipal Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, 100191 Beijing, China
3 Department of Neurology, Xuanwu Hospital of Capital Medical University, 100053 Beijing, China
4 Department of Pathology, Peking University Health Science Center, 100191 Beijing, China
5 Peking University Sixth Hospital, 100083 Beijing, China
6 Key Laboratory for Neuroscience, National Health Commission/Ministry of Education, Peking University, 100871 Beijing, China
Abstract
Amyotrophic lateral sclerosis (ALS) is a rare neurodegenerative disease that mostly presents as sporadic cases. Currently, no mitochondrial-related gene mutations have been identified as the cause of ALS. Mitochondrial gene mutations cause rare hereditary diseases, and the symptoms of pure muscle weakness and muscle atrophy are rarely observed.
We report the case of a young patient clinically diagnosed with ALS concurrently associated with a pathogenic mutation in the mitochondrially encoded nicotinamide adenine dinucleotide: ubiquinone oxidoreductase core subunit 6 (MT-ND6) gene. However, the pathogenic relationship between the MT-ND6 gene and ALS has not been confirmed.
We provide a case report and a literature review aimed at increasing the understanding of the connection between the two. It is essential to consider the potential modifying role of mitochondrial pathogenic genes in ALS.
Keywords
- amyotrophic lateral sclerosis
- muscle weakness
- m.14484T>C
- MT-ND6 gene
- Leber’s hereditary optic neuropathy
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of upper and lower motor neurons. Its typical clinical features include muscle weakness, muscle atrophy, dysarthria, and respiratory failure [1]. The peak age at onset is 58–63 years for sporadic disease and 47–52 years for familial disease [2]. While most ALS patients are classified as having sporadic ALS, up to 10% of ALS patients with a family history have familial ALS, and two-thirds carry ALS-related gene mutations [3].
Here, we report the case of a young female patient with ALS. Using mitochondrial
full-genome analysis, we identified a homoplasmic variation (m.14484T
A 36-year-old Chinese Han woman with a 7-month history of gradually progressive
distal left upper extremity weakness and atrophy as the chief complaint was
admitted to the hospital. She first exhibited weakness in the distal part of her
left hand 7 months prior, followed by progressive atrophy. She later noted muscle
twitches, particularly in the left upper limb. At the time of admission, the
other limbs remained unaffected. Motor examination revealed thenar atrophy in the
left hand, first interosseous muscle, and anterior forearms. The patient denied
any history of exercise intolerance or muscle pain. In her past medical history,
the patient underwent craniotomy surgery for a sellar mass (5
The family history was negative for neurological disorders. The patient had one sister and one brother, as well as a 13-year-old daughter and a 5-year-old son. The mother has two sisters and one brother. All of these individuals were healthy.
Strength testing revealed weakness in both upper limb movements on the Medical Research Council scale (on the left hand: finger abduction/adduction/flexion 3/5, finger extension 4/5, wrist flexion/extension 4/5, elbow flexion 4/5, elbow extension 4/5, and shoulder abduction/adduction/flexion/extension 4/5). On the right hand, her finger abduction/adduction/flexion/extension ratio was 4/5, and the strength of the other muscles was normal. Muscle strength in both lower limbs was normal. All the deep tendon reflexes were present and hyperactive, but there was no ankle clonus. Hoffmann’s and Rossolimo’s signs were present in both hands. Superficial abdominal reflexes and Babinski’s signs were absent. Cognitive, mental, cranial nerve, and sensory examinations were normal.
Routine blood test results were normal. The patient underwent lumbar puncture, and the cerebrospinal fluid (CSF) pressure was within the normal range. CSF analyses revealed normal levels of protein with no cells or oligoclonal bands. Additionally, the patient’s CSF was negative for both the ganglioside antibody spectrum and paraneoplastic syndrome antibodies.
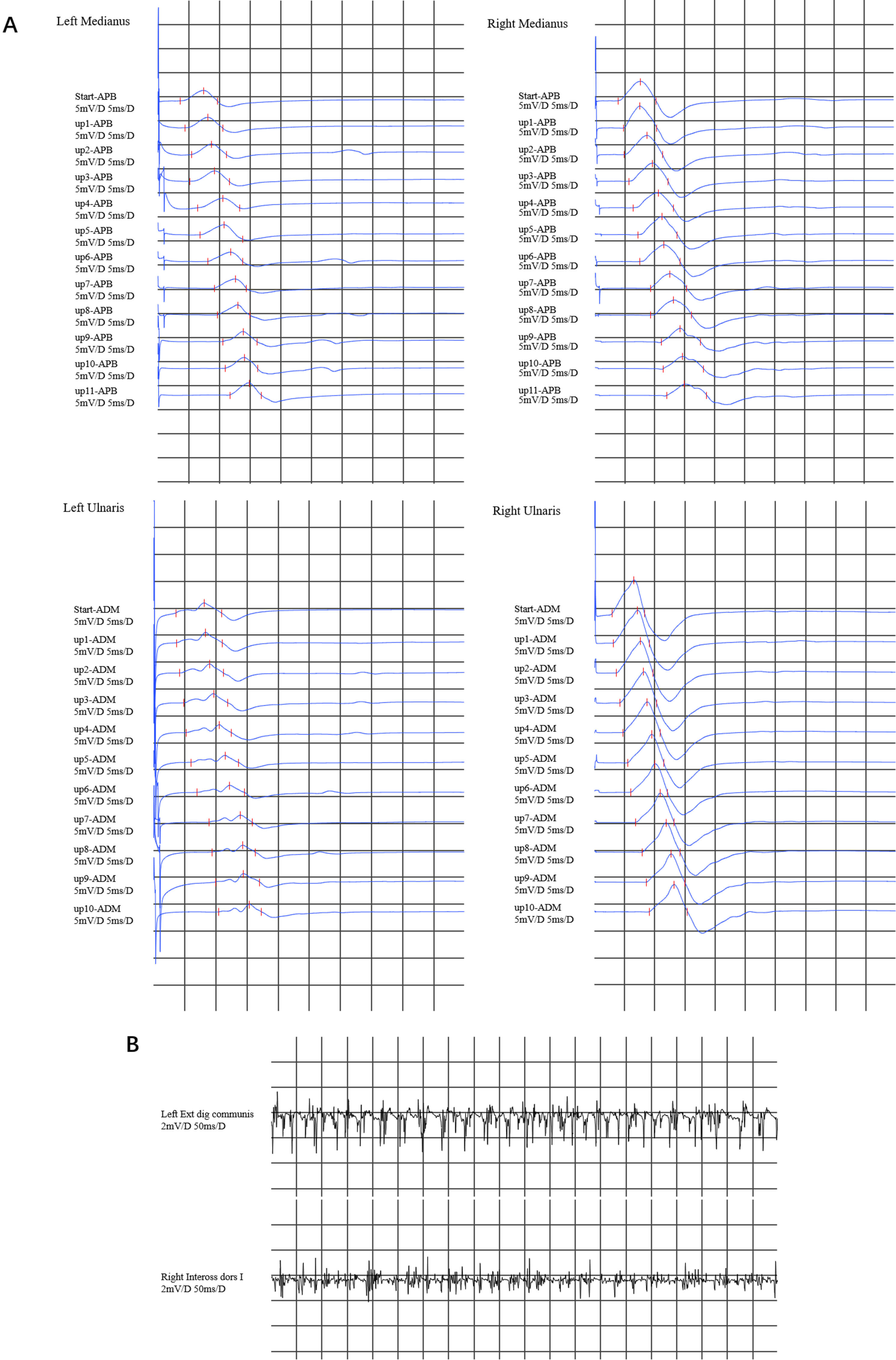
Nerve conduction studies revealed decreased compound muscle action potentials (CMAPs) in the left median nerve, ulnar nerve, and proximal part of the right median nerve, but we did not find conduction blockade during the inching test (Fig. 1A), with normal motor nerve conduction velocities or sensory nerve action potentials. Electromyographic (EMG) evaluation revealed high-amplitude polyphasic motor unit potentials, fibrillation and fasciculation potentials (FPs), and incomplete interference patterns, indicating neurogenic disorders in the upper (left extensor digitorum communis muscle and right first dorsal interosseous) limbs (Fig. 1B). No evidence of neurogenic damage was found in the muscles tested in the bulbar or lower limbs.
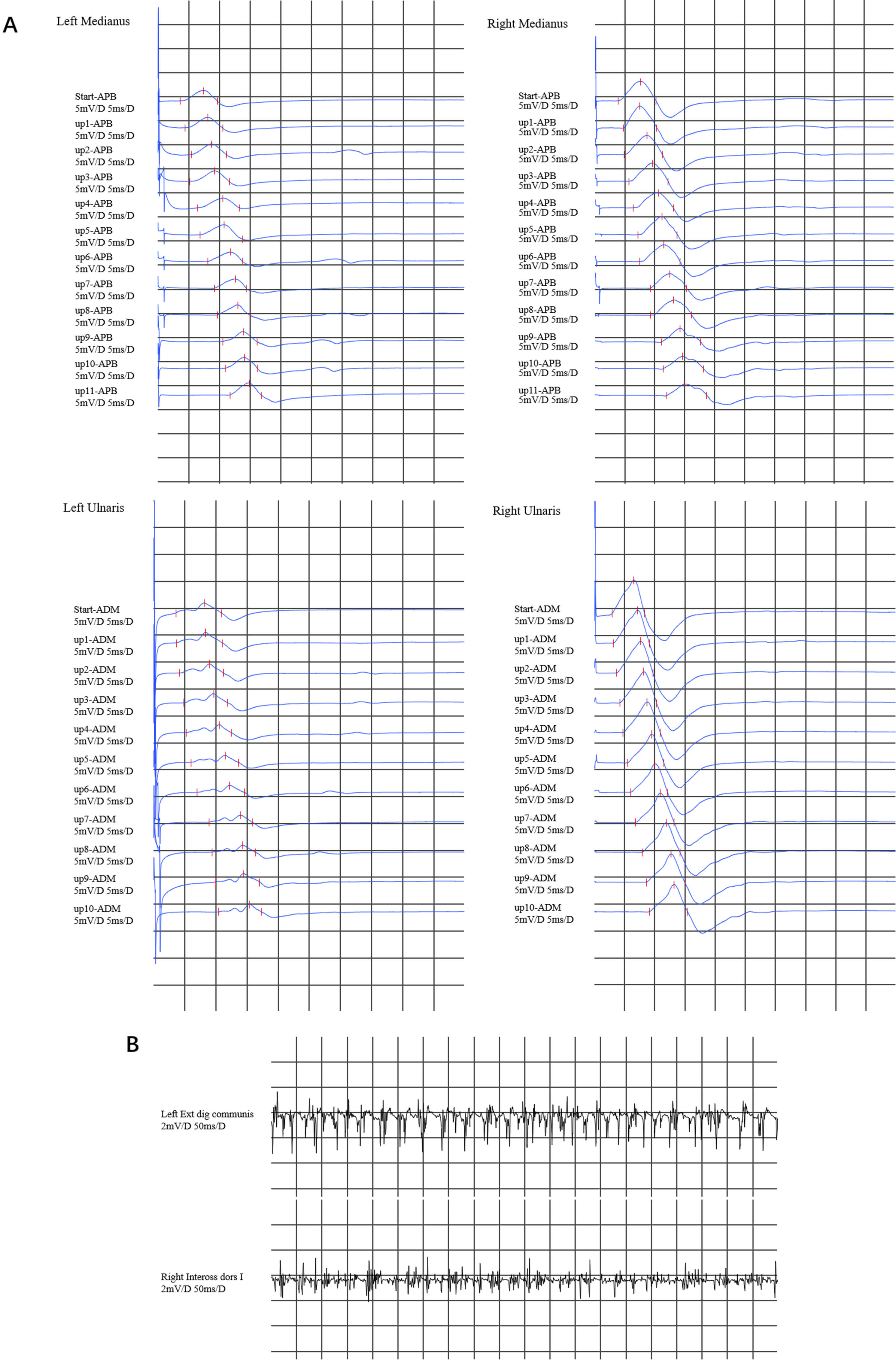 Fig. 1.
Fig. 1.
Electromyography results. (A) In the inching test of both the median nerve and the ulnar nerve, a decrease in compound muscle action potentials (CMAPs) was observed in the left median nerve and the ulnar nerve. The CMAP in the proximal part of the right median nerve decreased, whereas the CMAP in the right ulnar nerve was normal. There was no evidence of conduction block in any of the upper limb nerves. (B) Electromyographic (EMG) revealed that the left extensor digitorum communis muscle and right first dorsal interosseous muscle exhibited a simple mixed phase during vigorous contraction. APB, abductor pollicis brevis; ADM, abductor digiti minimi.
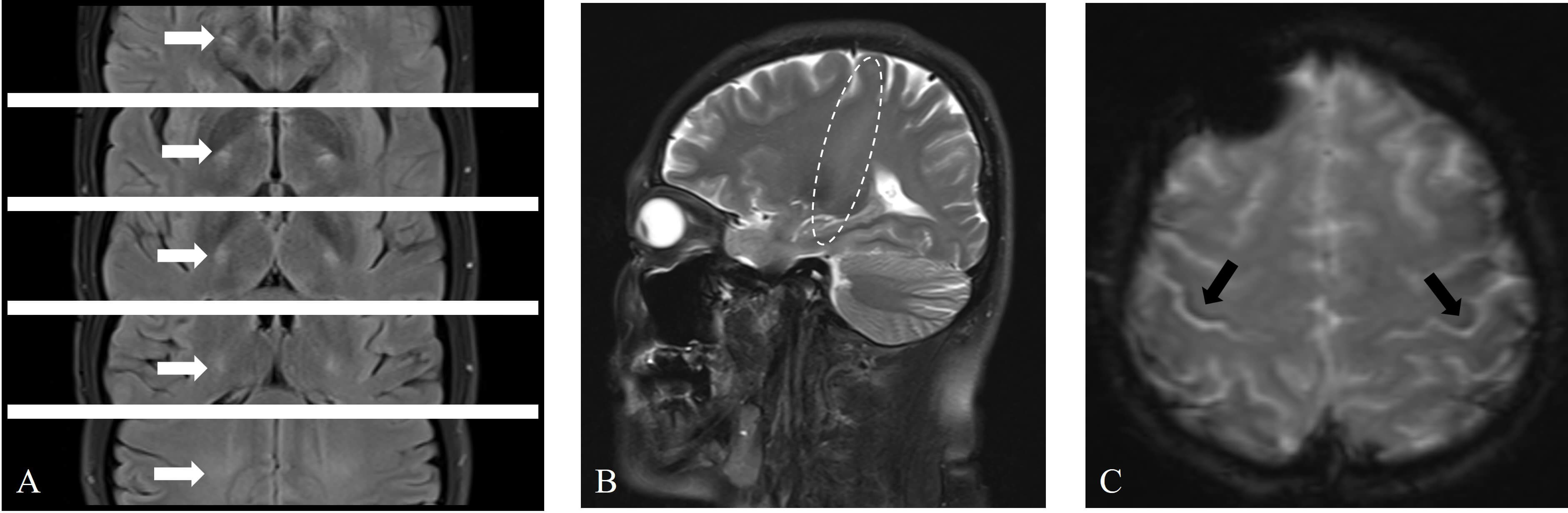
The patient underwent a series of imaging examinations, including brain magnetic resonance imaging (MRI) (Fig. 2), cervical spine MRI, brachial plexus MRI, and bilateral upper limb muscle MRI (Fig. 3A,B). High signal intensity was observed in the corticospinal tract (CST) on T2 and T2 fluid attenuated inversion recovery (FLAIR) sequences (Fig. 2A,B), and hypointensity was observed in the bilateral posterior part of the precentral gyrus on T2-star weighted imaging (T2*) sequences, known as the motor band sign (MBS) (Fig. 2C). MRI of other areas was normal. Additionally, ultrasonic cardiography, 24-hour Holter monitoring, and abdominal ultrasound revealed no significant abnormalities. The pulmonary function test indicated that the forced vital capacity was 113.6% of the predicted value.
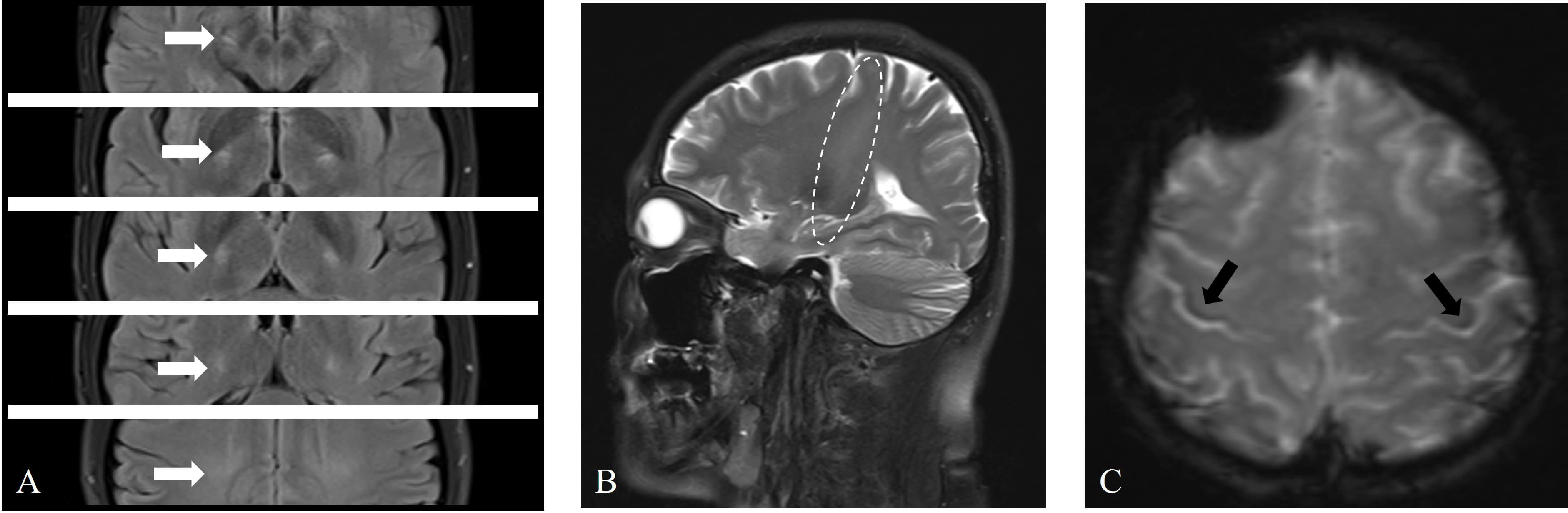 Fig. 2.
Fig. 2.
Brain magnetic resonance imaging (MRI) revealed abnormal signals in the bilateral corticospinal tract (CST) and bilateral precentral gyrus. (A) Axial T2 fluid attenuated inversion recovery (FLAIR) sequences showing symmetrical high signals in the CST bilaterally (white arrows). (B) Sagittal T2 sequences revealing long-segment high signals in the CST (elliptical circle). (C) Axial T2-star weighted imaging (T2*) sequences showing asymmetric curvilinear bands of low signals in the precentral gyrus (right more than left) (black arrows).
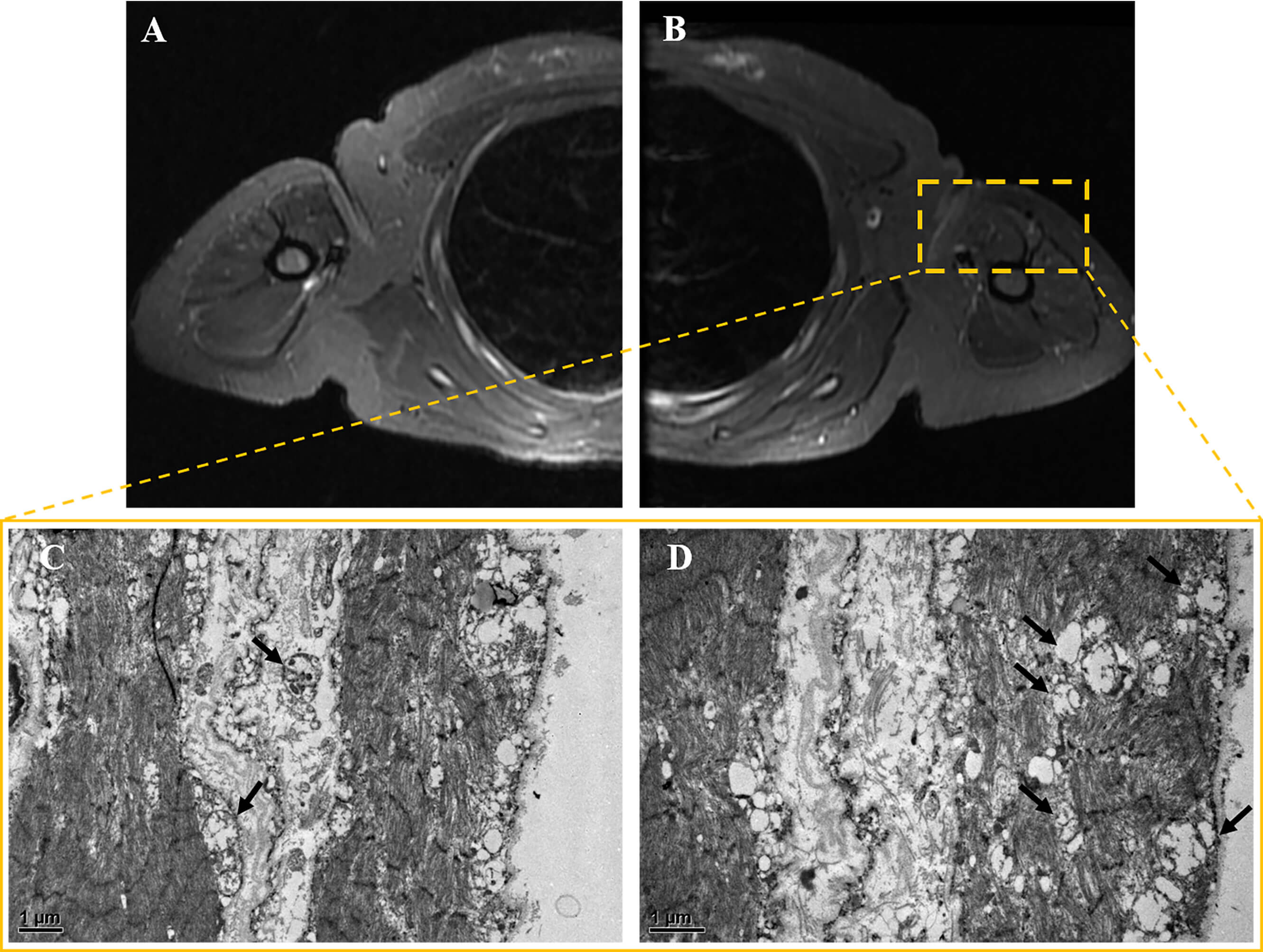
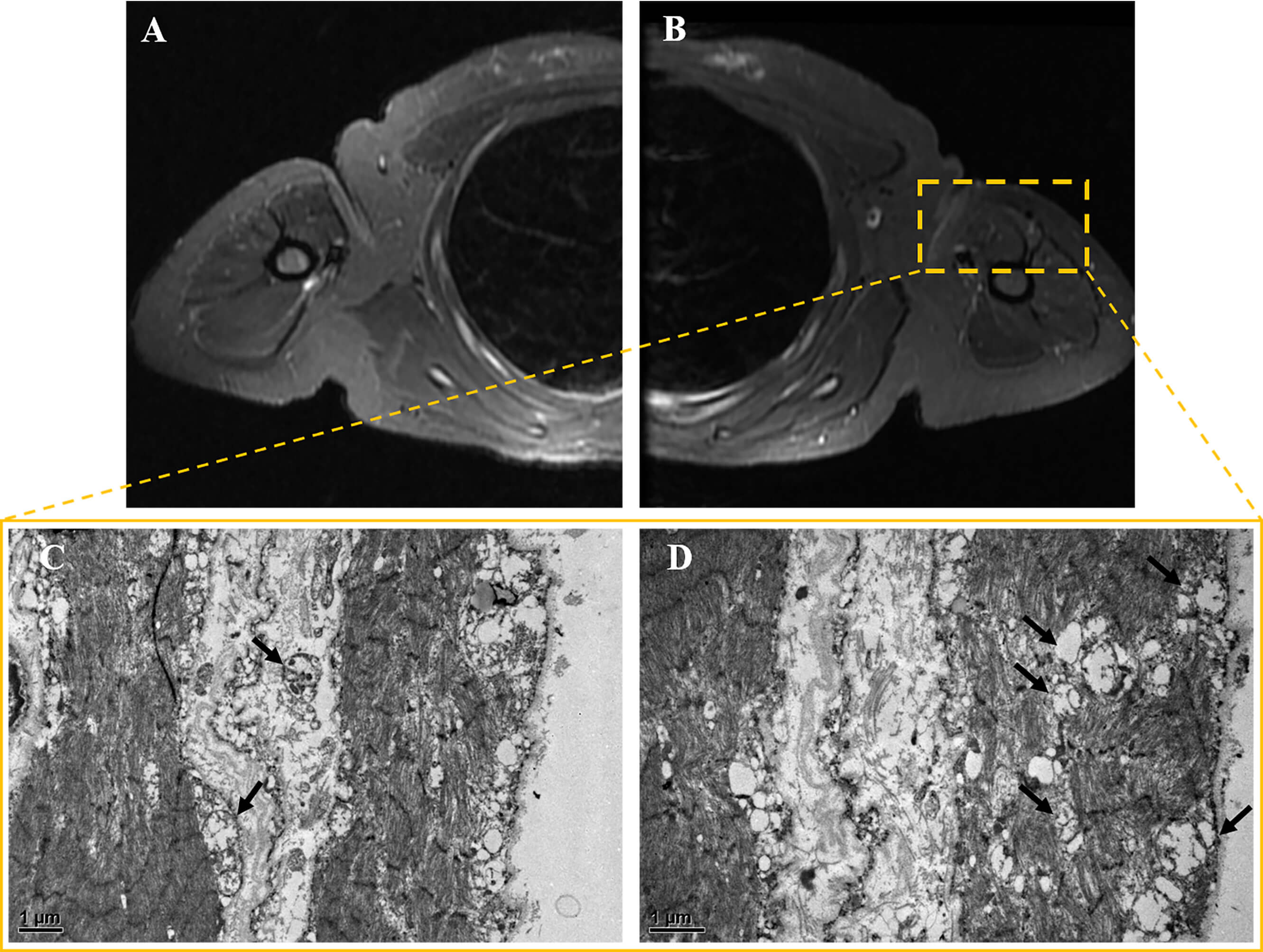 Fig. 3.
Fig. 3.
Muscle MRI and pathology results. (A,B) Bilateral
upper limb muscle MRI. The axial short TI inversion recovery (STIR) sequence
revealed no significant atrophy, hypertrophy, edema, or inflammatory changes in
either upper arm. (C,D) Muscle electron microscopy images. At magnifications of
(C) 12,000
Muscle biopsy was performed on the patient’s left biceps brachii muscle. These pathological findings suggest that approximately 30%–50% of muscle fibers exhibit bundle atrophy accompanied by compensatory hypertrophy. Muscle fiber grouping was observed via NADH staining. Modified Gomori Trichrome (MGT) staining revealed no typical ragged-red fibers (RRFs). Cytochrome oxidase (COX) and succinate dehydrogenase (SDH) staining revealed the deposition of small amounts of subsarcolemmal material in some muscle fibers (see Supplementary Material-2). No definite infiltration of inflammatory cells was observed. Electron microscopy revealed mitochondrial proliferation in some muscle fibers and abundant swollen mitochondria beneath the sarcolemma in a few fibers (Fig. 3C,D). Overall, the pathological diagnosis was consistent with moderate neurogenic muscle atrophy with mitochondrial abnormalities.
Peripheral blood and muscle samples were collected from patients at Peking University Third Hospital. Whole-exome sequencing, mitochondrial gene testing, and multiplex ligation-dependent probe amplification (MLPA) analysis of the peripheral myelin protein 22 (PMP22) gene were performed by Beijing Kangso Medical Inspection Co., Ltd. Additionally, Sanger sequencing of the identified abnormal genes was performed.
Mutational screening was negative for all known pathogenic genes except for
mitochondrial DNA (mtDNA) MT-ND6 (m.14484T
The MT-ND6 gene was screened from her asymptomatic mother and sister (Fig. 4A). The mother presented heteroplasmic variation at the same site, whereas the younger sister presented homoplasmic variation (Fig. 4B). Currently, the patient’s sister has no symptoms of visual impairment or muscle weakness.
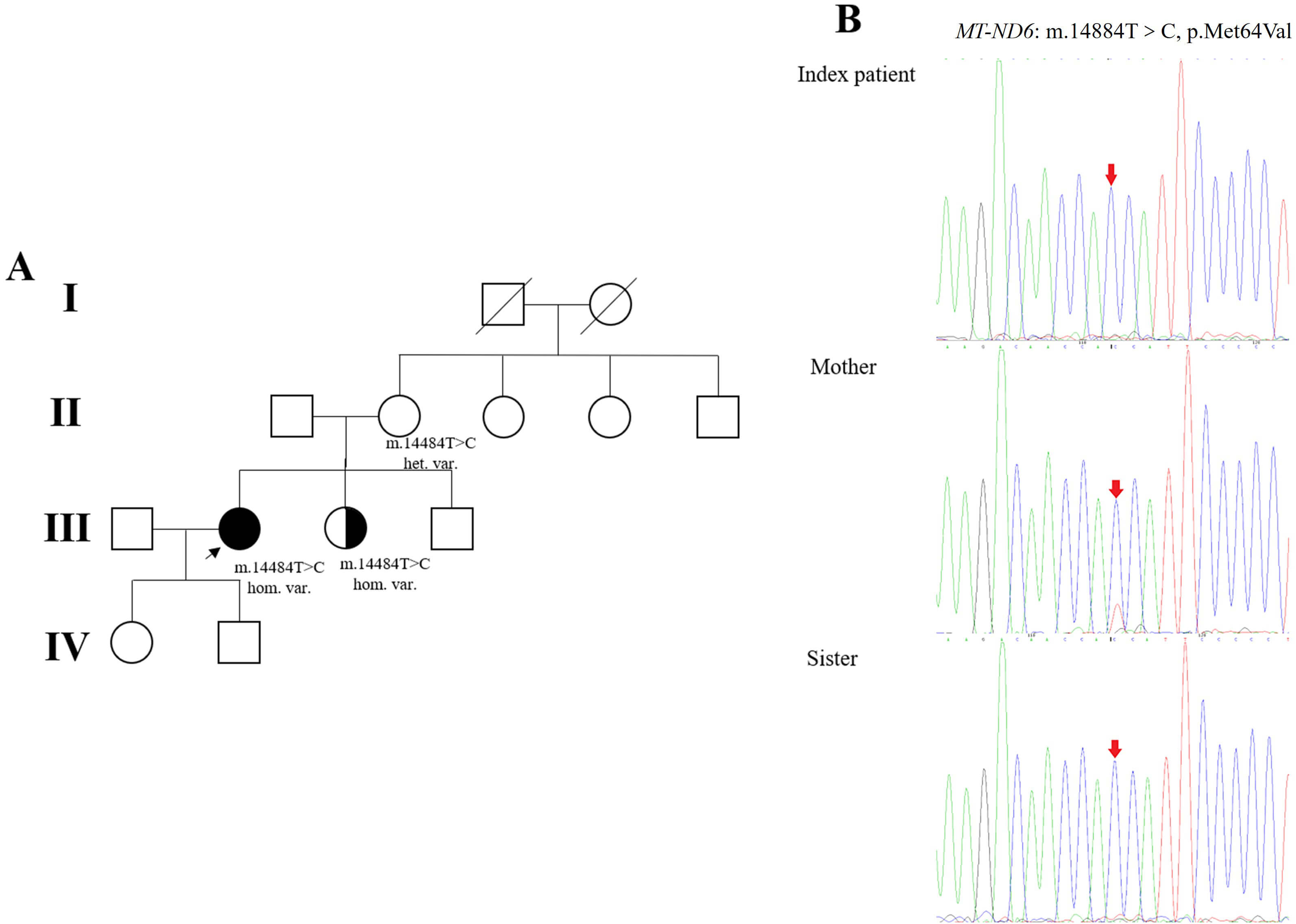 Fig. 4.
Fig. 4.
Pedigree of the patient’s family and genetic analysis. (A) The
proband is indicated by an arrow. Males and females are represented as squares
and circles, respectively. The patients’ parents, siblings, and children were all
healthy. The patient is indicated by black filled symbols, individuals with
asymptomatic homoplasmic m.14484T
The results of the mitochondrial gene analysis of muscle samples also revealed
homoplasmic variation in the MT-ND6 gene at position m.14484T
In addition to the LHON-associated mutation (homoplasmic
m.14484T
The next-generation sequencing (NGS) panel revealed variants of interest in this
patient. She carried the c.5225T
We excluded other mitochondrial syndromes (such as MELAS, Leigh, MERRF-like, and LHON-plus) on the basis of clinical presentation (absence of exercise intolerance, myoclonus, etc.), muscle biopsy (absence of ragged-red fibers), and brain imaging (absence of cortical or white matter lesions and normal structure of the visual pathways). At the time of clinical diagnosis, the young woman presented with pure motor involvement and met the revised El Escorial and Gold Coast criteria [10, 11]; this condition was confirmed as ALS (clinical-instrumental results are shown in Table 1). Additionally, on the basis of the patient’s genetic results along with her history of painlessness and progressive bilateral vision loss, she was confirmed to have LHON disease.
| Items | Available evidence |
| Neurological examination | LMN signs at cervical region, UMN signs at upper and lower limbs |
| EMG | LMN sign in one region (upper limbs) |
| Blood and CSF exams | Unremarkable |
| Clinical diagnosis | Laboratory-supported probable (El Escorial criteria) |
Abbreviations: LMN, lower motor neuron; UMN, upper motor neuron; EMG, electromyography; CSF, cerebral spinal fluid.
We mainly employ cocktail therapy for treating mitochondrial diseases, which primarily utilizes medications for energy and vitamin supplementation, and riluzole is used to treat ALS. Additionally, adequate nutrition and weight maintenance are essential. Regular evaluations to detect manifestations that can occur with time include neurologic deficits, psychiatric abnormalities, impaired respiratory function, and loss of vision.
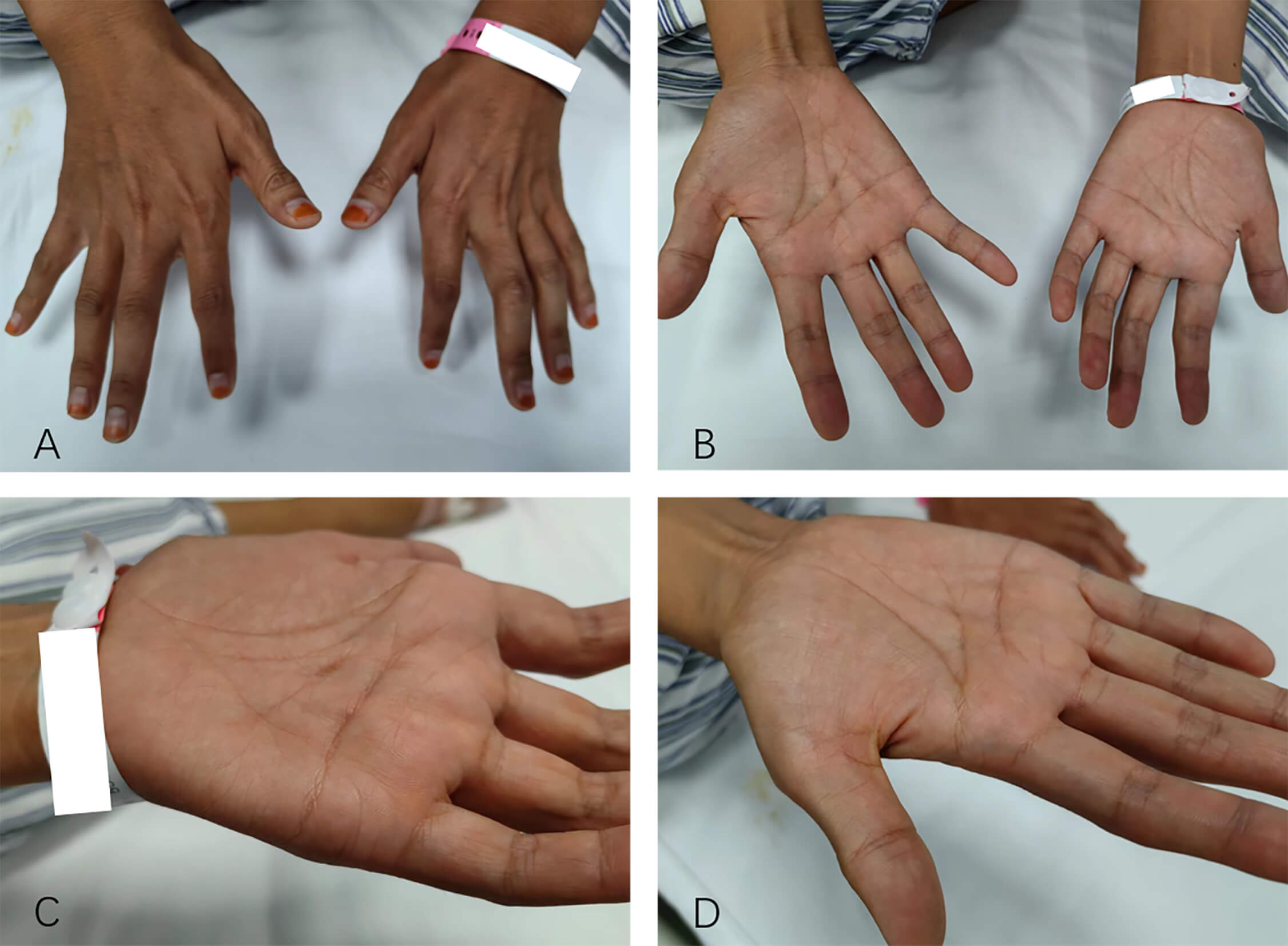
Three months after the follow-up, the patient reported a reduction in muscle fasciculations compared with before, but weakness in the right hand had also emerged. Over time, the weakness and atrophy in both hands gradually worsened (Fig. 5). The patient’s timeline of symptom onset and progression is illustrated in the Supplementary Material-2.
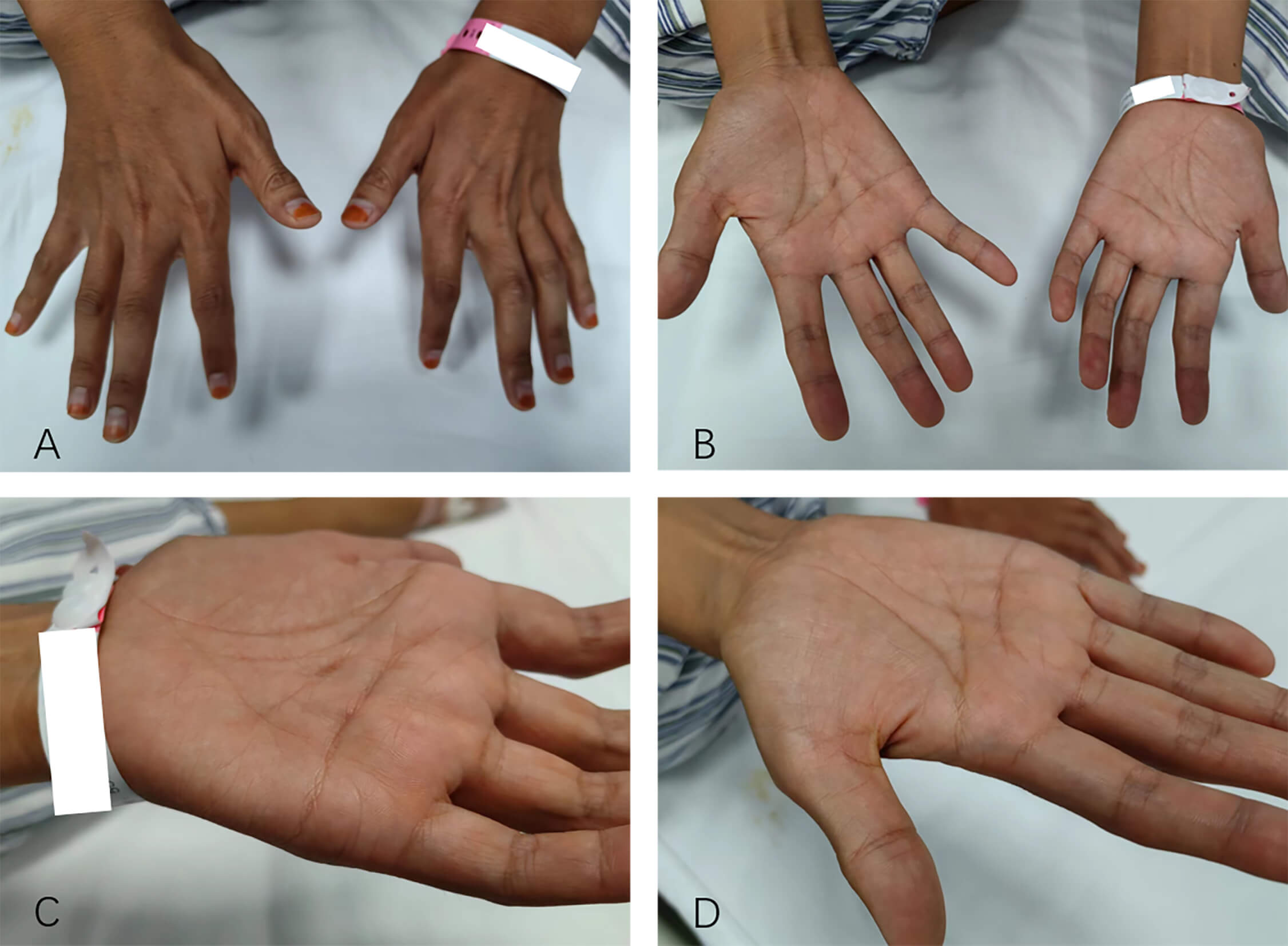 Fig. 5.
Fig. 5.
The patient had atrophy in the thenar muscles of both hands. (A) Back of both hands. (B) Palms of both hands. (C) Palm of the left hand. (D) Palm of the right hand.
Primary mitochondrial diseases are a group of inherited metabolic disorders caused by mutations in mtDNA or nuclear DNA [12]. Diseases associated with mtDNA mutations exhibit significant clinical heterogeneity, impact isolated or multiple organ systems, and can manifest at any age [13]. Furthermore, these diseases do not follow Mendelian inheritance.
Although ALS is generally considered a single disease entity, there are various classifications based on genetic and phenotypic patterns, and it is likely that it is more appropriate to consider this a syndrome of motor neuron degeneration with multiple causes.
In this report, the homoplasmic m.14484T
We tested the patient’s sister and confirmed that she carried the homoplasmic mutation in her mtDNA at the same position. Although she is asymptomatic, the penetrance of optic neuropathy in the Chinese LHON family ranges from 5.6% to 100% [15], and we suggest that this mutation can exhibit incomplete penetrance and variable expressivity. The impact of the mutation may be regulated or modified by other factors, including the influence of other genes (such as nuclear genes) and environmental factors, even mitochondrial haplogroups. These factors determine the level of phenotypic penetrance and the affected tissues, which in turn may determine the inheritance pattern of the disease as well as its onset and progression [16]. In this scenario, we assume that the patient and her sister share similar environmental factors. However, on the basis of nuclear genetic inconsistencies, factors potentially affecting mtDNA stability, energy metabolism, or repair mechanisms, thereby influencing disease manifestation, even though we currently have not identified any pathogenic mutations in the nuclear genes. Some mitochondrial diseases exhibit incomplete penetrance due to the influence of haplogroups. However, a single study of 700 patients and 462 controls in the European population did not reveal any association between mitochondrial haplogroups and ALS, suggesting that mitochondrial DNA haplogroup variations may not be the primary genetic risk factor for ALS [17]. Additionally, previous study has suggested that there is no significant correlation between the level of heteroplasmy associated with primary mtDNA LHON mutations and the severity of the clinical phenotype or the risk of visual loss [15]. These mutations may not have a deleterious synergistic effect.
According to LHON cohort analysis in China, all patients carried the
m.14484T
The MitPhen database (http://www.mitophen.org/) [18] of pathogenic mtDNA genes
and human phenotypic ontologies (HPOs) has been established. Among the 111 mtDNA
mutations, 89 met the pathogenicity criteria (4 insertions and deletions, 85 single nucleotide variants (SNVs)), 40 of which were located in the mtDNA coding region. The total number of
pathogenic mutations covered 26,348 HPOs. In the MitoPhen database 1.7, a total
of 530 MitoPhen patients carrying the m.14484T
| Family id | Sex | Onset age | Variant | Tissue | Variant presence | Heteroplasmy | PMID | Symptoms of motor system | Supported signs | Signs or symptoms of sensory system | Diagnosis |
| 10414484Fa | F | 32 | m.14484T |
Blood | Present | Null | 18344382 | Lower limb muscle weakness; Upper limb muscle weakness | Lower limb hyperreflexia; Hyperreflexia in upper limbs | Null | Harding’s syndrome |
| 1914484Ma | M | 15 | m.14484T |
Blood | Homoplasmic | 100 | 21685233 | Lower limb muscle weakness | Null | Paresthesia; Abnormality of peripheral somatosensory evoked potentials | Harding’s syndrome |
| 2114484Ma | M | 6 | m.14484T |
Blood | Homoplasmic | 100 | 29249004 | Proximal muscle weakness in lower limbs | Areflexia of lower limbs | Distal sensory impairment | Leber hereditary optic neuropathy and longitudinally extensive transverse myelitis |
| 2214484Fa | F | 64 | m.14484T |
NG | Present | Null | 21734595 | Lower limb muscle weakness | EMG: neuropathic changes | Sensory axonal neuropathy | Leber hereditary optic neuropathy |
| 2414484Ma | M | 33 | m.14484T |
Blood | Homoplasmic | 100 | 27486939 | Proximal muscle weakness in lower limbs | Lower limb hyperreflexia | Impaired vibration sensation in the lower limbs; Impaired distal tactile sensation; Abnormality of peripheral somatosensory evoked potentials | Leber’s ‘Plus’ |
| 3214484Ma | F | 15 | m.14484T |
Blood | Homoplasmic | 100 | 8470982 | Distal muscle weakness | Null | Paresthesia | Leber hereditary optic neuropathy |
| 3214484Ma | M | 17 | m.14484T |
Blood | Homoplasmic | 100 | 8470982 | Distal muscle weakness | Null | Paresthesia | Leber hereditary optic neuropathy |
| 3314484Ma | M | 33 | m.14484T |
Blood | Homoplasmic | 100 | 8470982 | Distal muscle weakness | Null | Paresthesia | Leber hereditary optic neuropathy |
| 4714484Fa | F | 21 | m.14484T |
Blood | Homoplasmic | 100 | 8470982 | Distal muscle weakness | Null | Paresthesia | Leber hereditary optic neuropathy |
| 9414484Ma | M | 36 | m.14484T |
Blood | Present | Null | 11450909 | Lower limb muscle weakness | Null | Back pain | Leber hereditary optic neuropathy |
| 9814484Fa | F | 18 | m.14484T |
Blood | Homoplasmic | 100 | 15483043 | Lower limb muscle weakness | Babinski sign | Paraparesis; Episodic pain | White matter disease in Leber’s hereditary optic neuropathy |
Abbreviations: F, female; M, male; NG, not given; PMID, PubMed unique identifier.
Recently, a large study on ALS pointed to the burden of multiple risk factors identified in the nuclear genome, but the impact of mtDNA variation was not considered [20]. We did not find any mtDNA-related information associated with ALS in the ALS Online Database (https://alsod.ac.uk/). In our case, the patient presented with young-onset ALS in the context of a confirmed diagnosis of mtDNA-related disorder. Although we cannot definitively confirm that this mtDNA site is the causative gene for ALS, it is worth considering that the mutation at this gene site may contribute to the early onset of ALS and confer genetic risk.
Frameshift mutations in genes encoding mitochondrial respiratory chain complex I have previously been reported to occur in individuals with ALS, but such mutations are rare [21]. Mutations in the nuclear genecoiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10) are pathogenic mutations in ALS, and CHCHD10 is a mitochondrial protein located in the intermembrane space. This gene mainly causes mtDNA instability disorders through the accumulation of multiple mtDNA deletions, but these mutations are mainly responsible for the clinical spectrum of frontotemporal dementia (FTD)-ALS [22]. In addition, other rarer mutations that affect mtDNA instability, such as DNA polymerase subunit gamma-1 (POLG), thymidine kinase 2 (TK2) or deoxyguanosine kinase (DGUOK), can cause ALS-like symptoms [23, 24, 25]. This evidence suggests that mitochondrial diseases may be the origin of some phenotypes of ALS, opening a new field in which to explore the pathogenesis of the clinical spectrum of ALS.
Research on ALS patients has revealed the following factors: the accumulation of mitochondria in proximal axons, mitochondrial injury caused by excessive reactive oxygen species (ROS), COX I mtDNA mutation, and RRF. These factors act mainly through increased ROS and altered mitochondrial structure [16]. mtDNA deletions are more common in individuals with sporadic ALS than in healthy controls [26]. In sporadic ALS patients, the presence of COX-negative muscle fibers in skeletal muscles is common, but no correlation has been found between the severity of oxidative defects and patient age or disease duration [27, 28].
ALS as a type of neuromuscular disorders (NMDs). There is evidence that any defects at the mitochondrial level could jeopardize the function of cells and tissues, forming the basis of NMDs [16]. Interestingly, mitochondria can also play a secondary role in the development of the remaining NMDs when the mutation or deficiency is not directly related or located in the mitochondria, since affected cells need additional adenosine triphosphate (ATP) to support homeostatic mechanism imbalance (antistress or antioxidant responses) while minimizing the production of ROS. If mitochondria are unable to counterbalance cell dysfunction, a secondary mitochondrial disease, such as ALS caused by a mutation in trans-activation response DNA-binding protein 43 (TDP-43) [29], can also occur in spinal muscular atrophy (usually caused by a mutation in the coding sequence of survival of motor neuron 1) [30]. Therefore, disregarding genetic origins, mitochondrial function is key in the onset or progression of most NMDs.
From a genetic variation perspective, impaired Complex I function increases electron leakage during the electron transfer process, leading to elevated production of ROS, which constitute one of the pathogenic factors in ALS. Additionally, mitochondrial dysfunction disrupts mitochondrial dynamics (including fission, fusion, and transport), thereby impairing axonal transport [31]. For example, in drosophila models [32], loss of mitochondrial Complex I causes mitochondrial transport defects characterized by drastically reduced velocity and flux of mitochondrial movement within axons.
However, research on the relationship between the m.14484T
In conclusion, we report the case of an ALS patient with concurrent LHON
disease. Her m.14484T
Study data are available from the corresponding author upon request.
JYW, SY, TLY, GWM, JYF, DFZ, and SZ contributed to the data acquisition. JYW and SY drafted the manuscript. DSF interpreted the data for the work and reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All the authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the Medical Science Research Ethics Committee of Peking University Third Hospital (approval number: M2017198) and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from the participant.
Not applicable.
Supported by the National Natural Sciences Foundation of China, No. 81873784, 82071426, and the Clinical Cohort Construction Program of Peking University Third Hospital, No. BYSYDL2019002.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/RN44110.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.